1. Introduction
Lung cancer is a noteworthy cause of cancer-related fatalities across the globe, with non-small-cell lung cancer accounting for 84% of cases and small-cell lung cancer comprising 13% of cases, distinguished by their size and type [
1]. The pathological features and treatment protocols of these two types of cancer vary significantly. In particular, advanced-stage non-small-cell lung cancer is usually treated by chemotherapy or immunotherapy [
2].
Cell death is categorized as regulated cell death (RCD) and accidental cell death (ACD), with RCD being able to regulate cell death through specific regulatory molecules. In recent years, various RCD pathways have been elucidated, including programmed cell death (PCD), intrinsic apoptosis, necroptosis, pyroptosis, NETosis, parthanatos, mitochondrial permeability transition (MPT)-regulated necrosis, and ferroptosis, through extensive studies spanning the last 10–20 years [
3,
4]. Ferroptosis is a type of cell death that depends on ferrous (Fe
2+) ions, and one of its key features is the buildup of excessive lipid peroxidation in the cell membrane. The process of lipid peroxidation can be initiated by either an enzymatic reaction catalyzed by lipoxygenase (LOX) or a non-enzymatic reaction. Once the lipid peroxide is produced, it can react with Fe
2+, oxygen, or surrounding unsaturated fatty acids, leading to the continuous generation of radicals that spread to surrounding areas and trigger cell death [
5]. Although unsaturated fatty acids in the cell membrane are constantly oxidized, even under normal conditions, glutathione peroxidase 4 (GPX4) works to prevent the accumulation of lipid peroxide and thus protect cells from ferroptosis. GPX4 is an enzyme that catalyzes the oxidation of 2GSH to produce GSSH while also reducing lipid peroxide to produce lipid alcohol. There are two major groups of drugs that induce ferroptosis. Class I FINs (Ferroptosis Inducing agents) include Erastin, Sulfasalazine, and Sorafenib, which block system xc
−. System xc
− is a cystine–glutamate antiporter that imports the amino acid cystine into cells. Treatment with class I FINs induces ferroptosis by causing cysteine and glutathione (GSH) deficiency in cells. Class II FINs, such as RSL3, ML210, and ML162, inhibit GPX4 and also induce ferroptosis through the accumulation of lipid peroxidation [
6,
7]. Ferroptosis is a newly recognized form of immunogenic cell death that triggers secondary apoptosis by releasing damage-associated molecular patterns (DAMPs) from the cell [
8,
9]. When cells undergo ferroptosis, they secrete HMGB1, a type of DAMP that activates a pattern recognition receptor (PRR)/NF-κB pathway, leading to an inflammatory response in peripheral macrophages [
10,
11].
The formation of a permeability transition pore complex (PTPC) between the inner and outer mitochondrial membranes is the cause of mitochondrial permeability transition (MPT)-driven necrosis due to changes in the intracellular microenvironment, such as oxidative stress or excess Ca
2+ [
12,
13]. The composition, regulation, and precise mechanism of PTPC are still unclear and controversial. The only known protein required to induce MPT-driven necrosis in vivo is peptidylprolyl isomerase F (PPIF; cyclophilin D) [
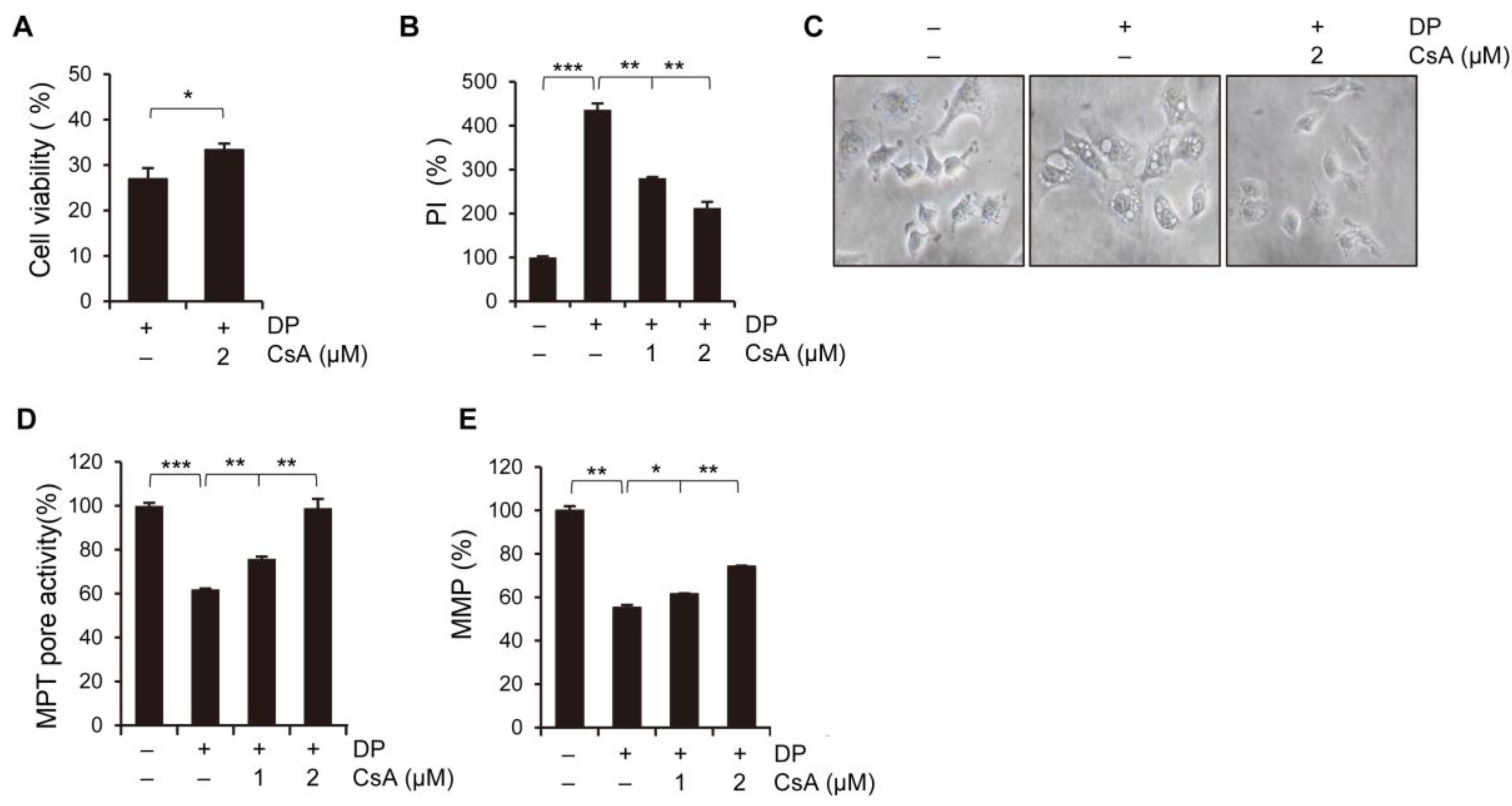
14]. Cyclosporin A, an inhibitor of cyclophilin D, can inhibit the development of MPT-driven necrosis in diseased animals with cardiac and cerebral ischemia [
15]. Based on these findings, we hypothesize that ferroptosis induces MPT-driven necrosis, which results in an increase in HMGB1 secretion in lung epithelial A549 cells, ultimately leading to greater cytotoxicity.
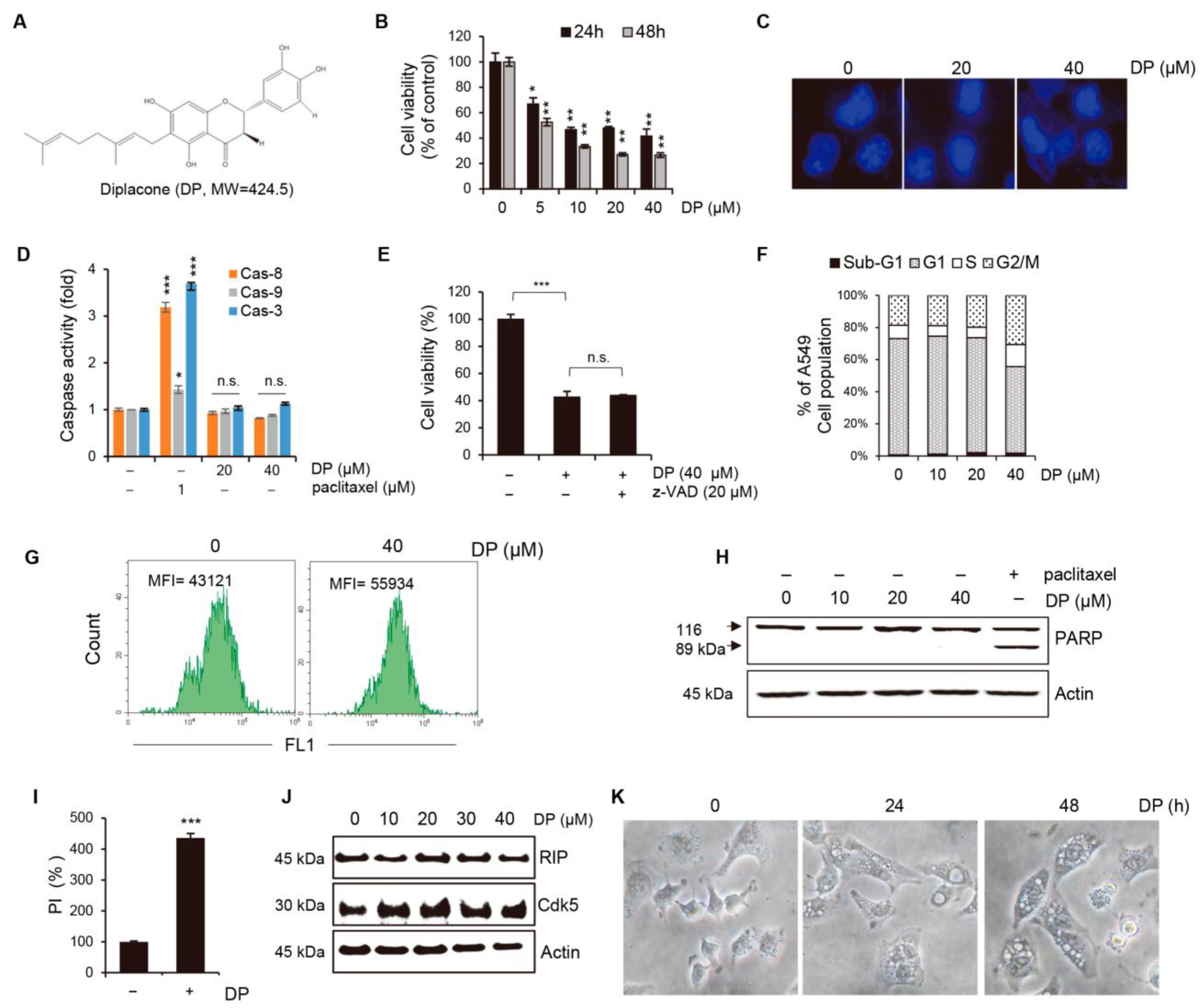
In the past, the fruit of
Paulownia tomentosa (Thunb.) Steud. (Paulowniaceae) has been used in Asia as a traditional remedy for various diseases, such as tonsillitis, bronchitis, asthmatic attacks, enteritis, and dysentery [
16]. In a previous study by our colleagues, diplacone (DP) was isolated from methanol extracts of dried
P. tomentosa fruits, along with 18 other dihydroflavonols and flavanone derivatives [
16]. DP has been reported to have anti-inflammatory [
17], antioxidant [
18], anticancer [
19], and antibacterial properties [
20,
21]. DP was found to have an antiproliferative effect on human ovarian cancer cell line A2780 [
22], but the cellular mechanism of the action of DP has not yet been fully elucidated. In this study, we observed that DP treatment of A549 human non-small lung carcinoma cells led to the formation of extensive cytoplasmic vacuoles and decreased cell growth, prompting us to investigate the underlying cellular mechanisms by which DP induces cancer cell death and causes vacuole formation in A549 cells.
3. Discussion
DP is a natural compound that is found in the fruit of the
P. tomentosa tree, also known as the empress tree. It is a type of flavanone, a class of flavonoids that are commonly found in plants and have been associated with various health benefits. DP has been studied for its potential anti-cancer properties, as it has been shown to inhibit the growth and proliferation of various types of cancer cells. It is believed to exert its anti-cancer effects by inhibiting the activity of the ERBB family of receptors, which are commonly overexpressed in many types of cancer [
30]. However, more research is required to comprehend the mechanisms of the action and potential therapeutic applications of DP.
First, we measured cell viability for 19 compounds obtained from the fruit of
P. tomentosa using A549 cells (
Supplementary Figure S3). A strong anticancer effect was observed with increasing numbers of compounds, indicating the presence of geranyl and hydroxyl groups. Hydroxyl groups possess binding affinity with the cellular plasma membrane, resulting in enhanced cell death. Moreover, vacuolation could be observed only in the cases of compounds 13, 14, 16, and 18 (
Supplementary Figure S4). To determine the difference in cytotoxicity according to the presence or absence of geranyl groups, we measured cytotoxicity using taxifolin (5,7,3′,4′-flavan-on-ol, also known as dihydroquercetin) treatment, in which the geranyl group was excluded from the DP structure. As a result of the experiment, vacuole formation and cytotoxicity could not be observed under the taxifolin treatment conditions (
Supplementary Figure S5).
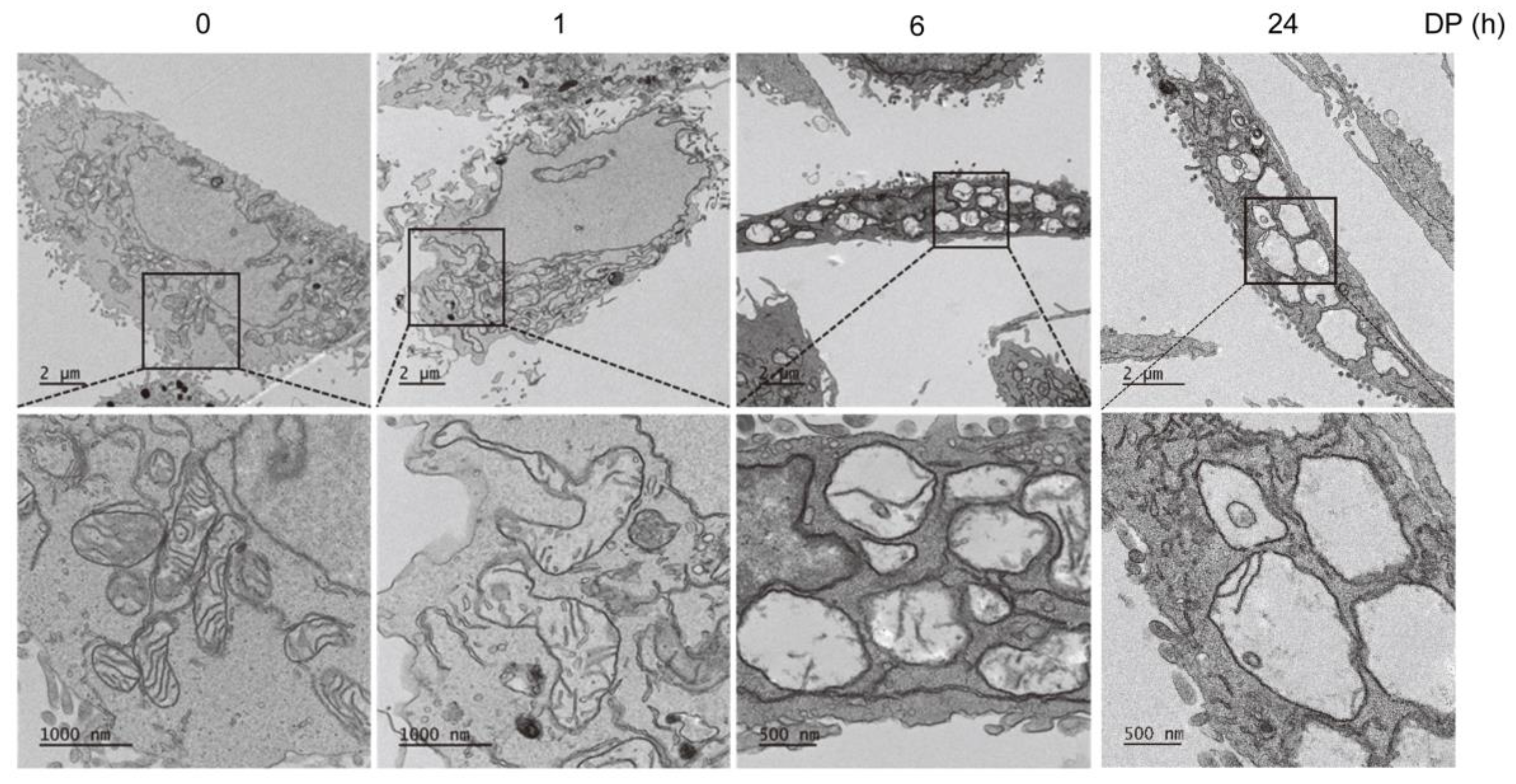
Programmed non-apoptotic cell death encompasses various mechanisms and phenotypes, including cell death with vacuoles (autophagy, entosis, methuosis, and paraptosis), cell death dependent on mitochondria (mitoptosis and parthanatos), iron-dependent cell death (ferroptosis), cell death with immune reactions (pyroptosis and NETosis), and other types, such as necroptosis [
31]. In this study, we aimed to investigate the involvement of MPT-mediated cell death and ferroptosis in the formation of vacuoles in A549 lung epithelial cells. MPT-mediated cell death is characterized by rapid increases in inner mitochondrial membrane permeability, and it involves the cyclophilin D-dependent opening of MPT pores. Although MPT-mediated cell death has been described in the context of neuronal excitotoxicity and ischemia-reperfusion injury (IRI), it has not been extensively studied. MPT-mediated cell death is characterized by cell swelling, the induction of an inflammatory response, increased intracellular Ca
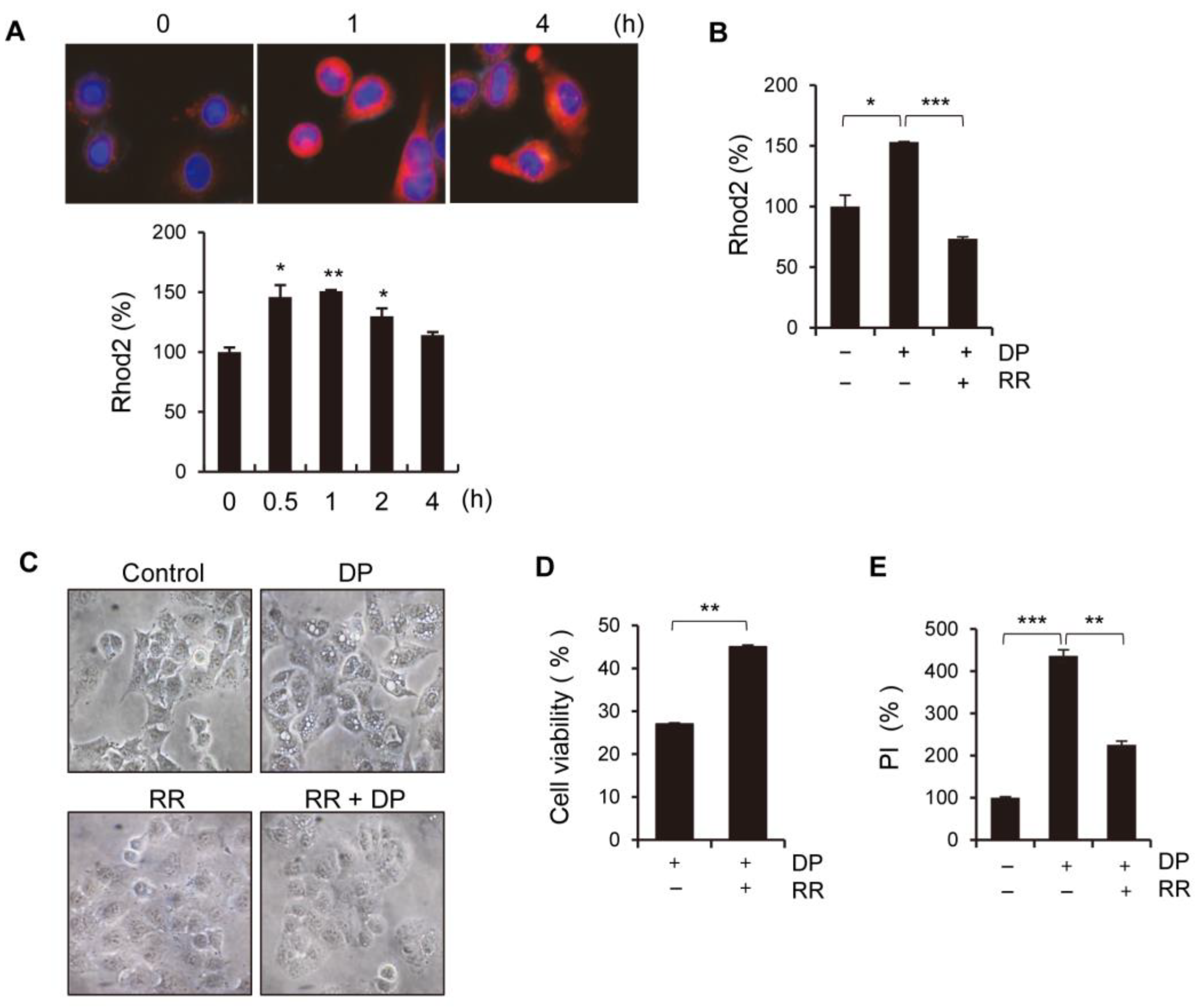
2+ levels, massive ATP depletion, and increased ROS levels. Ca
2+ is known to contribute to mitochondrial permeability disruption, which leads to reduced ATP production and increased ROS levels, and could also lead to the further perturbation of mitochondrial function and lysosomal permeability. Physiological mitochondrial Ca
2+ is attributed to the activation of either the MPT pore or MCU. The regulation of the mitochondrial Ca
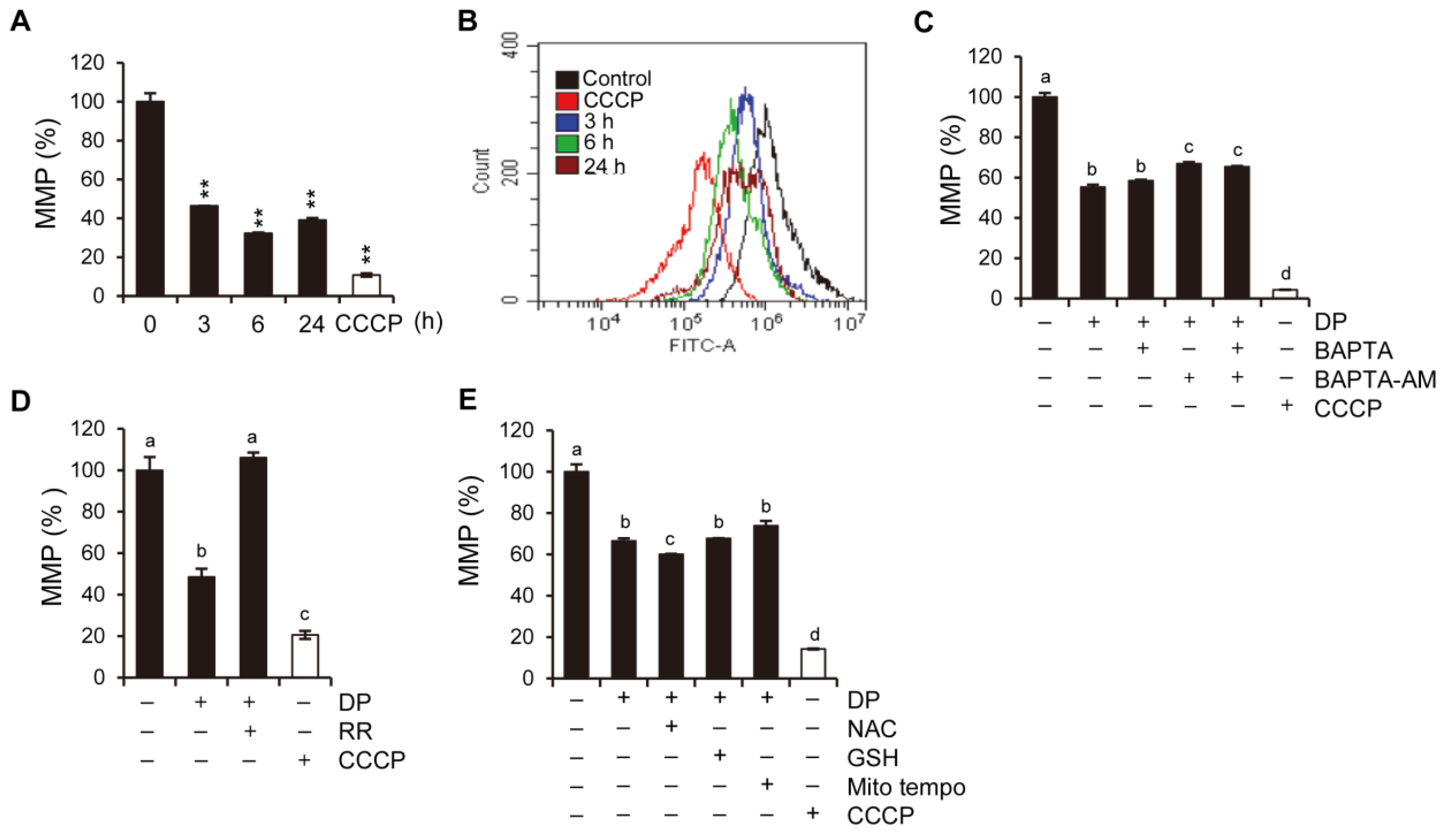
2+ levels is primarily dependent on the mitochondrial calcium uniporter (MCU) located on the inner mitochondrial membrane, while the cyclophilin D located in the mitochondrial matrix plays a crucial role in the MPT opening that leads to mitochondrial-dependent apoptosis or necrosis. Furthermore, ROS produced by the mitochondria and subsequent oxidative damage are strongly associated with MPT, and the selective inhibition of cyclophilin D using cyclosporine A can rescue cell death by preventing MPT. We speculate that the MCU may play a role in MPT regulation, and this regulatory relationship may be significant in MPT-mediated cell death. Although our results demonstrated that DP promoted MPT-mediated cell death, we believe that another regulated cell death mechanism could be a trigger of DP-induced cell growth inhibition, as DP can enhance immune responses.
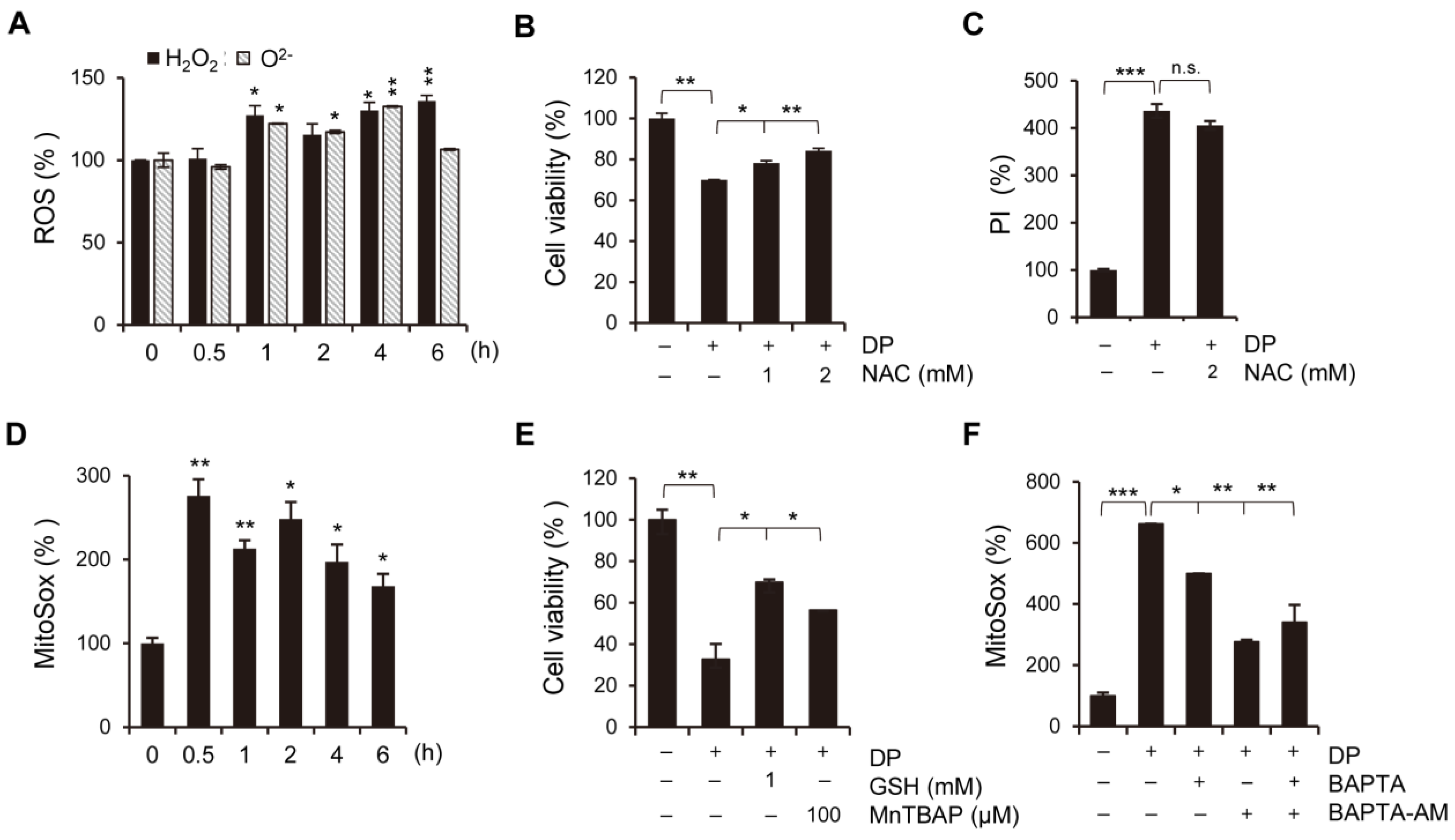
Ferroptosis, a newly discovered form of regulated cell death, is caused by the accumulation of lipid-based ROS. Current studies have shown that ferroptosis is associated with multiple pathological processes, including ischemia-reperfusion injury in the liver, heart, and kidney, as well as neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases [
32]. Other diseases, such as chronic obstructive pulmonary disease (COPD), acute lung injury, and liver fibrosis, are also linked to ferroptosis. The main morphological features of ferroptosis include increased mitochondrial membrane densities and ruptured outer mitochondrial membranes, while the nucleus remains unaffected. Ferroptosis is also characterized by iron accumulation, lipid peroxidation, and increased MMP. Recent studies have identified Frataxin, glutathione peroxidase 4 (GPX4), HSPB1, SLC7A1, and NCOA4 as key regulators of ferroptosis by modulating iron homeostasis and mitochondrial functioning [
33]. Specifically, System xc
− is responsible for transporting extracellular cystine into the cell, where it is converted into cysteine for the synthesis of glutathione (GSH). GSH peroxidase 4 (GPX4) is an enzyme that can directly catalyze the reaction between GSH and lipid hydroperoxides, reducing the cellular level of lipid peroxidation. However, the depletion of GSH or inhibition of GPX4 can lead to the accumulation of lipid hydroperoxides [
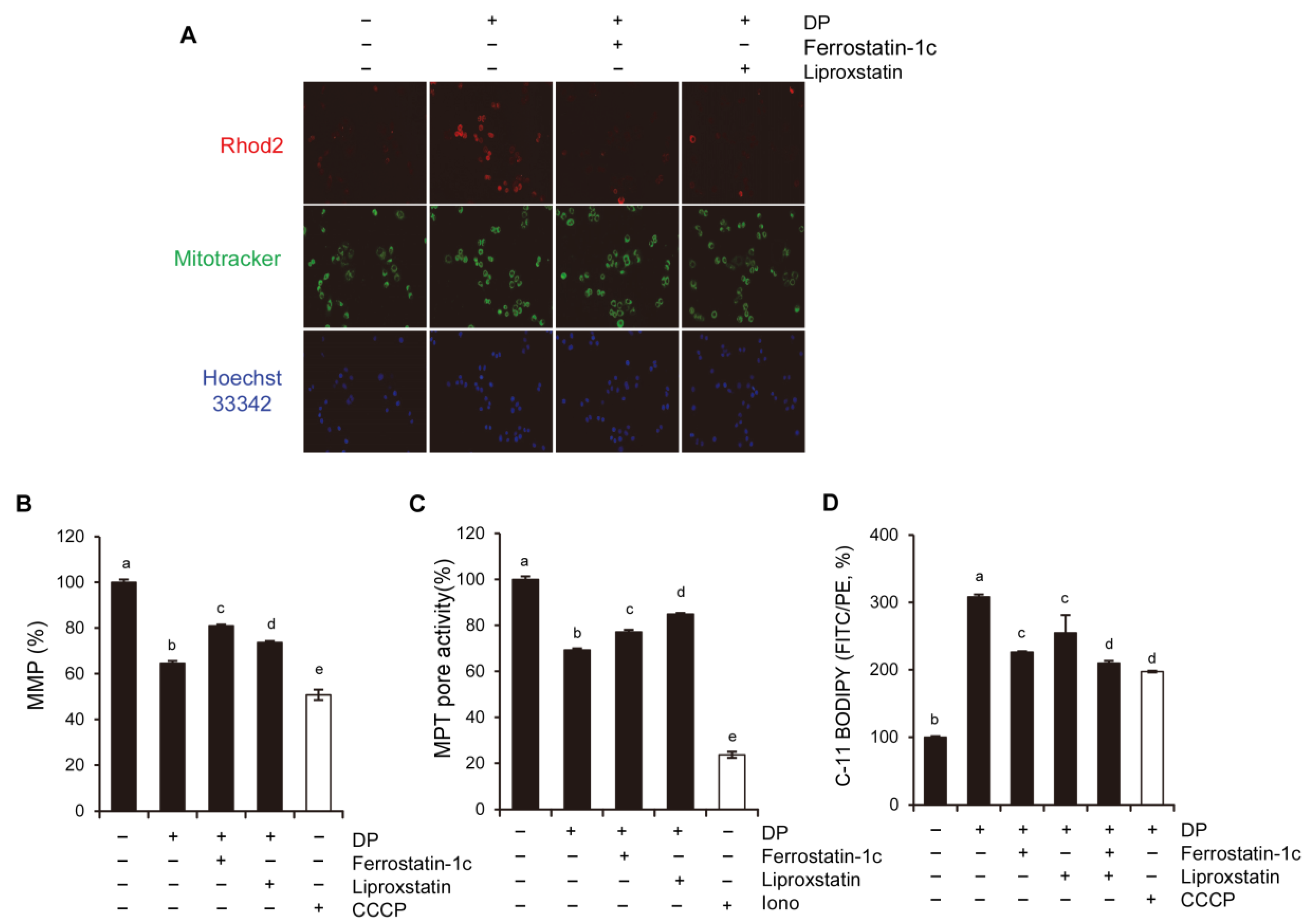
34]. In this study, we used ferroptosis inhibitors to investigate the relationship between DP and ferroptosis. We observed that treatment with ferroptosis inhibitors increased the MPT pore activity levels and significantly restored MMP induced by DP. Taken together, our results suggest that DP induces not only MPT-mediated cell death, but also ferroptosis, and that both of these mechanisms are influenced by each other.
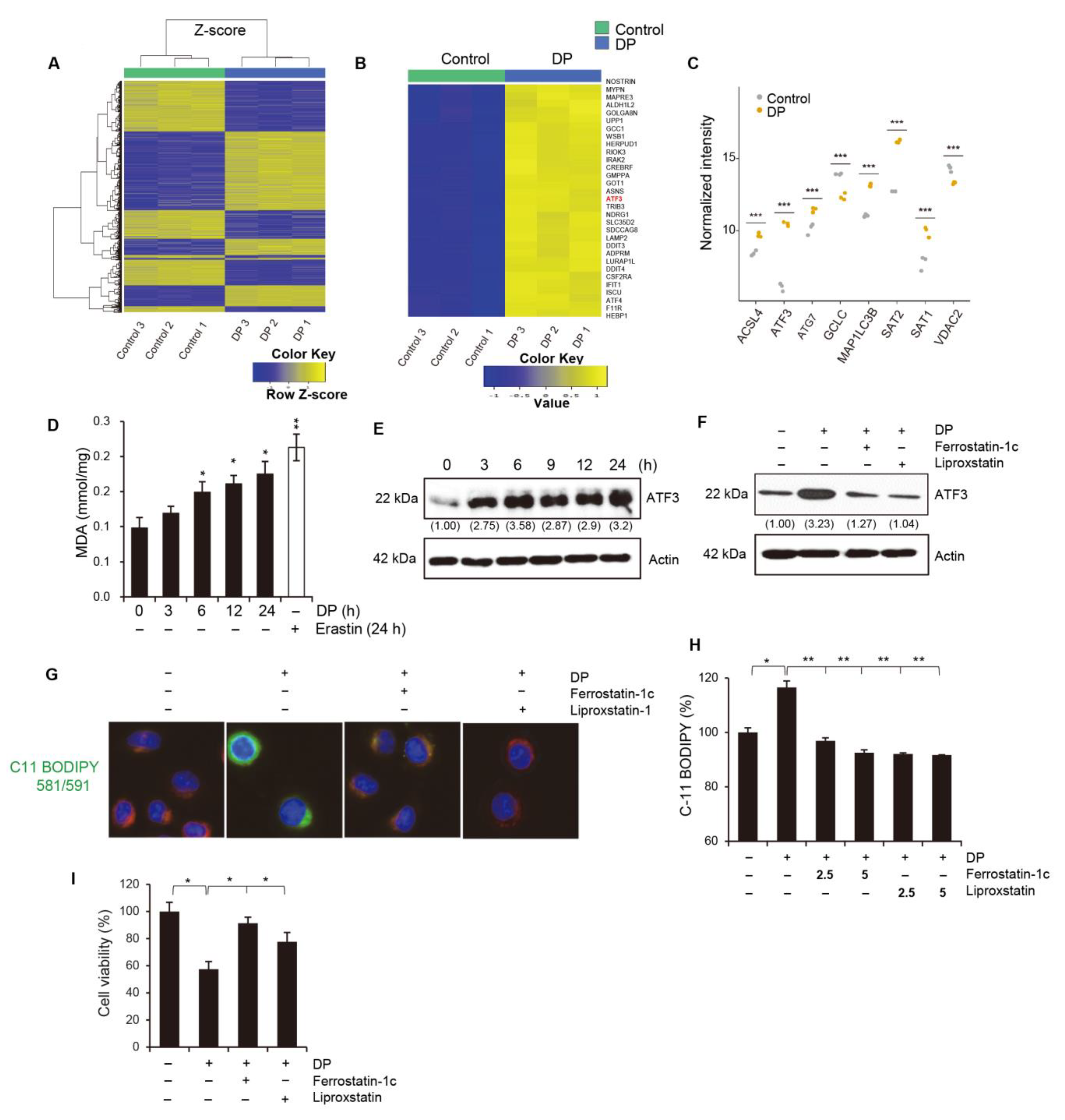
ATF3, activating transcription factor 3, has been shown to play a role in ferroptosis by regulating the expression of genes involved in iron metabolism and lipid peroxidation. ATF3 is activated in response to cellular stress, such as endoplasmic reticulum (ER) stress, amino acid deprivation, and oxidative stress. It has been shown to be involved in the regulation of several cellular processes, including the unfolded protein response (UPR), amino acid metabolism, and autophagy. In ferroptosis, ATF3 has been shown to upregulate the expression of several genes involved in iron metabolism, such as transferrin receptor 1 (TfR1) and divalent metal transporter 1 (DMT1), which are responsible for iron uptake. ATF3 also upregulates the expression of genes involved in lipid peroxidation, such as ACSL4, which is responsible for the esterification of polyunsaturated fatty acids (PUFAs) in phospholipids and subsequent lipid peroxidation. Our study demonstrated that the upregulation of ATF3 is significantly involved in DP-induced ferroptosis. In future studies, it is necessary to investigate the specific subgenes regulated by ATF3 and their respective functions.
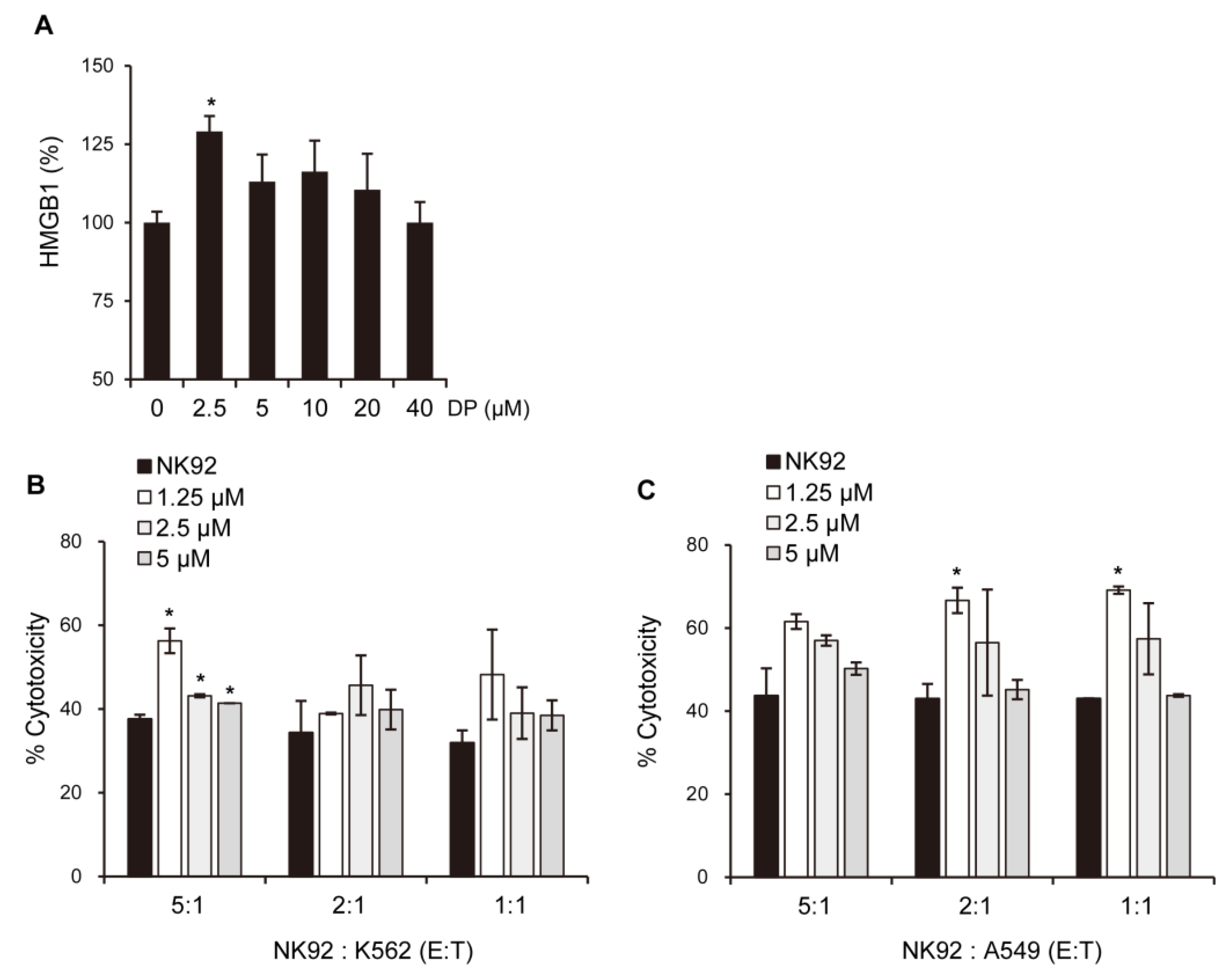
The immunological response depends on molecules presented or released by dying cells, and the HMGB1 protein, which is a nuclear DNA-binding factor and a secreted protein, plays a crucial role. Chemotherapeutics and cellular stress can promote the translocation of HMGB1 from the nucleus to the cytosol and subsequent release into the extracellular space. In the present study, DP was found to increase the secretion of HMGB1 in A549 cells (
Figure 9A). Interestingly, DP-induced supernatant with A549 cells enhanced the cytotoxic activity of NK-92 cells (
Figure 10B,C). Recent studies suggest that ferroptosis, through lipid peroxidation-mediated plasma membrane rupture, can lead to sterile inflammation by generating damage-associated molecular patterns (DAMPs) that stimulate pattern recognition receptor (PRR)-expressing immune cells, resulting in an inflammatory process called necroinflammation, if regulated necrosis initiated immune activation. While the precise mechanisms of this process are still unclear, Fer-1, a small-molecule inhibitor, has been shown to reduce immune cell infiltration in models of acute kidney injury and decrease cytokine and chemokine expression levels (such as C-X-C-motif chemokine 2, interleukin 6, p65 subunit of NF-κB, interleukin 33, TNF-α, and monocyte chemotactic protein 1), suggesting that ferroptotic DAMPs can induce secondary immune cell activation and cytokine production. Nevertheless, further research is required to determine which DP-induced factors in the A549 cells stimulated the cytotoxic ability of the NK-92 cells. Taken together, our findings suggest that DP induces MPT-derived cell death through ferroptosis and can increase the cytotoxic effect of NK-92 cells, providing a novel mechanism for ferroptosis involving MPT-mediated cell death. In summary, our findings reveal that DP treatment hinders cell proliferation by initiating ferroptosis and MPT-related cell death in A549 cells. We aim to provide valuable foundational research data for addressing non-small-cell lung cancer resistant to apoptosis through this study.
4. Materials and Methods
4.1. Reagents and Material
We purchased propidium iodide (PI), C11-BODIPY, MitoTracker, and 3,3′-Dihexyloxacarbocyanine iodide (DiOC6-3) from Thermo Fisher Scientific (Waltham, MA, USA), while chloroquine (CQ), N-acetyl-L-cysteine (NAC), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), carbonyl cyanide m-chlorophenylhydrazone (CCCP), ferrostatin-1c, liproxstatin, ionomycin, and MitoTEMPO were obtained from Sigma-Aldrich (Saint Louis, MO, USA). The caspase-8, caspase-9, and caspase-3 colorimetric assay kits and z-VAD-fmk (a pan-caspase inhibitor) were purchased from R&D systems (Minneapolis, MN, USA). We purchased Wortmannin and U0126 from Tocris (Bristol, UK), and 2′7′-dichlorofluorescein diacetate (H2DCF-DA), BAPTA-AM, CsA, and Ru360 were obtained from Calbiochem (San Diego, CA, USA). Additionally, we purchased dihydroethidium (Hydroethidine, HE), Fluo4-AM, MitoSOX, and BAPTA from Invitrogen (Carlsbad, CA, USA), Rhod-2 and Ruthenium Red from Abcam (Cambridge, UK), and glutathione (GSH) from Duchefa Biochemie (Haarlem, The Netherlands). The MitoProbe Transition Pore Assay Kit was purchased from Molecular Probes (Eugene, OR, USA), and the Human HMGB1/HMG-1 ELISA kit was obtained from Novus Biologicals (Centennial, CO, USA). Calcein-AM (ultra-pure) was purchased from Enzo Life Sciences (Farmingdale, NY, USA). The FITC-conjugated TUNEL Assay Kit (ab66108) and lipid peroxidation (MDA) assay kit (ab118972) were acquired from Abcam.
4.2. Plant Material
Sample collection for this study was as follows: the same Paulownia tomentosa mature fruit as sample KRIB 0059121-0059123 collected in Sancheng, Republic of Korea, in June 2015 was recollected in June 2021 by Doo-Young Kim (Natural Product Central Bank, Korea Research Institute of Bioscience and Biotechnology).
4.3. Extraction and Isolation
The target compound was isolated from dried mature fruit of
P. tomentosa as previously described [
16]. To obtain DP, the plant material containing the mature fruit was dried and ground into a fine powder. The compound was then extracted from the material using 100% CH
3OH, concentrated in vacuo at 40 °C, and purified using methods such as column chromatography (silica gel, reversed-phase silica gel, and prep-HPLC). Brief, the CHCl
3 layer (30.0 g) was fractionated on a silica gel column (10 × 30 cm, 230–400 mesh, 700 g) and eluted using hexane−EtOAc mixtures (15:1 (1.5 L)→1:1 (1 L), and pure EtOAc (2 L)) to produce 20 pooled fractions (Fr. 1–20), which were combined based on a comparison of their LC profiles. Subfraction Fr. 18 (0.8 g) enriched with DP was separated with a Kinetex Biphenyl column using a gradient of CH
3OH−H
2O (71%→100%) to produce DP (89.2 mg). The purity (>98%) of each isolated DP was confirmed by UPLC-PDA and MS.
4.4. Cell Culture
The human lung cancer A549 and human leukemia cell lines were obtained from the American Type Culture Collection (ATCC). The cells were cultured in DMEM medium (Welgene, Korea) supplemented with 10% fetal bovine serum (Gibco, Rockville, MD, USA) and 1% penicillin–streptomycin (Gibco, Rockville, MD, USA) at 37 °C under 5% CO2 atmosphere and with 100% humidity. The human NK-92 cell line was cultured in α-MEM (without ribo- and deoxyribonucleosides) supplemented with 2 mM L-glutamine, 1.5 g/L sodium bicarbonate, 0.2 mM inositol, 0.02 mM folic acid, 0.1 mM 2-mercaptoethanol (Sigma, St. Louis, MO, USA), 12.5% FBS (Gibco, Rockville, MD, USA), 12.5% horse serum (Gibco, Rockville, MD, USA), and 200 U/mL IL-2 (PeproTech, Rocky Hill, NJ, USA). NK-92-sensitive target cell line K562 is a human chronic myelogenous leukemia (CML) cell line. The K562 cell line was cultured RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
4.5. Cell Viability
A549 cells were seeded at a density of 1 × 105 cells/mL in 24-well plates and incubated for 24 h before being treated with various concentrations of DP for designated periods of time. The cells were incubated with an MTT solution (final concentration of 0.5 mg/mL) for 30 min after treatment. Next, the resulting purple formazan was dissolved in DMSO, and the absorbance was measured at 540 nm using an ELISA plate reader (Epoch BioTek, Winooski, VT, USA).
4.6. Caspase Activity
Commercially available total colorimetric caspase-3, caspase-8, and caspase-9 assay kits were used to assess their activity in A549 cells treated with different concentrations of DP. A549 cells were first cultured overnight, treated with varying concentrations of DP, lysed, and equal amounts of protein were incubated with each substrate (DEVE-PNA, IETD-AFC, or LEHD-PNA). The reaction mixtures were then analyzed on a microplate reader with absorbance measured at 405 nm. Each sample was tested in duplicate according to the manufacturer’s instructions.
4.7. TUNEL Assay
Following a 24 h treatment with 40 μM DP, A549 cells were fixed onto glass slides to retain their structure and keep the DNA fragments intact. The cells were then exposed to a permeabilizing solution, Triton X-100. The TdT enzyme was added to the A549 cells along with the FITC-labeled dUTPs. The TdT enzyme catalyzes the incorporation of the labeled dUTPs into the 3’-OH ends of the fragmented DNA. Cells containing fluorescein-labeled dUTPs were directly examined under a fluorescence microscope.
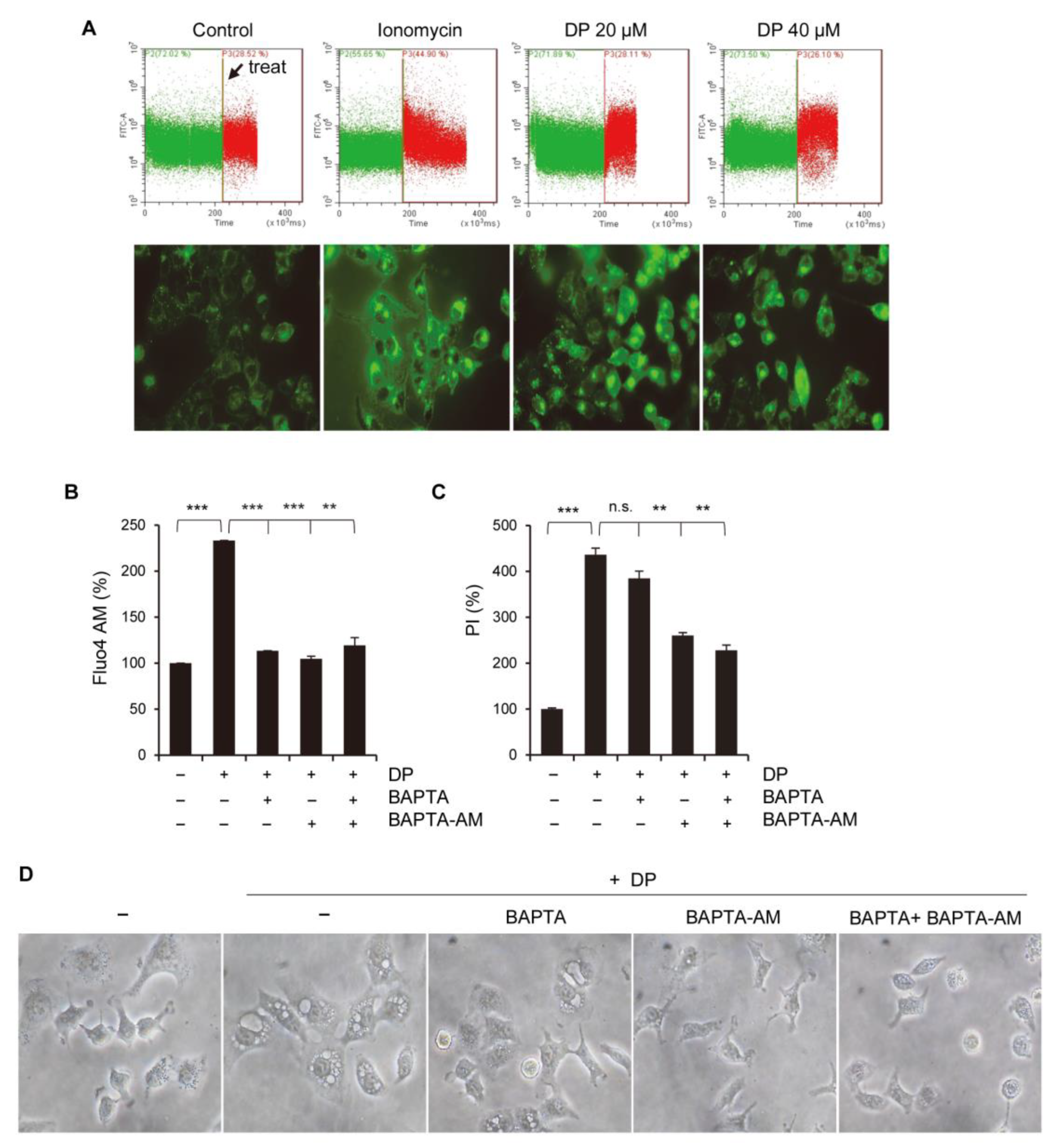
4.8. Measurement of Cytosolic and Mitochondrial Ca2+ Levels
Once harvested, A549 cells were exposed to either Fluo-4 AM or Rhod-2 AM at a concentration of 1 µg/mL, followed by incubation at 37 °C for 15 min, washing with HBSS, and immediate analysis using the FITC or PE channels on a flow cytometer (CytoFLEX, Beckman Coulter, Brea, CA, USA).
4.9. Transmission Electron Microscopy
A total of 1 × 105 A549 cells/mL were cultured in 6-well plates for 1 and 6 h and subsequently treated with 30 µM DP. The cells were then collected and fixed with a primary solution containing 2.5% paraformaldehyde and 2.5% glutaraldehyde. The sections were then stained with uranyl acetate and were visualized by Cryo-TEM (JEM-1400 Plus, 120 kV) (Jeol, Tokyo, Japan).
4.10. Intracellular ROS Level Measurement
The levels of ROS were measured using H2DCF-DA, HE, and MitoSOX (mitochondrial superoxide indicator) probes. Approximately 5 × 104 cells/mL were seeded onto a 24-well plate and treated with DP. Following treatment, the cells were collected by trypsinization, suspended in 0.6 mL of PBS, and incubated with 2 μM He, 4 μM H2DCF-DA, or 1 μM MitoSOX for 20 min in the dark at 37 °C. The fluorescence was measured using a flow cytometer (CytoFLEX, Beckman Coulter, CA, USA) on the FL-2 channel for HE and the FL-1 channel for H2DCF-DA and MitoSOX.
4.11. PI Flow Cytometric Detection
After the indicated treatment, A549 cells were incubated with PI dye (1 mg/mL) in the dark at 37 °C for 20 min. The cells were then washed with PBS and analyzed using the PE channel on a flow cytometer (CytoFLEX, Beckman Coulter, CA, USA).
4.12. Hoechst 33342, Rhod-2, and MitoTracker Stainings
A549 cells were seeded in 24-well plates on coverslips. Next, the cells were treated with DP for 24 h. After treatment, the cells were washed with PBS and incubated with pre-warmed Hoechst for 20 min at 37 °C in the dark. The cells were then washed with PBS again and visualized under a fluorescence microscope (ZEISS, Jena, Germany).
4.13. Live Cell Imaging
A549 cells were seeded in an 8µ-chamber and stained with Rhod-2 and MitoTracker green to measure the mitochondrial calcium levels. The images were taken using a fluorescence microscope (ZEISS, Germany) at ×60 magnification.
4.14. Lipid Peroxidation Measurement
To measure lipid oxidation, we used the C11-BODIPY 581/591 probe. A549 cells were seeded in a 12-well microplate at a density of 1 × 105 cells/well and incubated with C11-BODIPY 581/591 (1 µM) in RPMI-1640 for 30 min. The fluorescence signals of green (484/510 nm) and red (581/610 nm) C11-BODIPY were recorded using CytoFLEX flow cytometry (Beckman Coulter, Brea, CA, USA). MDA assays were performed according to the supplier’s instructions. RFU was measured at Ex/Em = 532/553 nm using a microplate reader.
4.15. Mitochondrial Membrane Potential Assay
After the indicated treatment, A549 cells were incubated with DiOC6(3) dye (40 nM) for 20 min at 37 °C in the dark. The cells were then washed with PBS and observed under a flow cytometer (CytoFLEX, Beckman Coulter, CA, USA) using the FITC channel. To serve as a positive control, CCCP, a mitochondrial oxidative phosphorylation uncoupler, was used.
4.16. Western Blotting
The cells were washed with ice-cold PBS and subsequently lysed on ice for 30 min in lysis buffer (Pro-Prep, iNtRON) containing a protease inhibitor. After the collected supernatants were quantified using the Bradford method following centrifugation at 13,200 rpm for 25 min at 4 °C, the lysates were boiled for 5 min at 95 °C and separated using SDS-PAGE gels. The separated proteins were then transferred onto polyvinylidene difluoride membranes (PVDF, Millipore, MA, USA), which were blocked with 3% skim milk for 30 min to prevent nonspecific binding before incubating with specific primary and secondary antibodies. After washing the membrane three times with TBS-T for 30 min, immune complex signals were detected using enhanced chemiluminescence (ECL, Thermo, USA), and equal protein amounts were assessed using anti-actin as an internal control.
4.17. MPT Pore Activity Measurement
The MitoProbe transition pore assay kit (Molecular Probes, Eugene, OR, USA) was used to determine MPT by utilizing calcein acetoxymethyl ester (AM) and CoCl2. The technique involves calcein-AM, which is a nonfluorescent molecule that accumulates in the cytosol and mitochondria, and is cleaved by intracellular esterases to produce the fluorescent calcein dye that does not cross the plasma or mitochondrial membranes. The addition of CoCl2 quenches the fluorescence of cytosolic, but not mitochondrial calcein in healthy cells. However, in cells with MPT, CoCl2 enters the mitochondria to quench the mitochondrial calcein fluorescence. The calcein-AM/CoCl2 method is restricted to vital cells as it requires the activity of intracellular esterases. For this assay, the cells (1 × 105) were labeled with 10 nM calcein-AM, 400 μM CoCl2, and 0.5 μM ionomycin for 15 min at 37 °C. After washing the cells with HBSS/Ca, they were resuspended in HBSS/Ca and analyzed using CytoFLEX flow cytometry. Calcein-AM fluorescence was expressed as a percentage of its initial value by the following calculation: (mean calcein fluorescence of cells treated with Calcein-AM and CoCl2/mean calcein fluorescence of cells treated with Calcein-AM) × 100. The positive control used for this assay was ionomycin, which is known to induce complete MPT.
4.18. HMGB1 ELISA Assay
The experimental procedures were carried out following the guidelines provided in the assay kit’s manual. Briefly, A549 cells were treated with various concentrations of DP (ranging from 0 to 40 μM) and the resulting supernatant was collected. Next, the supernatant was added to the wells of the microplate, followed by the addition of HRP-conjugated anti-HMGB1 antibody and overnight incubation. The wells were then aspirated and washed several times with a wash buffer, after which TMB substrate was added and incubated. Stop solution was then added, and the contents of the wells were mixed by gentle shaking. Finally, the optical density was measured at 450 nm using a microplate reader immediately after completing the assay.
4.19. Measurement of NK Cell Cytotoxicity
To assess the effect of HMGB1-containing supernatant from DP-treated A549 cells on NK cell activity, A549 cells were treated with 1.25, 2.5, and 5 μM DP for 24 h, and the resulting supernatant was collected. The supernatant was then incubated with NK-92 cells for 4 h before co-culturing with K562 cells and A549 cells at E:T ratios of 5:1, 2:1, and 1:1. After 24 h of co-culturing, cytotoxicity was evaluated using Calcein-AM staining. The fluorescence intensity emitted from the lysed target cells was measured by a fluorescence plate reader.
4.20. Statistical Analysis
All data are presented as the mean ± standard deviation of three independent experiments unless otherwise described. The differences were determined by two-way ANOVA with Tukey’s multiple comparison test using GraphPad Prism (version 6). The statistical significance compared with the control group or between the two groups is indicated as follows: p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). In case there were three or more comparison groups, they were written in lowercase letters. Different lowercase letters above the bar graphs indicate significant statistical differences (p < 0.05).


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









