
Dual Blockade of TGF-β Receptor and Endothelin Receptor Synergistically Inhibits Angiotensin II-Induced Myofibroblast Differentiation: Role of AT1R/Gαq-Mediated TGF-β1 and ET-1 Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Treatment with Ang II, TGF-β1, or ET-1 Promotes Myofibroblast Differentiation in a Dose-Dependent Manner in Adult Human Cardiac Fibroblasts (HCFs)
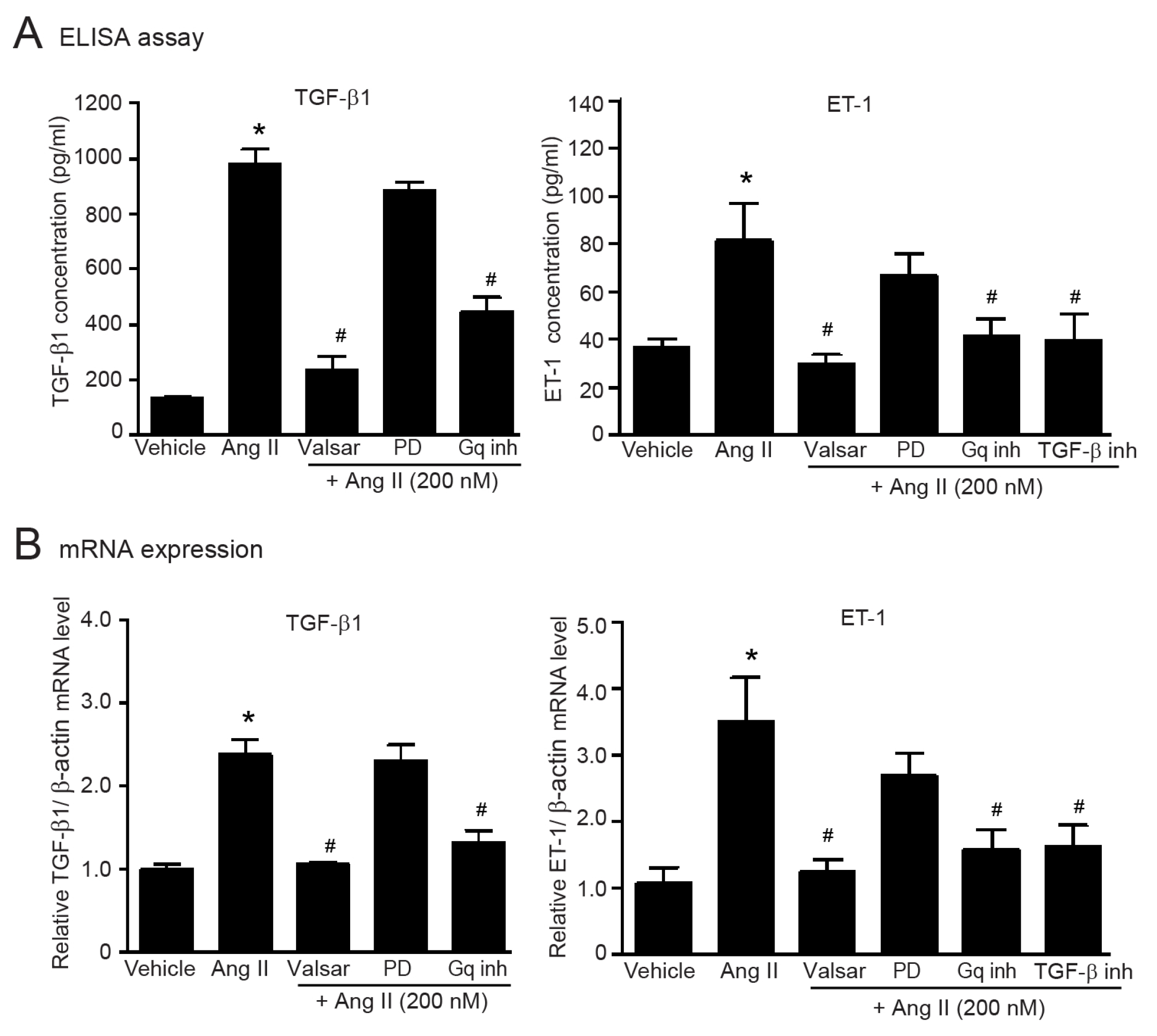
2.2. Ang II Induces Myofibroblast Differentiation through AT1R/Gαq Axis
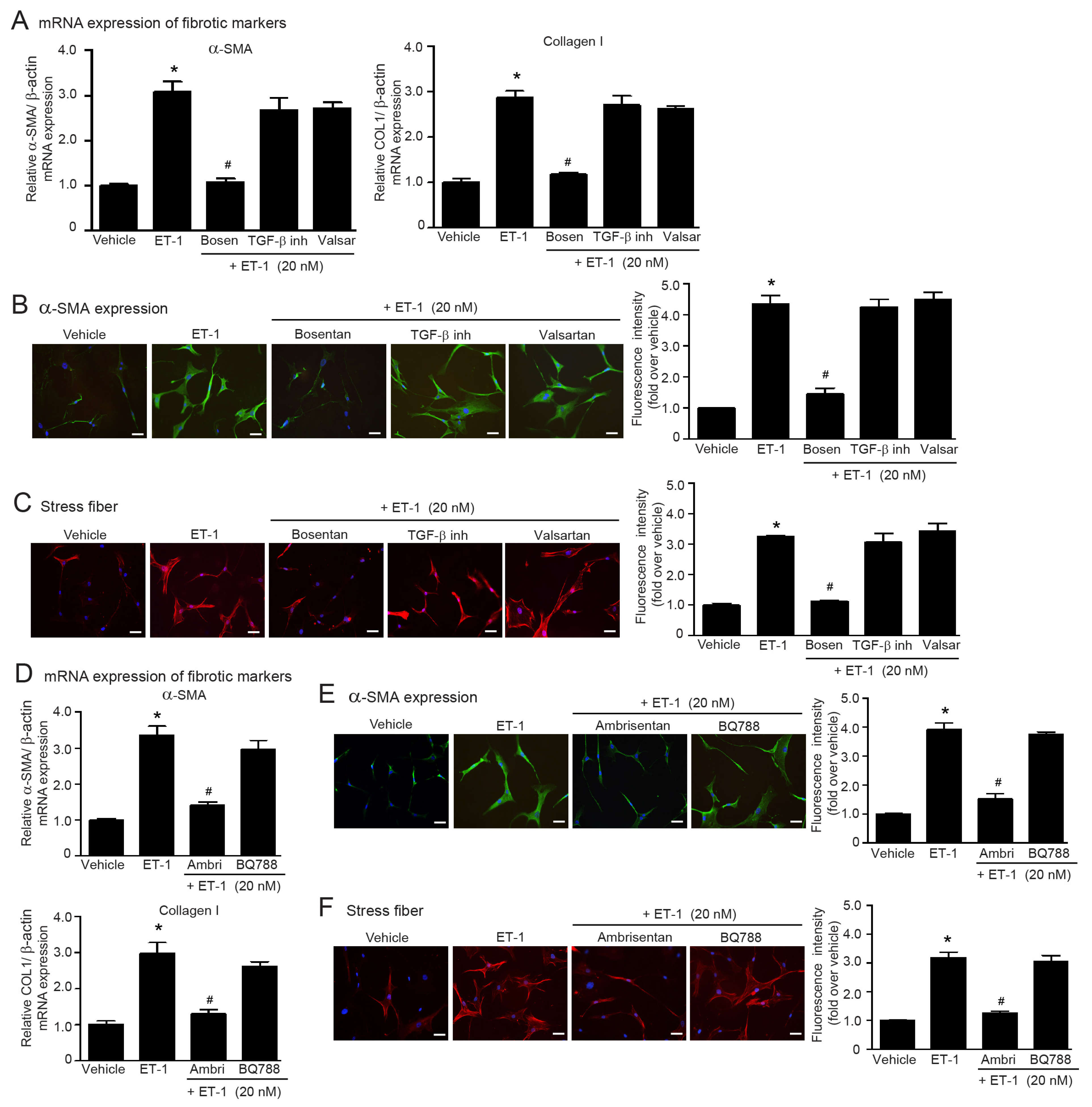
2.3. Dual Blockade of TGF-β Receptor and Endothelin Receptor Inhibits Ang II-Induced Myofibroblast Differentiation
2.4. Blockade of ETRs, Not ATRs, Inhibits TGF-β1-Induced Myofibroblast Differentiation
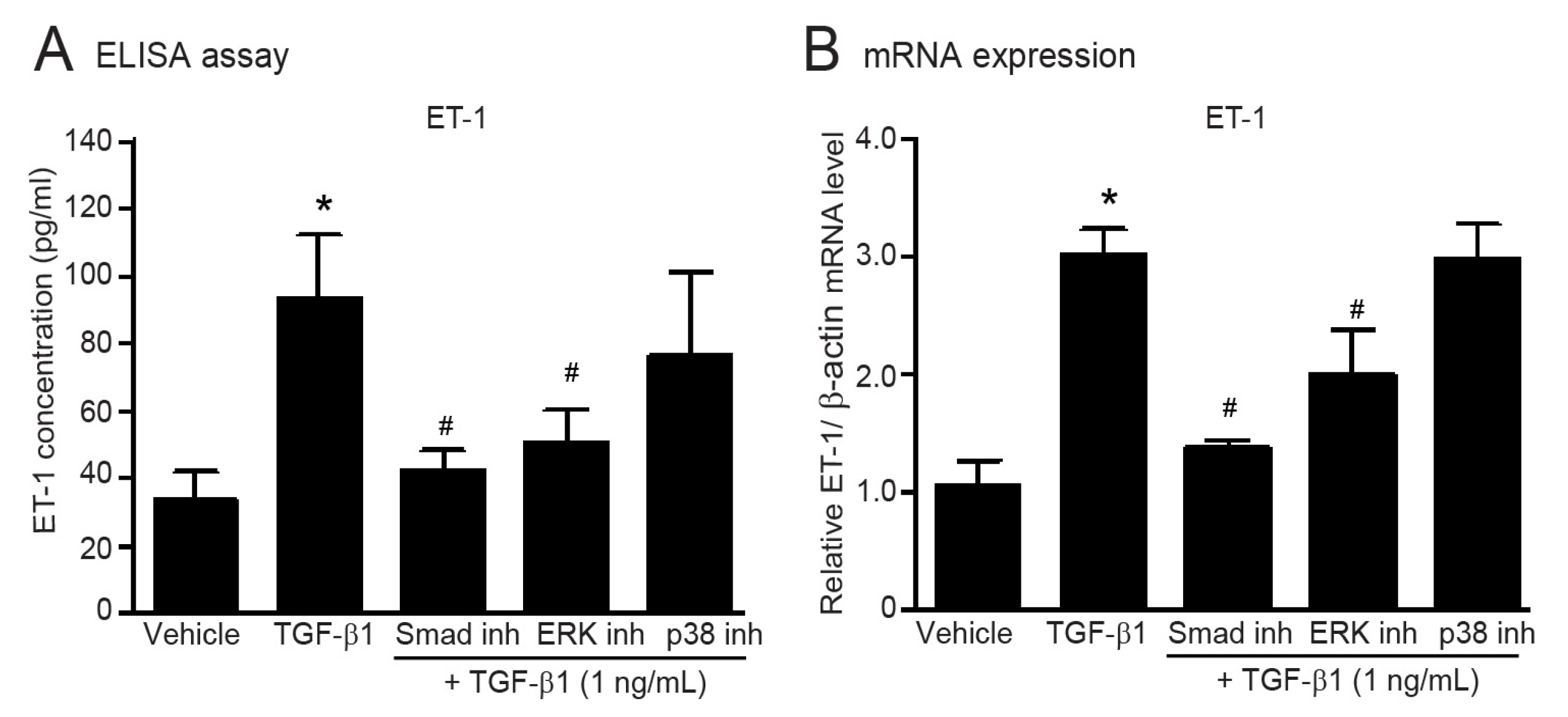
2.5. Smad and ERK Are Required for TGF-β1-Induced Myofibroblast Differentiation in Adult HCFs
2.6. ET-1 Is a Downstream Effector of Ang II/TGF-β Axis That Stimulates Myofibroblast Differentiation via the ETAR Subtype in Adult HCFs
2.7. Gαq-Dependent Activation of AT1Rs Is Necessary for the Upregulation of TGF-β1 and ET-1 in Adult HCFs
2.8. TGF-β1-Induced Upregulation of ET-1 Is Dependent of Smad and ERK1/2 in Adult HCFs
2.9. The Reversibility of Ang II-Induced Myofibroblast Differentiation by Dual Inhibition of TGF-β Receptor and ETR
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Fibroblast Cultures
4.3. Immunofluorescence Staining for the Detection of α-SMA Expression
4.4. Immunofluorescence Staining for Detection of Actin Stress Fiber
4.5. Quantitative Real-Time RT-PCR
4.6. Enzyme-Linked Immunosorbent Assays (ELISA)
4.7. Quantification and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aoki, T.; Fukumoto, Y.; Sugimura, K.; Oikawa, M.; Satoh, K.; Nakano, M.; Nakayama, M.; Shimokawa, H. Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure-Comparison between preserved and reduced ejection fraction heart failure. Circ. J. 2011, 75, 2605–2613. [Google Scholar] [CrossRef]
- Kato, S.; Saito, N.; Kirigaya, H.; Gyotoku, D.; Iinuma, N.; Kusakawa, Y.; Iguchi, K.; Nakachi, T.; Fukui, K.; Futaki, M.; et al. Prognostic significance of quantitative assessment of focal myocardial fibrosis in patients with heart failure with preserved ejection fraction. Int. J. Cardiol. 2015, 191, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; Do, Y.S.; Kawano, Y.; Starnes, V.; Barr, M.; Law, R.E.; Hsueh, W.A. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation 2000, 101, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.M.; Wong, A.L.; Bezuhly, M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair 2015, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Kurose, H.; Mangmool, S. Myofibroblasts and inflammatory cells as player of cardiac fibrosis. Arch. Pharm. Res. 2016, 39, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Schnee, J.M.; Hsueh, W.A. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc. Res. 2000, 46, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Matavelli, L.C.; Siragy, H.M. AT2 receptor activities and pathophysiological implications. J. Cardiovasc. Pharmacol. 2015, 65, 226. [Google Scholar] [CrossRef]
- Lopez, J.; Lorell, B.; Ingelfinger, J.; Weinberg, E.; Schunkert, H.; Diamant, D.; Tang, S.S. Distribution and function of cardiac angiotensin AT1-and AT2-receptor subtypes in hypertrophied rat hearts. Am. J. Physiol. Heart Circ. Physiol. 1994, 267, H844–H852. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Matsubara, H.; Ohkubo, N.; Mori, Y.; Nozawa, Y.; Murasawa, S.; Kijima, K.; Maruyama, K.; Masaki, H.; Moriguchi, Y.; et al. Angiotensin II type 2 receptor is upregulated in human heart with interstitial fibrosis, and cardiac fibroblasts are the major cell type for its expression. Circ. Res. 1998, 83, 1035–1046. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Duangrat, R.; Parichatikanond, W.; Morales, N.P.; Pinthong, D.; Mangmool, S. Sustained AT1R stimulation induces upregulation of growth factors in human cardiac fibroblasts via Gαq/TGF-β/ERK signaling that influences myocyte hypertrophy. Eur. J. Pharmacol. 2022, 937, 175384. [Google Scholar] [CrossRef]
- Rosenkranz, S. TGF-β1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 2004, 63, 423–432. [Google Scholar] [CrossRef]
- Flores-Vergara, R.; Olmedo, I.; Aránguiz, P.; Riquelme, J.A.; Vivar, R.; Pedrozo, Z. Communication between cardiomyocytes and fibroblasts during cardiac ischemia/reperfusion and remodeling: Roles of TGF-β, CTGF, the renin angiotensin axis, and non-coding RNA molecules. Front. Physiol. 2021, 12, 716721. [Google Scholar] [CrossRef]
- Kurihara, H.; Yoshizumi, M.; Sugiyama, T.; Takaku, F.; Yanagisawa, M.; Masaki, T.; Hamaoki, M.; Kato, H.; Yazaki, Y. Transforming growth factor-β stimulates the expression of endothelin mRNA by vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 159, 1435–1440. [Google Scholar] [CrossRef]
- Lee, S.D.; Lee, D.S.; Chun, Y.G.; Paik, S.H.; Kim, W.S.; Kim, D.S.; Kim, W.D.; Tuder, R.M.; Voelkel, N.F. Transforming growth factor-β1 induces endothelin-1 in a bovine pulmonary artery endothelial cell line and rat lungs via cAMP. Pulm. Pharmacol. Ther. 2000, 13, 257–265. [Google Scholar] [CrossRef]
- Rodríguez-Pascual, F.; Reimunde, F.M.; Redondo-Horcajo, M.; Lamas, S. Transforming growth factor-β induces endothelin-1 expression through activation of the Smad signaling pathway. J. Cardiovasc. Pharmacol. 2004, 44, S39–S42. [Google Scholar] [CrossRef]
- Gallo, E.M.; Loch, D.C.; Habashi, J.P.; Calderon, J.F.; Chen, Y.; Bedja, D.; van Erp, C.; Gerber, E.E.; Parker, S.J.; Sauls, K.; et al. Angiotensin II–dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J. Clin. Invest. 2014, 124, 448–460. [Google Scholar] [CrossRef]
- Cheng, T.H.; Cheng, P.Y.; Shih, N.L.; Chen, I.B.; Wang, D.L.; Chen, J.J. Involvement of reactive oxygen species in angiotensin II-induced endothelin-1 gene expression in rat cardiac fibroblasts. J Am. Coll. Cardiol. 2003, 42, 1845–1854. [Google Scholar] [CrossRef]
- Barton, M.; Shaw, S.; Moreau, P.; Lüscher, T.F. Angiotensin II increases vascular and renal endothelin-1 and functional endothelin converting enzyme activity in vivo: Role of ETA-receptors for endothelin regulation. Biochem. Biophys. Res. Commun. 1997, 238, 861–865. [Google Scholar] [CrossRef]
- d’Uscio, L.V.; Shaw, S.; Barton, M.; Lüscher, T.F. Losartan but not verapamil inhibits angiotensin II–induced tissue endothelin-1 increase: Role of blood pressure and endothelial function. Hypertension 1998, 31, 1305–1310. [Google Scholar] [CrossRef]
- Davenport, A.P.; Hyndman, K.A.; Dhaun, N.; Southan, C.; Kohan, D.E.; Pollock, J.S.; Pollock, D.M.; Webb, D.J.; Maguire, J.J. Endothelin. Pharmacol. Rev. 2016, 68, 357–418. [Google Scholar] [CrossRef]
- Shi-Wen, X.; Chen, Y.; Denton, C.P.; Eastwood, M.; Renzoni, E.A.; Bou-Gharios, G.; Pearson, J.D.; Dashwood, M.; du Bois, R.M.; Black, C.M.; et al. Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol. Biol. Cell 2004, 15, 2707–2719. [Google Scholar] [CrossRef]
- Ammarguellat, F.; Larouche, I.; Schiffrin, E.L. Myocardial fibrosis in DOCA-salt hypertensive rats: Effect of endothelin ETA receptor antagonism. Circulation 2001, 103, 319–324. [Google Scholar] [CrossRef]
- Cambrey, A.D.; Harrison, N.K.; Dawes, K.E.; Southcott, A.M.; Black, C.M.; du Bois, R.M.; Laurent, G.J.; McAnulty, R.J. Increased levels of endothelin-1 in bronchoalveolar lavage fluid from patients with systemic sclerosis contribute to fibroblast mitogenic activity in vitro. Am. J. Respir. Cell Mol. Biol. 1994, 11, 439–445. [Google Scholar] [CrossRef]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming growth factor-β function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 2002, 106, 130–135. [Google Scholar] [CrossRef]
- Engebretsen, K.V.; Skårdal, K.; Bjørnstad, S.; Marstein, H.S.; Skrbic, B.; Sjaastad, I.; Christensen, G.; Bjørnstad, J.L.; Tønnessen, T. Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J. Mol. Cell. Cardiol. 2014, 76, 148–157. [Google Scholar] [CrossRef]
- Singh, A.D.; Amit, S.; Kumar, O.S.; Rajan, M.; Mukesh, N. Cardioprotective effects of bosentan, a mixed endothelin type A and B receptor antagonist, during myocardial ischaemia and reperfusion in rats. Basic. Clin. Pharmacol. Toxicol. 2006, 98, 604–610. [Google Scholar] [CrossRef]
- Adiarto, S.; Heiden, S.; Vignon-Zellweger, N.; Nakayama, K.; Yagi, K.; Yanagisawa, M.; Emoto, N. ET-1 from endothelial cells is required for complete angiotensin II-induced cardiac fibrosis and hypertrophy. Life Sci. 2012, 91, 651–657. [Google Scholar] [CrossRef]
- Abraham, D. Role of endothelin in lung fibrosis. Eur. Respir. Rev. 2008, 17, 145–150. [Google Scholar]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Rodríguez-Pascual, F.; Busnadiego, O.; González-Santamaría, J. The profibrotic role of endothelin-1: Is the door still open for the treatment of fibrotic diseases? Life Sci. 2014, 118, 156–164. [Google Scholar] [CrossRef]
- Yang, X.; Chen, B.; Liu, T.; Chen, X. Reversal of myofibroblast differentiation: A review. Eur. J. Pharmacol. 2014, 734, 83–90. [Google Scholar] [CrossRef]
- Parichatikanond, W.; Luangmonkong, T.; Mangmool, S.; Kurose, H. Therapeutic targets for the treatment of cardiac fibrosis and cancer: Focusing on TGF-β signaling. Front. Cardiovasc. Med. 2020, 7, 34. [Google Scholar] [CrossRef]
- Schieffer, B.; Wirger, A.; Meybrunn, M.; Seitz, S.; Holtz, J.; Riede, U.N.; Drexler, H. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation 1994, 89, 2273–2282. [Google Scholar] [CrossRef]
- Regan, C.P.; Anderson, P.G.; Bishop, S.P.; Berecek, K.H. Pressure-independent effects of AT1-receptor antagonism on cardiovascular remodeling in aortic-banded rats. Am. J. Physiol. Heart. Circ. Physiol. 1997, 272, H2131–H2138. [Google Scholar] [CrossRef]
- Luchtefeld, M.; Drexler, H.; Schieffer, B. Role of Gbeta-subunit in angiotensin II-type 1 receptor signaling. Biochem. Biophys. Res. Commun. 2001, 280, 756–760. [Google Scholar] [CrossRef]
- Schultz, J.E.J.; Witt, S.A.; Glascock, B.J.; Nieman, M.L.; Reiser, P.J.; Nix, S.L.; Kimball, T.R.; Doetschman, T. TGF-β1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Invest. 2002, 109, 787–796. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Padgett, R.W. Matters of context guide future research in TGFβ superfamily signaling. Sci. Signal. 2015, 8, re10. [Google Scholar] [CrossRef]
- Gao, X.; He, X.; Luo, B.; Peng, L.; Lin, J.; Zuo, Z. Angiotensin II increases collagen I expression via transforming growth factor-beta1 and extracellular signal-regulated kinase in cardiac fibroblasts. Eur. J. Pharmacol. 2009, 606, 115–120. [Google Scholar] [CrossRef]
- Yamamoto, K.; Masuyama, T.; Sakata, Y.; Mano, T.; Nishikawa, N.; Kondo, H.; Akehi, N.; Kuzuya, T.; Miwa, T.; and Hori, M. Roles of renin-angiotensin and endothelin systems in development of diastolic heart failure in hypertensive hearts. Cardiovasc. Res. 2000, 47, 274–283. [Google Scholar] [CrossRef]
- Abraham, D.J.; Vancheeswaran, R.; Dashwood, M.R.; Rajkumar, V.S.; Pantelides, P.; Xu, S.W.; du Bois, R.M.; Black, C.M. Increased levels of endothelin-1 and differential endothelin type A and B receptor expression in scleroderma-associated fibrotic lung disease. Am. J. Pathol. 1997, 151, 831. [Google Scholar]
- Davie, N.; Haleen, S.J.; Upton, P.D.; Polak, J.M.; Yacoub, M.H.; Morrell, N.W.; Wharton, J. ETA and ETB receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am. J. Respir. Crit. Care Med. 2002, 165, 398–405. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.Q.; Zhang, J.; Ramires, F.J. Angiotensin II, transforming growth factor-β1 and repair in the infarcted heart. J. Mol. Cell. Cardiol. 1998, 30, 1559–1569. [Google Scholar] [CrossRef]
- Imai, T.; Hirata, Y.; Emori, T.; Yanagisawa, M.; Masaki, T.; Marumo, F. Induction of endothelin-1 gene by angiotensin and vasopressin in endothelial cells. Hypertension 1992, 19, 753–757. [Google Scholar] [CrossRef]
- Sung, C.P.; Arleth, A.J.; Storer, B.L.; Ohlstein, E.H. Angiotensin type 1 receptors mediate smooth muscle proliferation and endothelin biosynthesis in rat vascular smooth muscle. J. Pharmacol. Exp. Ther. 1994, 271, 429–437. [Google Scholar]
- Shi-Wen, X.; Rodríguez-Pascual, F.; Lamas, S.; Holmes, A.; Howat, S.; Pearson, J.D.; Dashwood, M.R.; du Bois, R.M.; Denton, C.P.; Black, C.M.; et al. Constitutive ALK5-independent c-Jun N-terminal kinase activation contributes to endothelin-1 overexpression in pulmonary fibrosis: Evidence of an autocrine endothelin loop operating through the endothelin A and B receptors. Molecular and cellular biology. Mol. Cell. Biol. 2006, 26, 5518–5527. [Google Scholar] [CrossRef]
- Garrison, G.; Huang, S.K.; Okunishi, K.; Scott, J.P.; Kumar Penke, L.R.; Scruggs, A.M.; Peters-Golden, M. Reversal of myofibroblast differentiation by prostaglandin E2. Am. J. Respir. Cell. Mol. Biol. 2013, 48, 550–558. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, K.; Locy, M.L.; Ravi, S.; Deshane, J.; Mannon, R.B.; et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef]
- Duangrat, R.; Parichatikanond, W.; Likitnukul, S.; Mangmool, S. Endothelin-1 induces cell proliferation and myofibroblast differentiation through the ETAR/Gαq/ERK signaling pathway in human cardiac fibroblasts. Int. J. Mol. Sci. 2023, 24, 4475. [Google Scholar] [CrossRef]
- Phosri, S.; Bunrukchai, K.; Parichatikanond, W.; Sato, V.H.; Mangmool, S. Epac is required for exogenous and endogenous stimulation of adenosine A2B receptor for inhibition of angiotensin II-induced collagen synthesis and myofibroblast differentiation. Purinergic Signal 2018, 14, 141–156. [Google Scholar] [CrossRef]
- Mangmool, S.; Kyaw, E.T.H.; Nuamnaichati, N.; Pandey, S.; Parichatikanond, W. Stimulation of adenosine A1 receptor prevents oxidative injury in H9c2 cardiomyoblasts: Role of Gβγ-mediated Akt and ERK1/2 signaling. Toxicol. Appl. Pharmacol. 2022, 451, 116175. [Google Scholar] [CrossRef]
- Nuamnaichati, N.; Sato, V.H.; Moongkarndi, P.; Parichatikanond, P.; Mangmool, S. Sustained β-AR stimulation induces synthesis and secretion of growth factors in cardiac myocytes that affect on cardiac fibroblast activation. Life Sci. 2018, 193, 257–269. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duangrat, R.; Parichatikanond, W.; Mangmool, S. Dual Blockade of TGF-β Receptor and Endothelin Receptor Synergistically Inhibits Angiotensin II-Induced Myofibroblast Differentiation: Role of AT1R/Gαq-Mediated TGF-β1 and ET-1 Signaling. Int. J. Mol. Sci. 2023, 24, 6972. https://doi.org/10.3390/ijms24086972
Duangrat R, Parichatikanond W, Mangmool S. Dual Blockade of TGF-β Receptor and Endothelin Receptor Synergistically Inhibits Angiotensin II-Induced Myofibroblast Differentiation: Role of AT1R/Gαq-Mediated TGF-β1 and ET-1 Signaling. International Journal of Molecular Sciences. 2023; 24(8):6972. https://doi.org/10.3390/ijms24086972
Chicago/Turabian StyleDuangrat, Ratchanee, Warisara Parichatikanond, and Supachoke Mangmool. 2023. "Dual Blockade of TGF-β Receptor and Endothelin Receptor Synergistically Inhibits Angiotensin II-Induced Myofibroblast Differentiation: Role of AT1R/Gαq-Mediated TGF-β1 and ET-1 Signaling" International Journal of Molecular Sciences 24, no. 8: 6972. https://doi.org/10.3390/ijms24086972
APA StyleDuangrat, R., Parichatikanond, W., & Mangmool, S. (2023). Dual Blockade of TGF-β Receptor and Endothelin Receptor Synergistically Inhibits Angiotensin II-Induced Myofibroblast Differentiation: Role of AT1R/Gαq-Mediated TGF-β1 and ET-1 Signaling. International Journal of Molecular Sciences, 24(8), 6972. https://doi.org/10.3390/ijms24086972