
Carborane-Containing Hydroxamate MMP Ligands for the Treatment of Tumors Using Boron Neutron Capture Therapy (BNCT): Efficacy without Tumor Cell Entry
 , ,
, ,  , ,
, ,  ,
,  ,
, 
Abstract
1. Introduction
2. Results
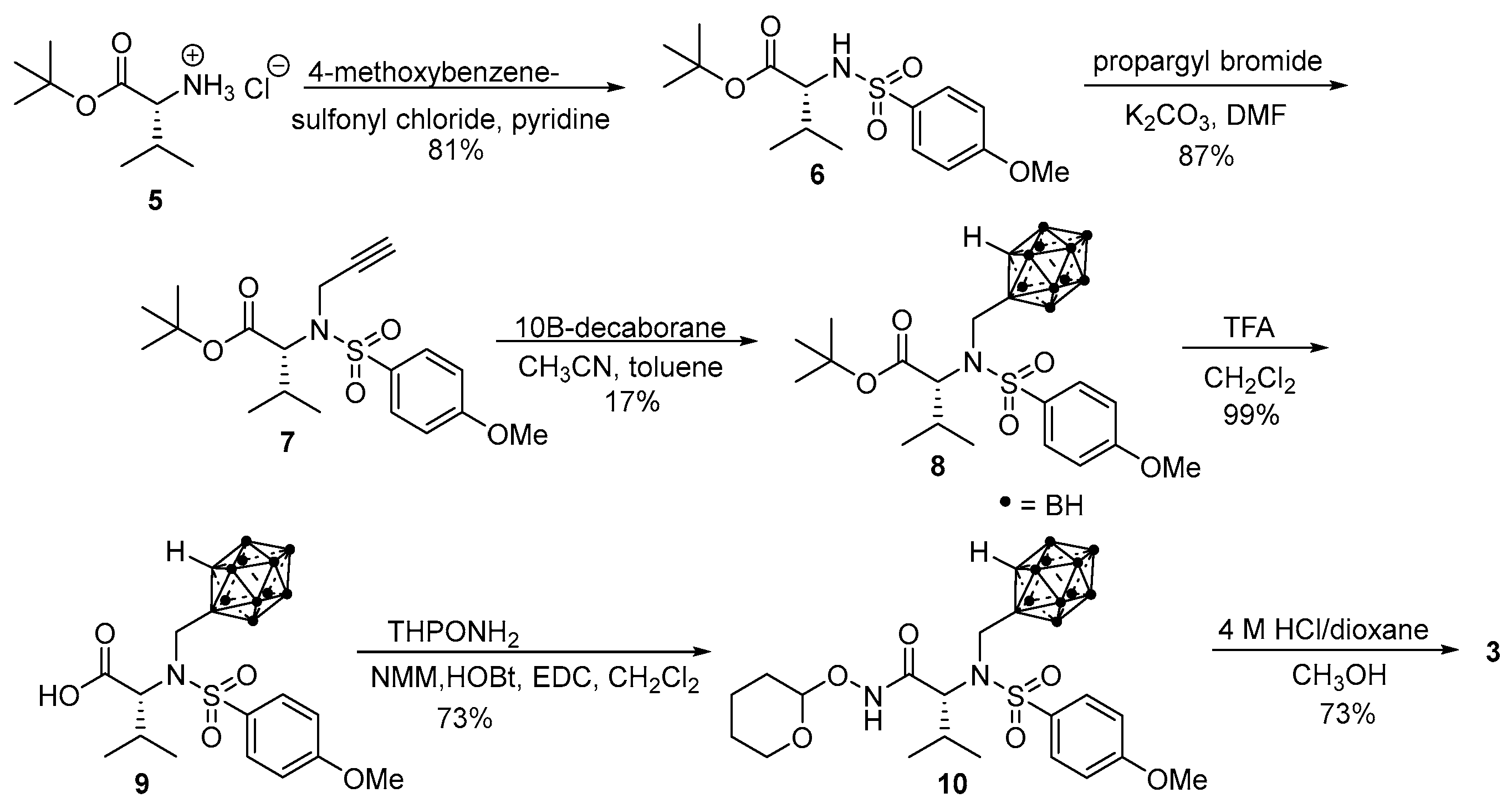
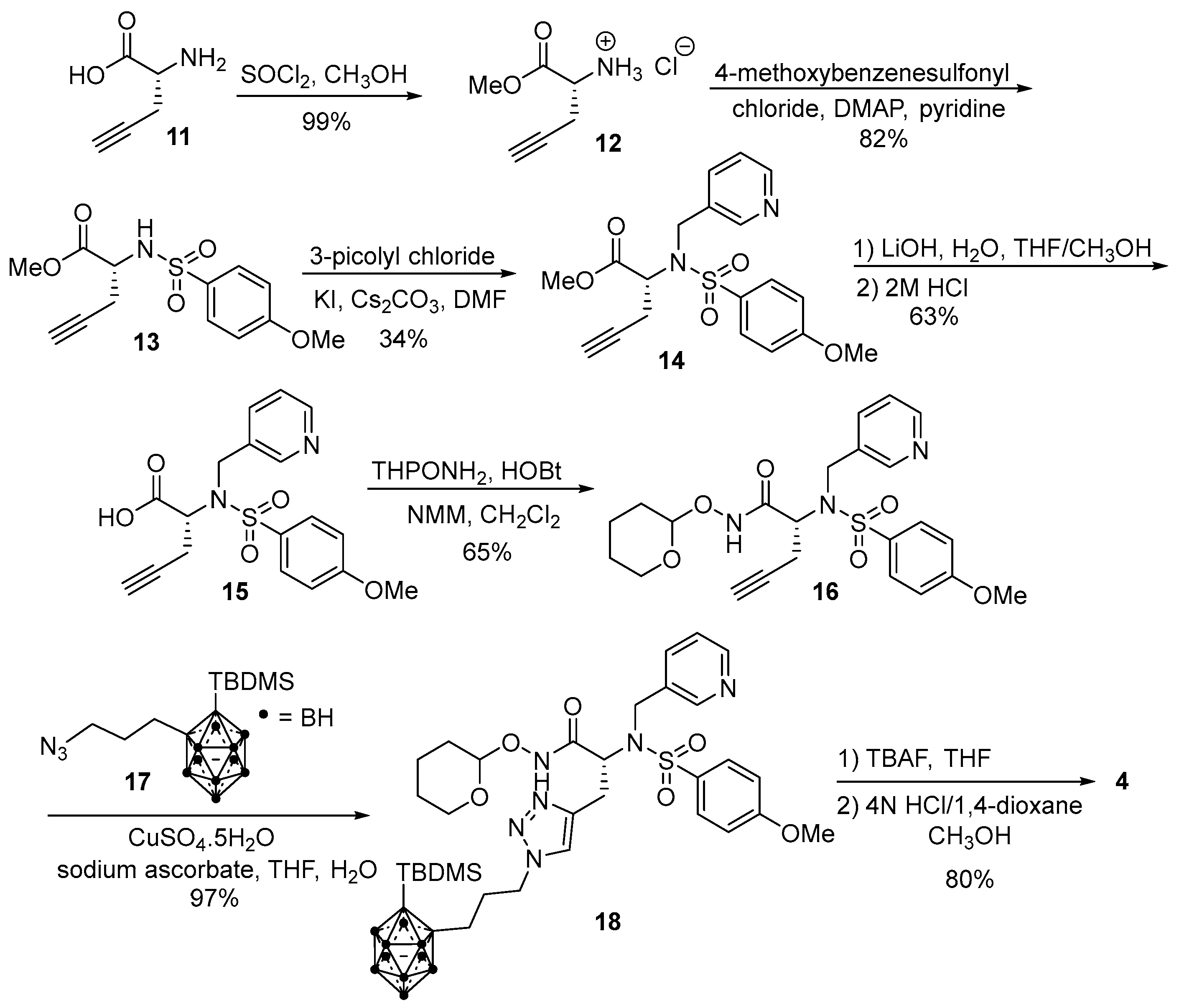
2.1. Synthesis
2.2. MMP Inhibition
2.3. Molecular Docking
2.4. BNCT Results In Vitro BNCT Effect
2.4.1. Cytotoxicity: Evaluation of CGS-27023A Analog 3
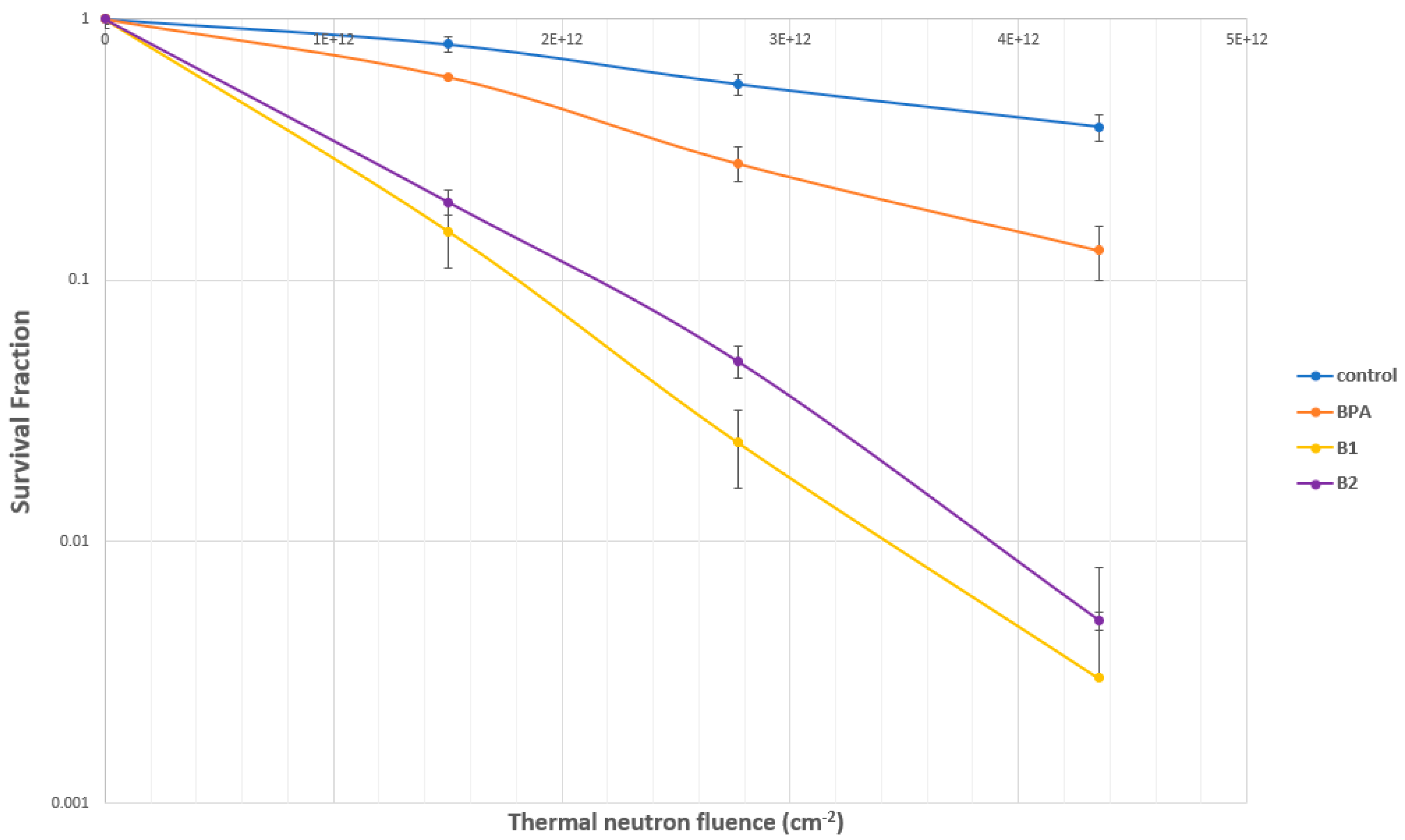
2.4.2. Evaluation of SC-78080/SC-77964 Analogs 1 (B1) and 2 (B2) for BNCT Efficacy
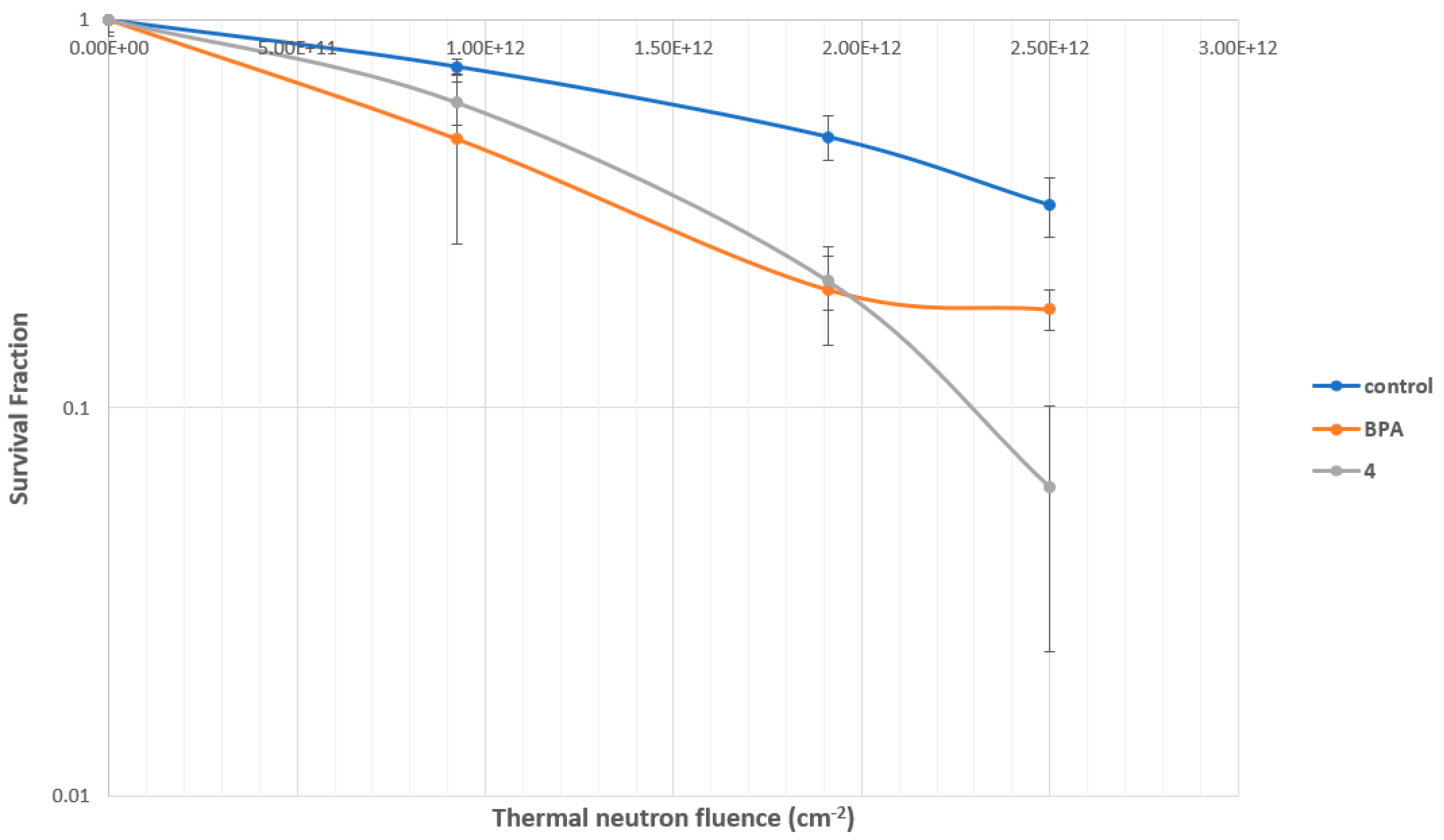
2.4.3. Evaluation of MMP Carborane 4 for BNCT Efficacy
2.5. Boron Cell Concentration Determination
2.6. Water Solubility Determinations
3. Discussion
4. Materials and Methods
4.1. General Experimental Protocols
4.2. Enzyme Inhibition Assays
4.3. Kinetic Analysis of Inhibition Data

4.4. Molecular Docking Protocol
4.5. Cytotoxicity: IC50
4.6. Survival Study: Colony Formation Assay
4.7. Statistical Analysis
4.8. Water Solubility
4.9. Intracellular Uptake of Boron Compounds into SCC VII Cells and U87MG Delta EGFR Cells Evaluated with ICP-OES
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barth, R.F.; Vicente, M.H.; Harling, O.K.; Kiger, W.; Riley, K.J.; Binns, P.J.; Wagner, F.M.; Suzuki, M.; Aihara, T.; Kato, I. Current status of boron neutron capture therapy of high grade gliomas and recurrent head and neck cancer. Radiat. Oncol. 2012, 7, 146. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Fu, R.; Hosmane, N.S. Nanomaterials for boron and gadolinium neutron capture therapy for cancer treatment. Pure Appl. Chem. 2015, 87, 123–134. [Google Scholar] [CrossRef]
- Barth, R.F.; Coderre, J.A.; Vicente, M.G.; Blue, T.E. Boron neutron capture therapy of cancer: Current status and future prospects. Clin. Cancer Res. 2005, 11, 3987–4002. [Google Scholar] [CrossRef] [PubMed]
- Grimes, R.N. Boron clusters come of age. J. Chem. Educ. 2004, 81, 657. [Google Scholar] [CrossRef]
- Valliant, J.F.; Guenther, K.J.; King, A.S.; Morel, P.; Schaffer, P.; Sogbein, O.O.; Stephenson, K.A. The medicinal chemistry of carboranes. Coord. Chem. Rev. 2002, 232, 173–230. [Google Scholar] [CrossRef]
- Hu, K.; Yang, Z.; Zhang, L.; Xie, L.; Wang, L.; Xu, H.; Josephson, L.; Liang, S.H.; Zhang, M. Boron agents for neutron capture therapy. Coord. Chem. Rev. 2020, 405, 213139. [Google Scholar] [CrossRef]
- Soloway, A.H.; Tjarks, W.; Barnum, B.A.; Rong, F.; Barth, R.F.; Codogni, I.M.; Wilson, J.G. The Chemistry of Neutron Capture Therapy. Chem. Rev. 1998, 98, 2389–2390. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakai, K.; Matsumura, A. Boron neutron capture therapy for glioblastoma. Cancer Lett. 2008, 262, 143–152. [Google Scholar] [CrossRef]
- Barth, R.F.; Grecula, J.C. Boron neutron capture therapy at the crossroads-Where do we go from here? Radiat. Isot. 2020, 160, 109029. [Google Scholar] [CrossRef]
- Barth, R.F.; Zhang, Z.; Liu, T. A realistic appraisal of boron neutron capture therapy as a cancer treatment modality. Cancer Commun. 2018, 38, 36. [Google Scholar] [CrossRef]
- Birkedal-Hansen, H.; Moore, W.; Bodden, M.; Windsor, L.; Birkedal-Hansen, B.; DeCarlo, A.; Engler, J. Matrix metalloproteinases: A review. Crit. Rev. Oral Biol. Med. 1993, 4, 197–250. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Welsch, D.J.; Pelletier, J. Metalloproteases and inhibitors in arthritic diseases. Best Pract. Res. Clin. Rheumatol. 2001, 15, 805–829. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.P.; Barta, T.E.; Bedell, L.J.; Boehm, T.L.; Bond, B.R.; Carroll, J.; Carron, C.P.; De Crescenzo, G.A.; Easton, A.M.; Freskos, J.N.; et al. Orally Active MMP-1 Sparing α-Tetrahydropyranyl and α-Piperidinyl Sulfone Matrix Metalloproteinase (MMP) Inhibitors with Efficacy in Cancer, Arthritis, and Cardiovascular Disease. J. Med. Chem. 2010, 53, 6653–6680. [Google Scholar] [CrossRef]
- Becker, D.P.; Villamil, C.I.; Barta, T.E.; Bedell, L.J.; Boehm, T.L.; DeCrescenzo, G.A.; Freskos, J.N.; Getman, D.P.; Hockerman, S.; Heintz, R.; et al. Synthesis and structure-activity relationships of beta- and alpha-piperidine sulfone hydroxamic acid matrix metalloproteinase inhibitors with oral antitumor efficacy. J. Med. Chem. 2005, 48, 6713–6730. [Google Scholar] [CrossRef] [PubMed]
- Skiles, J.W.; Gonnella, N.C.; Jeng, A.Y. The design, structure, and clinical update of small molecular weight matrix metalloproteinase inhibitors. Curr. Med. Chem. 2004, 11, 2911–2977. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.T. Matrix metalloproteinase inhibitor development and the remodeling of drug discovery. Heart Fail. Rev. 2004, 9, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Nagase, H. Progress in matrix metalloproteinase research. Mol. Asp. Med. 2008, 29, 290–308. [Google Scholar] [CrossRef]
- Overall, C.M.; Kleifeld, O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227. [Google Scholar] [CrossRef]
- Fisher, J.F.; Mobashery, S. Recent advances in MMP inhibitor design. Cancer Metastasis Rev. 2006, 25, 115–136. [Google Scholar] [CrossRef]
- Klomp, D.W.; van Laarhoven, H.W.; Kentgens, A.P.; Heerschap, A. Optimization of localized 19F magnetic resonance spectroscopy for the detection of fluorinated drugs in the human liver. Magn. Reson. Med. 2003, 50, 303–308. [Google Scholar] [CrossRef]
- Beck-Sickinger, A.G.; Becker, D.P.; Chepurna, O.; Das, B.; Flieger, S.; Hey-Hawkins, E.; Hosmane, N.; Jalisatgi, S.S.; Nakamura, H.; Patil, R. New Boron Delivery Agents. In Cancer Biotherapy & Radiopharmaceuticals; Mary Ann Liebert, Inc.: New Rochelle, NY, USA, 2022. [Google Scholar]
- Ahrens, V.M.; Frank, R.; Boehnke, S.; Schütz, C.L.; Hampel, G.; Iffland, D.S.; Bings, N.H.; Hey-Hawkins, E.; Beck-Sickinger, A.G. Receptor-mediated uptake of boron-rich neuropeptide y analogues for boron neutron capture therapy. Chem. Med. Chem. 2015, 10, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Isono, A.; Tsuji, M.; Sanada, Y.; Matsushita, A.; Masunaga, S.; Hirayama, T.; Nagasawa, H. Design, Synthesis, and Evaluation of Lipopeptide Conjugates of Mercaptoundecahydrododecaborate for Boron Neutron Capture Therapy. Chem. Med. Chem. 2019, 14, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Hermawan, A.; Susidarti, R.A.; Ramadani, R.D.; Qodria, L.; Utomo, R.Y.; Ishimura, M.; Hattori, Y.; Ohta, Y.; Kirihata, M.; Meiyanto, E. Cellular uptake evaluation of pentagamaboronon-0 (PGB-0) for boron neutron capture therapy (BNCT) against breast cancer cells. Investig. New Drugs 2019, 37, 1292–1299. [Google Scholar] [CrossRef]
- Kanemitsu, T.; Kawabata, S.; Fukumura, M.; Futamura, G.; Hiramatsu, R.; Nonoguchi, N.; Nakagawa, F.; Takata, T.; Tanaka, H.; Suzuki, M. Folate receptor-targeted novel boron compound for boron neutron capture therapy on F98 glioma-bearing rats. Radiat. Environ. Biophys. 2019, 58, 59–67. [Google Scholar] [CrossRef]
- Li, R.; Zhang, J.; Guo, J.; Xu, Y.; Duan, K.; Zheng, J.; Wan, H.; Yuan, Z.; Chen, H. Application of Nitroimidazole–Carbobane-Modified Phenylalanine Derivatives as Dual-Target Boron Carriers in Boron Neutron Capture Therapy. Mol. Pharm. 2019, 17, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Worm, D.J.; Els-Heindl, S.; Beck-Sickinger, A.G. Targeting of peptide-binding receptors on cancer cells with peptide-drug conjugates. Pept. Sci. 2020, 112, e24171. [Google Scholar] [CrossRef]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-drug conjugates and their targets in advanced cancer therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef] [PubMed]
- Couto, M.; Alamón, C.; Sánchez, C.; Dávila, B.; Fernández, M.; Lecot, N.; Cabral, P.; Teixidor, F.; Viñas, C.; Cerecetto, H. Carboranylanilinoquinazoline EGFR-inhibitors: Toward ‘lead-to-candidate’stage in the drug-development pipeline. Future Med. Chem. 2019, 11, 2273–2285. [Google Scholar] [CrossRef]
- Couto, M.; Mastandrea, I.; Cabrera, M.; Cabral, P.; Teixidor, F.; Cerecetto, H.; Viñas, C. Small-molecule kinase-inhibitors-loaded boron cluster as hybrid agents for glioma-cell-targeting therapy. Chem. A Eur. J. 2017, 23, 9233–9238. [Google Scholar] [CrossRef]
- Lutz, M.R., Jr.; Flieger, S.; Colorina, A.; Wozny, J.; Hosmane, N.S.; Becker, D.P. Carborane-Containing Matrix Metalloprotease (MMP) Ligands as Candidates for Boron Neutron-Capture Therapy (BNCT). Chem. Med. Chem. 2020, 15, 1897–1908. [Google Scholar] [CrossRef]
- MacPherson, L.J.; Bayburt, E.K.; Capparelli, M.P.; Carroll, B.J.; Goldstein, R.; Justice, M.R.; Zhu, L.; Hu, S.; Melton, R.A.; Fryer, L. Discovery of CGS 27023A, a non-peptidic, potent, and orally active stromelysin inhibitor that blocks cartilage degradation in rabbits. J. Med. Chem. 1997, 40, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.I.; Grochow, L.B. Clinical potential of matrix metalloprotease inhibitors in cancer therapy. Drugs 2000, 59, 1043–1055. [Google Scholar] [CrossRef]
- Gonnella, N.C.; Li, Y.; Zhang, X.; Paris, C.G. Bioactive conformation of a potent stromelysin inhibitor determined by X-nucleus filtered and multidimensional NMR spectroscopy. Bioorg. Med. Chem. 1997, 5, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Giovenzana, G.B.; Lay, L.; Monti, D.; Palmisano, G.; Panza, L. Synthesis of carboranyl derivatives of alkynyl glycosides as potential BNCT agents. Tetrahedron 1999, 55, 14123–14136. [Google Scholar] [CrossRef]
- Kliegel, W.; Nanninga, D. Borchelate von N-substituierten Hydroxamsäuren. Eur. J. Inorg. Chem. 1983, 116, 2616–2629. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Bode, B.P.; Reidl, C.T.; Sawicki, J.W.; Mason, A.J.; Maguire, J.A.; Hosmane, N.S. “Click” Chemistry-Mediated Phenylene-Cored Carborane Dendrimers. Organometallics 2012, 31, 2931–2935. [Google Scholar] [CrossRef]
- Rossello, A.; Nuti, E.; Orlandini, E.; Carelli, P.; Rapposelli, S.; Macchia, M.; Minutolo, F.; Carbonaro, L.; Albini, A.; Benelli, R. New N-arylsulfonyl-N-alkoxyaminoacetohydroxamic acids as selective inhibitors of gelatinase A (MMP-2). Bioorg. Med. Chem. 2004, 12, 2441–2450. [Google Scholar] [CrossRef]
- Lesnikowski, Z.J. Challenges and opportunities for the application of boron clusters in drug design. J. Med. Chem. 2016, 59, 7738–7758. [Google Scholar] [CrossRef]
- Tiwari, R.; Mahasenan, K.; Pavlovicz, R.; Li, C.; Tjarks, W. Carborane clusters in computational drug design: A comparative docking evaluation using AutoDock, FlexX, Glide, and Surflex. J. Chem. Inf. Model. 2009, 49, 1581–1589. [Google Scholar] [CrossRef]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in drug discovery: Carboranes as unique pharmacophores in biologically active compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef]
- Wilkinson, S.M.; Gunosewoyo, H.; Barron, M.L.; Boucher, A.; McDonnell, M.; Turner, P.; Morrison, D.E.; Bennett, M.R.; McGregor, I.S.; Rendina, L.M. The first CNS-active carborane: A novel P2X7 receptor antagonist with antidepressant activity. ACS Chem. Neurosci. 2014, 5, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Stockmann, P.; Gozzi, M.; Kuhnert, R.; Sárosi, M.B.; Hey-Hawkins, E. New keys for old locks: Carborane-containing drugs as platforms for mechanism-based therapies. Chem. Soc. Rev. 2019, 48, 3497–3512. [Google Scholar] [CrossRef]
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, W.; Crawford Jr, B.; Lipscomb, W.N. The valence structure of the boron hydrides. J. Chem. Phys. 1954, 22, 989–1001. [Google Scholar] [CrossRef]
- Lee Jr, M.W.; Sevryugina, Y.V.; Khan, A.; Ye, S.Q. Carboranes increase the potency of small molecule inhibitors of nicotinamide phosphoribosyltranferase. J. Med. Chem. 2012, 55, 7290–7294. [Google Scholar] [PubMed]
- Endo, Y.; Iijima, T.; Yaguchi, K.; Kawachi, E.; Inoue, N.; Kagechika, H.; Kubo, A.; Itai, A. Structure–activity study of retinoid agonists bearing substituted dicarba-closo-dodecaborane. Relation between retinoidal activity and conformation of two aromatic nuclei. Bioorg. Med. Chem. Lett. 2001, 11, 1307–1311. [Google Scholar] [CrossRef]
- Ogawa, T.; Ohta, K.; Yoshimi, T.; Yamazaki, H.; Suzuki, T.; Ohta, S.; Endo, Y. m-Carborane bisphenol structure as a pharmacophore for selective estrogen receptor modulators. Bioorg. Med. Chem. Lett. 2006, 16, 3943–3946. [Google Scholar] [CrossRef]
- Goto, T.; Ohta, K.; Suzuki, T.; Ohta, S.; Endo, Y. Design and synthesis of novel androgen receptor antagonists with sterically bulky icosahedral carboranes. Bioorg. Med. Chem. 2005, 13, 6414–6424. [Google Scholar] [CrossRef]
- Julius, R.L.; Farha, O.K.; Chiang, J.; Perry, L.J.; Hawthorne, M.F. Synthesis and evaluation of transthyretin amyloidosis inhibitors containing carborane pharmacophores. Proc. Natl. Acad. Sci. USA 2007, 104, 4808–4813. [Google Scholar] [CrossRef]
- Page, M.F.; Jalisatgi, S.S.; Maderna, A.; Hawthorne, M.F. Design and synthesis of a candidate α-human thrombin irreversible inhibitor containing a hydrophobic carborane pharmacophore. Synthesis 2008, 2008, 555–563. [Google Scholar]
- Reynolds, R.C.; Campbell, S.R.; Fairchild, R.G.; Kisliuk, R.L.; Micca, P.L.; Queener, S.F.; Riordan, J.M.; Sedwick, W.D.; Waud, W.R.; Leung, A.K. Novel boron-containing, nonclassical antifolates: Synthesis and preliminary biological and structural evaluation. J. Med. Chem. 2007, 50, 3283–3289. [Google Scholar] [CrossRef]
- Feng, Y.; Likos, J.J.; Zhu, L.; Woodward, H.; Munie, G.; McDonald, J.J.; Stevens, A.M.; Howard, C.P.; De Crescenzo, G.A.; Welsch, D. Solution structure and backbone dynamics of the catalytic domain of matrix metalloproteinase-2 complexed with a hydroxamic acid inhibitor. Biochim. Biophys. Acta Proteins Proteom. 2002, 1598, 10–23. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the Molecular Operating Environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Hugenberg, V.; Breyholz, H.; Riemann, B.; Hermann, S.; Schober, O.; Schäfers, M.; Gangadharmath, U.; Mocharla, V.; Kolb, H.; Walsh, J. A new class of highly potent matrix metalloproteinase inhibitors based on triazole-substituted hydroxamates:(radio) synthesis and in vitro and first in vivo evaluation. J. Med. Chem. 2012, 55, 4714–4727. [Google Scholar] [CrossRef] [PubMed]
- Ourailidou, M.E.; Lenoci, A.; Zwergel, C.; Rotili, D.; Mai, A.; Dekker, F.J. Towards the development of activity-based probes for detection of lysine-specific demethylase-1 activity. Bioorg. Bioorg. Med. Chem. 2017, 25, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Creighton, C.J.; Yan, Z.; Gauthier, D.A.; Dahl, J.P.; Zhao, B.; Belkowski, S.M.; Reitz, A.B. The synthesis and evaluation of 10-and 12-membered ring benzofused enediyne amino acids. Bioorg. Med. Chem. 2005, 13, 5936–5948. [Google Scholar] [CrossRef]
- Olson, M.W.; Gervasi, D.C.; Mobashery, S.; Fridman, R. Kinetic analysis of the binding of human matrix metalloproteinase-2 and-9 to tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2. J. Biol. Chem. 1997, 272, 29975–29983. [Google Scholar] [CrossRef]
- Lim, N.H.; Meinjohanns, E.; Bou-Gharios, G.; Gompels, L.L.; Nuti, E.; Rossello, A.; Devel, L.; Dive, V.; Meldal, M.; Nagase, H. In vivo imaging of matrix metalloproteinase 12 and matrix metalloproteinase 13 activities in the mouse model of collagen-induced arthritis. Arthritis Rheumatol. 2014, 66, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Knäuper, V.; López-Otin, C.; Smith, B.; Knight, G.; Murphy, G. Biochemical characterization of human collagenase-3. J. Biol. Chem. 1996, 271, 1544–1550. [Google Scholar] [CrossRef]
- Caswell, R.; Coyne, J.; Randolph, M. Kerma factors for neutron energies below 30 MeV. Radiat. Res. 1980, 83, 217–254. [Google Scholar] [CrossRef]
- Snyder, W.S.; Cook, M.J.; Nasset, E.S.; Karhausen, L.R.; Howells, G.P.; Tipton, I. Cross and Elemental Content of Reference Man; Report of the Task Group on Reference Man; Snyder, W.S., Ed.; Pergamon Press: Oxford, UK, 1975; pp. 273–324. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (nM) a | |||
|---|---|---|---|---|
| MMP-1 | MMP-2 | MMP-9 | MMP-13 | |
| 1,4-triazole 1 b | >10,000 | 37 ± 1.3 | 46 ± 4.6 | - |
| 1,5-triazole 2 b | >10,000 | 9.8 ± 0.96 | 13 ± 1.2 | - |
| CGS-27023A analog 3 | - | 5.4 ± 2.0 | 125 ± 100 | 64 ± 70 |
| CGS-27023A analog 1,4-triazole 4 | - | 5.2 ± 2.0 | 5.1 ± 0.2 | 3.1 ± 0.6 |
| NNGH | 163 ± 2.8 | 8.5 ± 1.1 | 2.8 ± 0.014 | - |
| BNCT Agent | Drug Water Solubility (µg/mL) |
|---|---|
| 1,4-triazole 1 | 6.2 ± 0.93 |
| 1,5-triazole 2 | 6.9 ± 7.1 |
| CGS-27023A analog 3 | 5.3 ± 5.6 |
| CGS-27023A analog 1,4-triazole 4 | 96 ± 9.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flieger, S.; Takagaki, M.; Kondo, N.; Lutz, M.R., Jr.; Gupta, Y.; Ueda, H.; Sakurai, Y.; Moran, G.; Kempaiah, P.; Hosmane, N.; et al. Carborane-Containing Hydroxamate MMP Ligands for the Treatment of Tumors Using Boron Neutron Capture Therapy (BNCT): Efficacy without Tumor Cell Entry. Int. J. Mol. Sci. 2023, 24, 6973. https://doi.org/10.3390/ijms24086973
Flieger S, Takagaki M, Kondo N, Lutz MR Jr., Gupta Y, Ueda H, Sakurai Y, Moran G, Kempaiah P, Hosmane N, et al. Carborane-Containing Hydroxamate MMP Ligands for the Treatment of Tumors Using Boron Neutron Capture Therapy (BNCT): Efficacy without Tumor Cell Entry. International Journal of Molecular Sciences. 2023; 24(8):6973. https://doi.org/10.3390/ijms24086973
Chicago/Turabian StyleFlieger, Sebastian, Mao Takagaki, Natsuko Kondo, Marlon R. Lutz, Jr., Yash Gupta, Hiroki Ueda, Yoshinori Sakurai, Graham Moran, Prakasha Kempaiah, Narayan Hosmane, and et al. 2023. "Carborane-Containing Hydroxamate MMP Ligands for the Treatment of Tumors Using Boron Neutron Capture Therapy (BNCT): Efficacy without Tumor Cell Entry" International Journal of Molecular Sciences 24, no. 8: 6973. https://doi.org/10.3390/ijms24086973
APA StyleFlieger, S., Takagaki, M., Kondo, N., Lutz, M. R., Jr., Gupta, Y., Ueda, H., Sakurai, Y., Moran, G., Kempaiah, P., Hosmane, N., Suzuki, M., & Becker, D. P. (2023). Carborane-Containing Hydroxamate MMP Ligands for the Treatment of Tumors Using Boron Neutron Capture Therapy (BNCT): Efficacy without Tumor Cell Entry. International Journal of Molecular Sciences, 24(8), 6973. https://doi.org/10.3390/ijms24086973