

Microscopic and Biochemical Hallmarks of BICD2-Associated Muscle Pathology toward the Evaluation of Novel Variants

 , , , and
, , , and
Abstract
1. Introduction
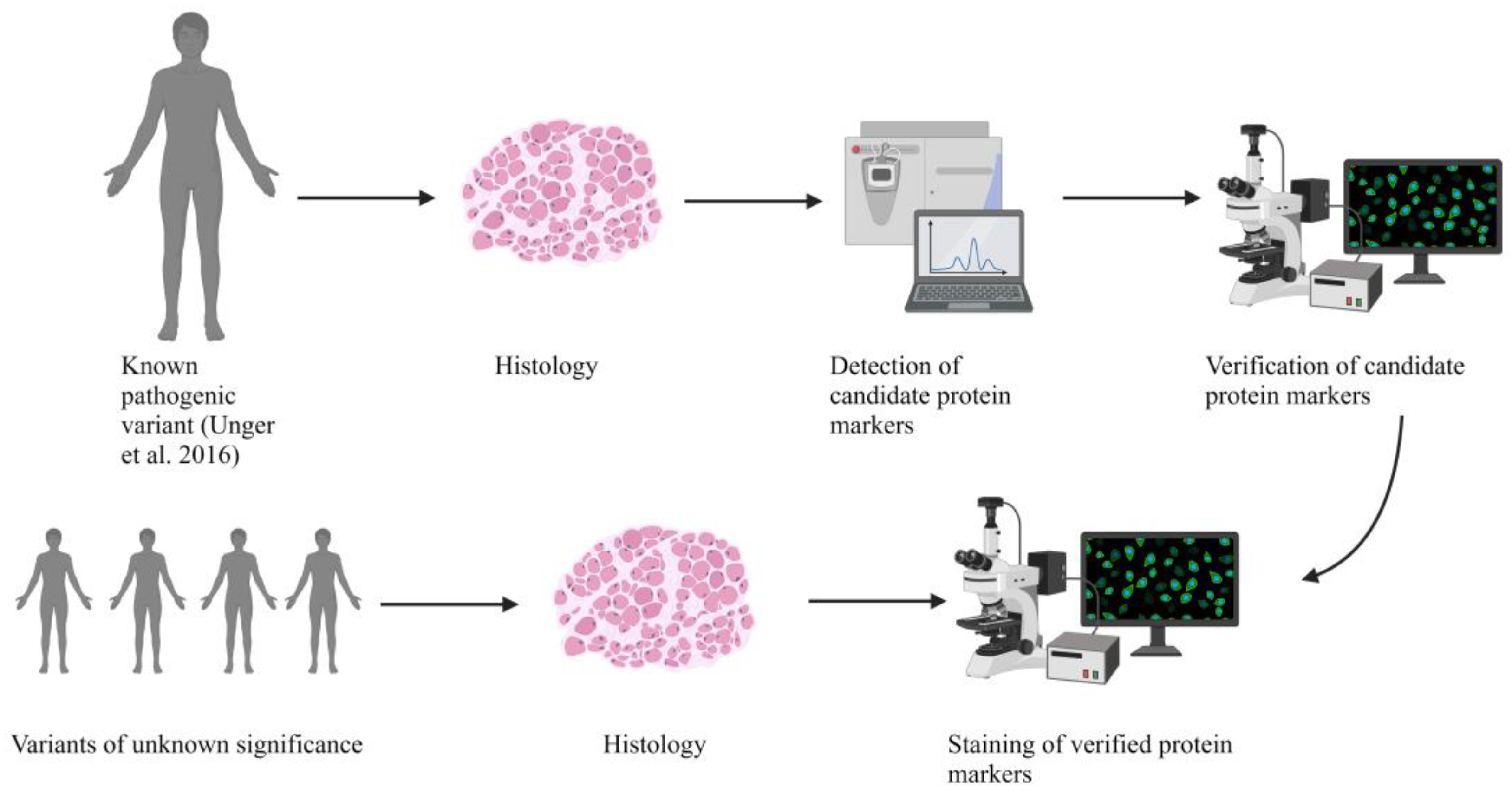
2. Results
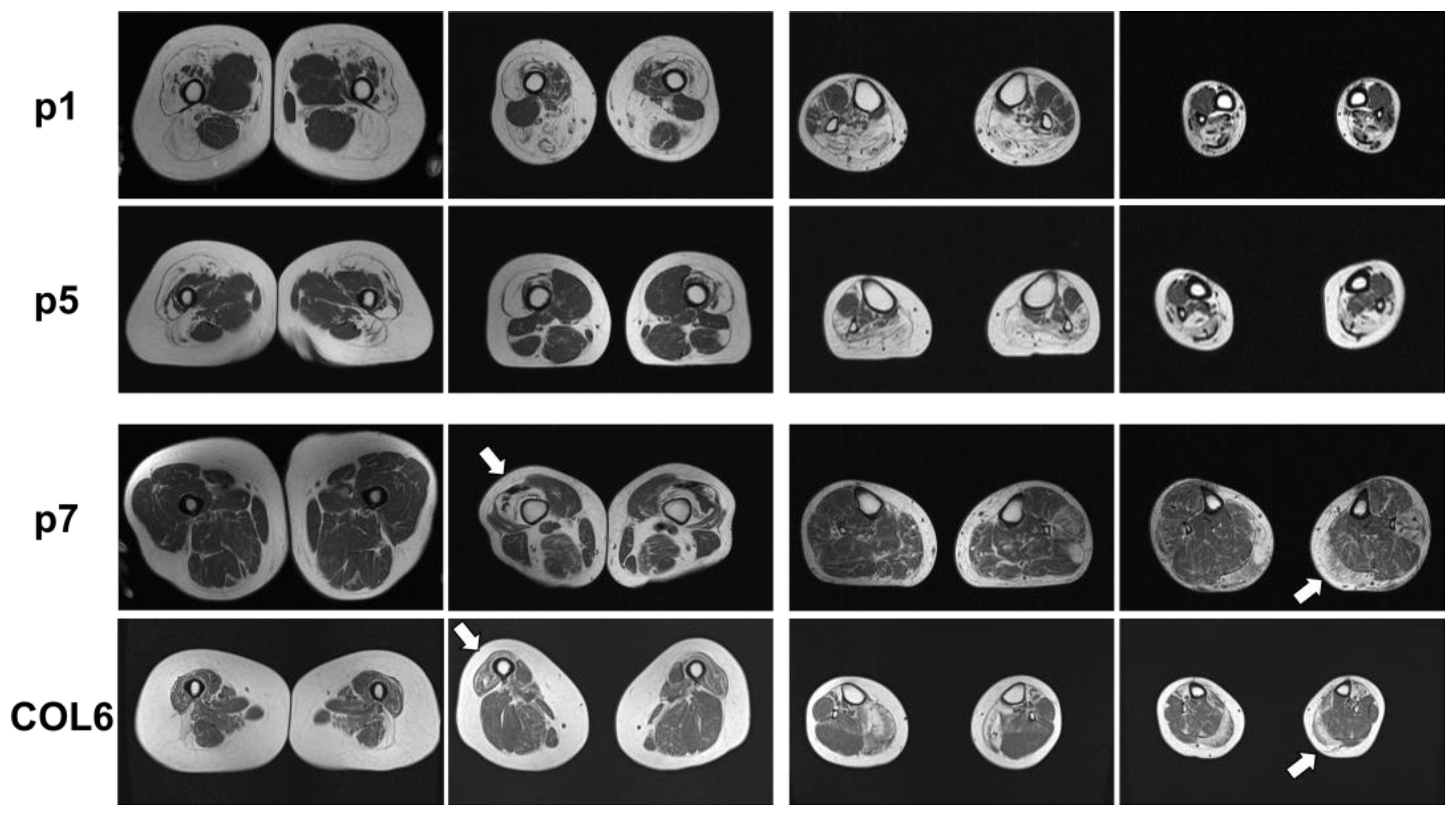
2.1. Clinical and MRI Features of Patients with BICD2-Associated Myopathy Reveal Typical Pattern of Muscle Weakness of the Legs (with Sparing of Adductor Muscles)
2.2. Genetic Analysis Revealed a Known Pathogenic and Four Novel Sequence Variants in BICD2
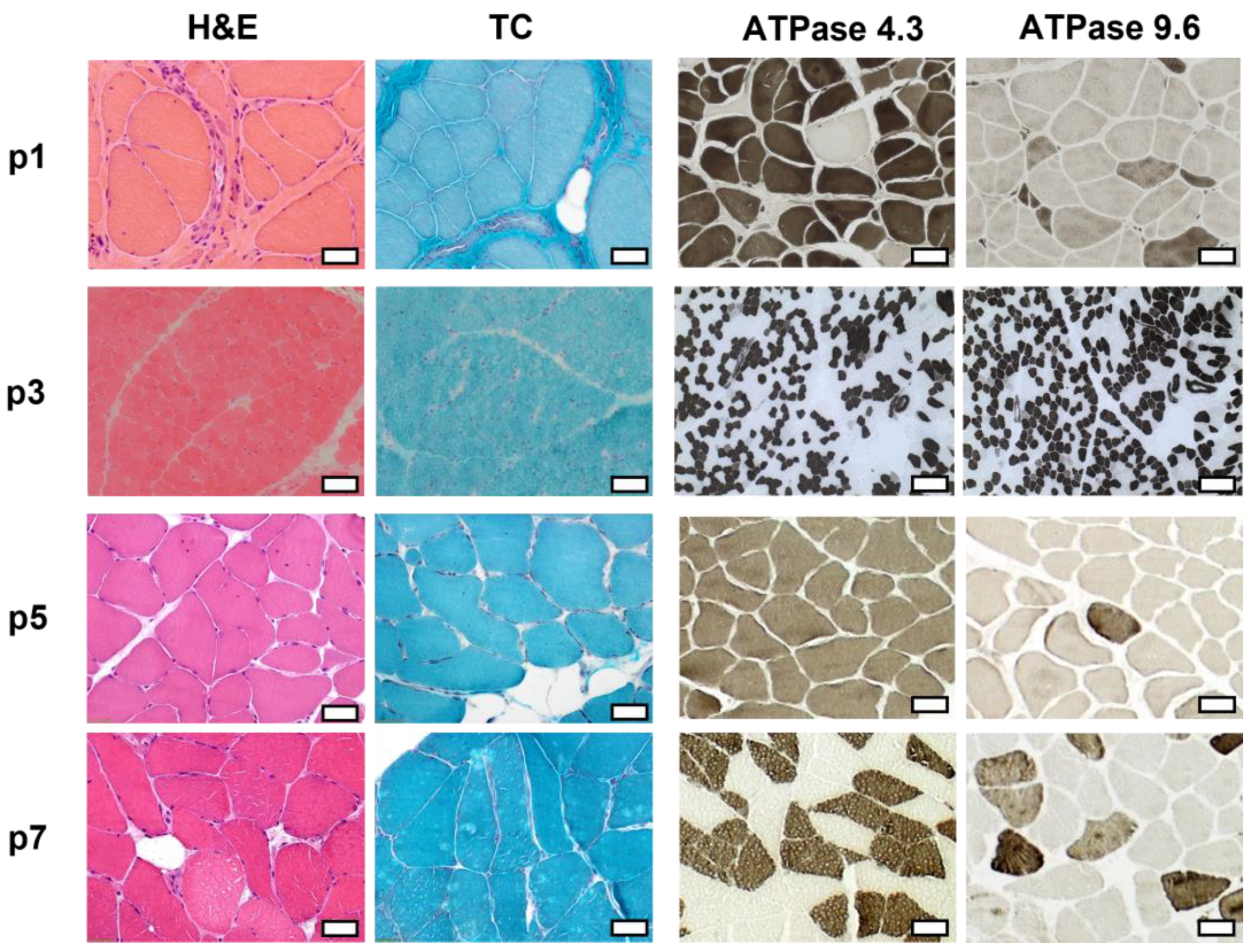
2.3. Histopathologic Analysis Reveals Slight Myopathic Changes in Novel BICD2-Patients
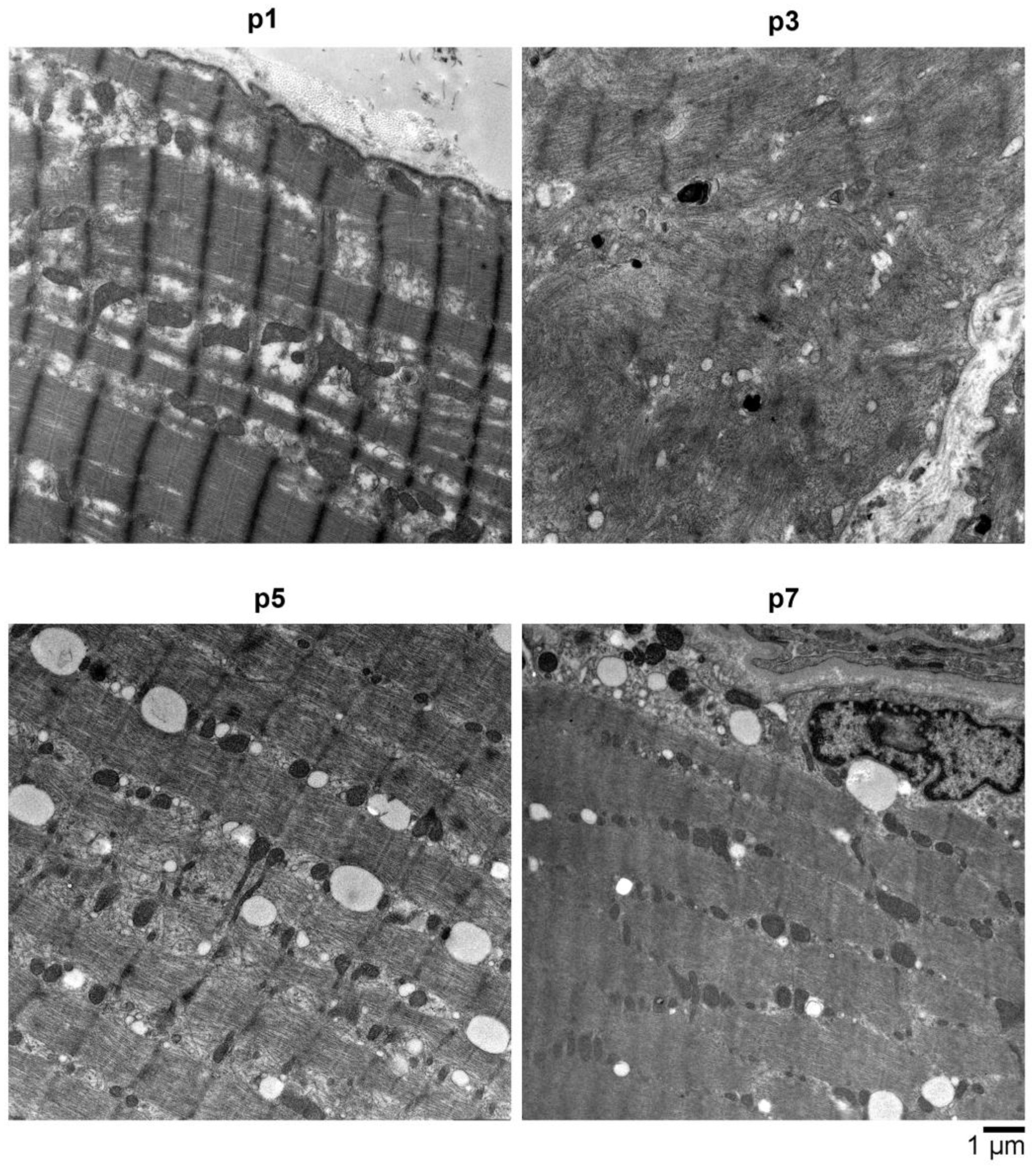
2.4. Electron Microscopy (EM) Showed Myopathic Changes including Abundant Autophagic Vacuoles
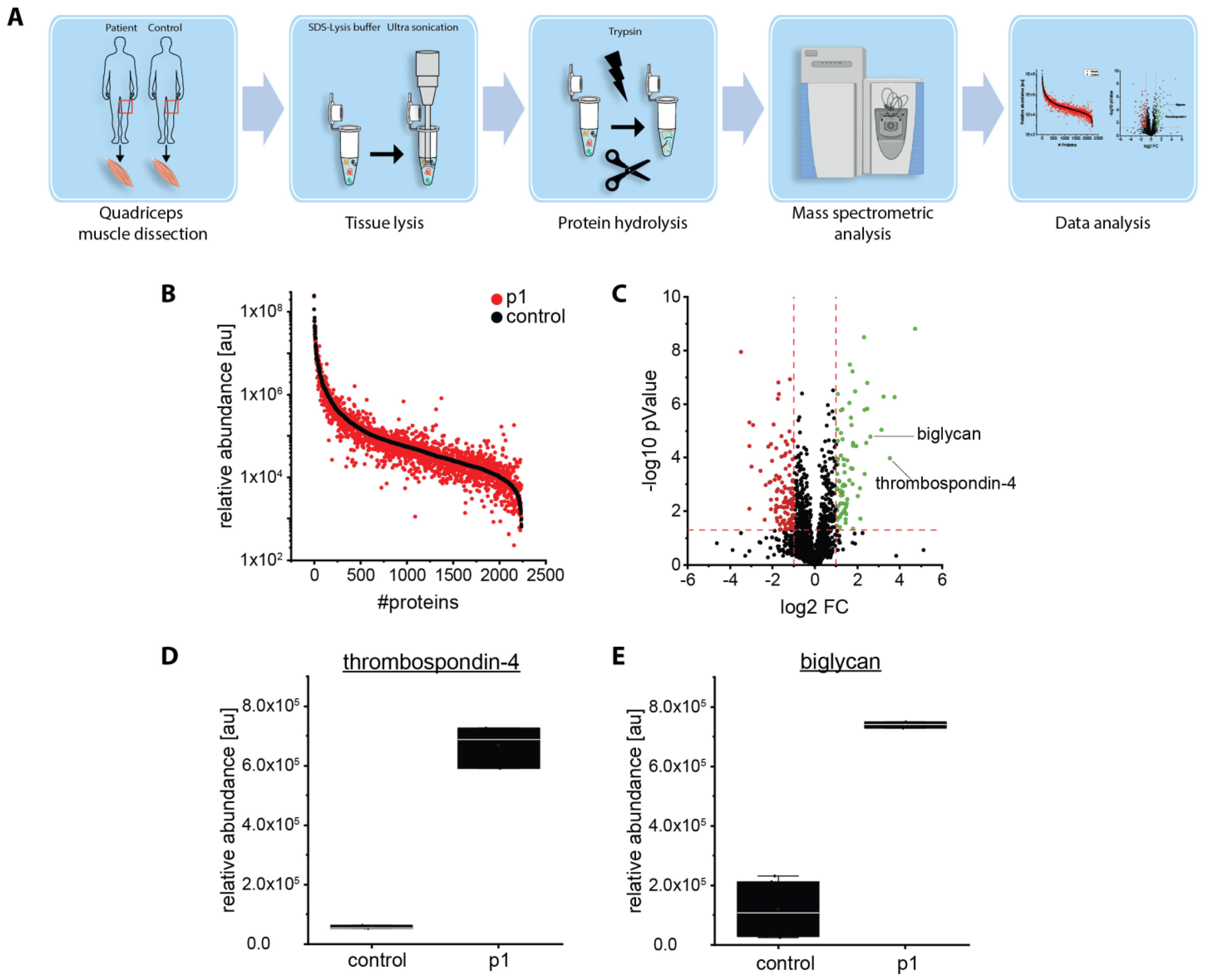
2.5. Proteomic Analysis Revealed an Increase of Proteins Associated with Perturbed Vesicular Transport
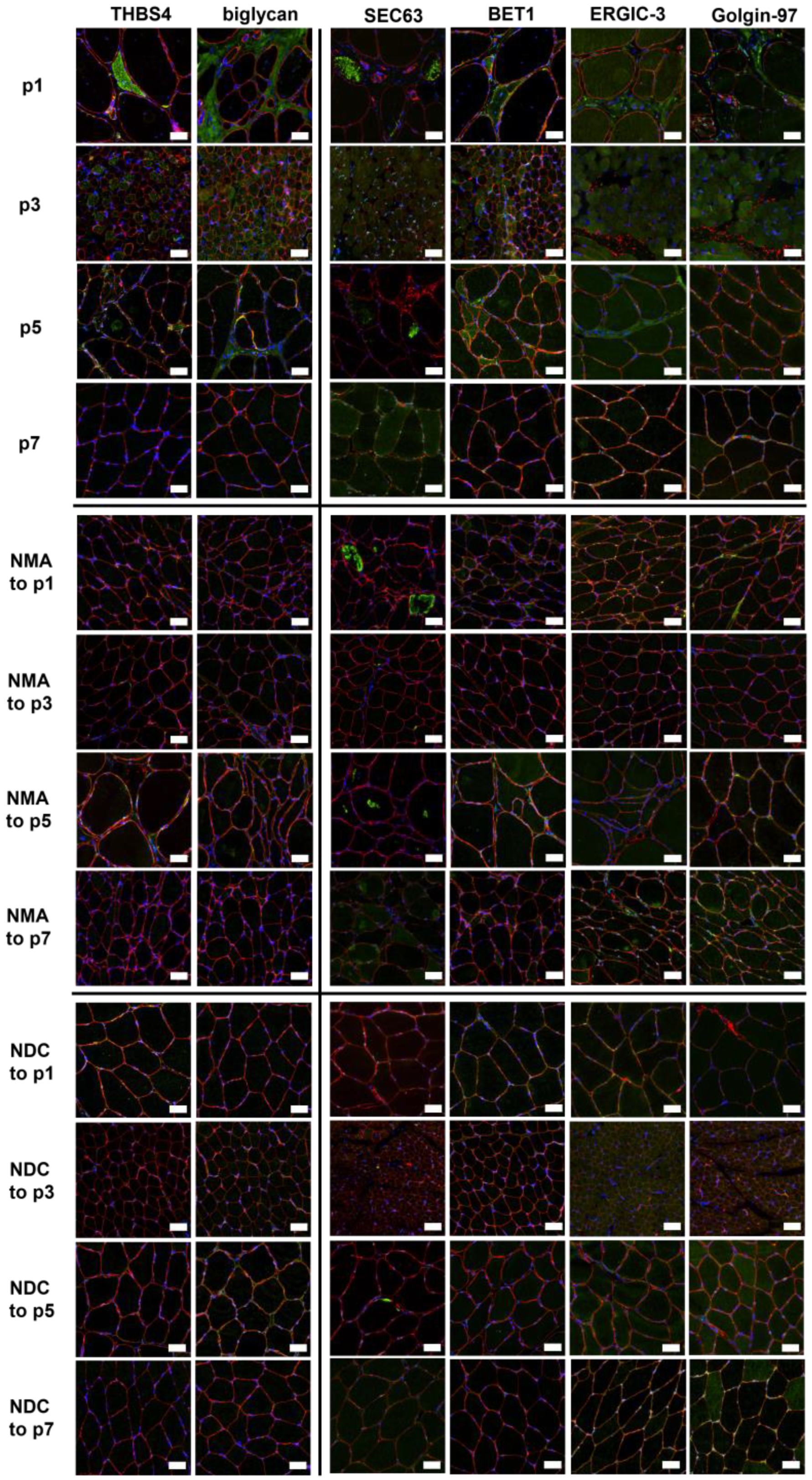
2.6. Verification of Proteomic Data by Immunofluorescence Analyses
3. Discussion
4. Materials and Methods
4.1. Patients and Muscle Biopsies
4.2. Muscle MRI
4.3. Genetic Analyses
4.4. Histological Studies and Immunofluorescence Stainings
4.5. Electron Microscopy (EM)
4.6. Proteomic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matanis, T.; Akhmanova, A.; Wulf, P.; Del Nery, E.; Weide, T.; Stepanova, T.; Galjart, N.; Grosveld, F.; Goud, B.; de Zeeuw, C.I.; et al. Bicaudal-D regulates COPI-independent Golgi-ER transport by recruiting the dynein-dynactin motor complex. Nat. Cell Biol. 2002, 4, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Peeters, K.; Litvinenko, I.; Asselbergh, B.; Almeida-Souza, L.; Chamova, T.; Geuens, T.; Ydens, E.; Zimoń, M.; Irobi, J.; De Vriendt, E.; et al. Molecular Defects in the Motor Adaptor BICD2 Cause Proximal Spinal Muscular Atrophy with Autosomal-Dominant Inheritance. Am. J. Hum. Genet. 2013, 92, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Hoogenraad, C.C.; Akhmanova, A.; Howell, S.A.; Dortland, B.R.; de Zeeuw, C.I.; Willemsen, R.; Visser, P.; Grosveld, F.; Galjart, N. Mammalian Golgi-associated Bicaudal-D2 functions in the dynein–dynactin pathway by interacting with these complexes. EMBO J. 2001, 20, 4041–4054. [Google Scholar] [CrossRef] [PubMed]
- Jaarsma, D.; Hoogenraad, C.C. Cytoplasmic dynein and its regulatory proteins in Golgi pathology in nervous system disorders. Front. Neurosci. 2015, 9, 397. [Google Scholar] [CrossRef] [PubMed]
- Schlager, M.A.; Kapitein, L.C.; Grigoriev, I.; Burzynski, G.M.; Wulf, P.S.; Keijzer, N.; de Graaff, E.; Fukuda, M.; Shepherd, I.T.; Akhmanova, A.; et al. Pericentrosomal targeting of Rab6 secretory vesicles by Bicaudal-D-related protein 1 (BICDR-1) regulates neuritogenesis. EMBO J. 2010, 29, 1637–1651. [Google Scholar] [CrossRef]
- Short, B.; Preisinger, C.; Schaletzky, J.; Kopajtich, R.; Barr, F.A. The Rab6 GTPase regulates recruitment of the dynactin complex to Golgi membranes. Curr. Biol. 2002, 12, 1792–1795. [Google Scholar] [CrossRef]
- Neveling, K.; Martinez-Carrera, L.A.; Hölker, I.; Heister, A.; Verrips, A.; Hosseini-Barkooie, S.M.; Gilissen, C.; Vermeer, S.; Pennings, M.; Meijer, R.; et al. Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am. J. Hum. Genet. 2013, 92, 946–954. [Google Scholar] [CrossRef]
- Oates, E.C.; Rossor, A.M.; Hafezparast, M.; Gonzalez, M.; Speziani, F.; MacArthur, D.G.; Lek, M.; Cottenie, E.; Scoto, M.; Foley, A.R.; et al. Mutations in BICD2 Cause Dominant Congenital Spinal Muscular Atrophy and Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2013, 92, 965–973. [Google Scholar] [CrossRef]
- Rossor, A.M.; Oates, E.C.; Salter, H.K.; Liu, Y.; Murphy, S.M.; Schule, R.; Gonzalez, M.A.; Scoto, M.; Phadke, R.; Sewry, C.A.; et al. Phenotypic and molecular insights into spinal muscular atrophy due to mutations in BICD2. Brain 2014, 138, 293–310. [Google Scholar] [CrossRef]
- Synofzik, M.; Martinez-Carrera, L.A.; Lindig, T.; Schöls, L.; Wirth, B. Dominant spinal muscular atrophy due to BICD2: A novel mutation refines the phenotype. J. Neurol. Neurosurg. Psychiatry 2014, 85, 590–592. [Google Scholar] [CrossRef]
- Martinez Carrera, L.A.; Gabriel, E.; Donohoe, C.D.; Hölker, I.; Mariappan, A.; Storbeck, M.; Uhlirova, M.; Gopalakrishnan, J.; Wirth, B. Novel insights into SMALED2: BICD2 mutations increase microtubule stability and cause defects in axonal and NMJ development. Hum. Mol. Genet. 2018, 27, 1772–1784. [Google Scholar] [CrossRef]
- Unger, A.; Dekomien, G.; Güttsches, A.; Dreps, T.; Kley, R.; Tegenthoff, M.; Ferbert, A.; Weis, J.; Heyer, C.; Linke, W.A.; et al. Expanding the phenotype of BICD2 mutations toward skeletal muscle involvement. Neurology 2016, 87, 2235–2243. [Google Scholar] [CrossRef]
- Rossor, A.M.; Sleigh, J.N.; Groves, M.; Muntoni, F.; Reilly, M.M.; Hoogenraad, C.C.; Schiavo, G. Loss of BICD2 in muscle drives motor neuron loss in a developmental form of spinal muscular atrophy. Acta Neuropathol. Commun. 2020, 8, 34. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Huynh, W.; Vale, R.D. Disease-associated mutations in human BICD2 hyperactivate motility of dynein-dynactin. J. Cell Biol. 2017, 216, 3051–3060. [Google Scholar] [CrossRef]
- Grigoriev, I.; Splinter, D.; Keijzer, N.; Wulf, P.S.; Demmers, J.; Ohtsuka, T.; Modesti, M.; Maly, I.V.; Grosveld, F.; Hoogenraad, C.C.; et al. Rab6 regulates transport and targeting of exocytotic carriers. Dev. Cell 2007, 13, 305–314. [Google Scholar] [CrossRef]
- Splinter, D.; Razafsky, D.S.; Schlager, M.A.; Serra-Marques, A.; Grigoriev, I.; Demmers, J.; Keijzer, N.; Jiang, K.; Poser, I.; Hyman, A.A.; et al. BICD2, dynactin, and LIS1 cooperate in regulating dynein recruitment to cellular structures. Mol. Biol. Cell 2012, 23, 4226–4241. [Google Scholar] [CrossRef]
- Reid, E.; Kloos, M.; Ashley-Koch, A.; Hughes, L.; Bevan, S.; Svenson, I.K.; Graham, F.L.; Gaskell, P.C.; Dearlove, A.; Pericak-Vance, M.A.; et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J. Hum. Genet. 2002, 71, 1189–1194. [Google Scholar] [CrossRef]
- Arber, S.; Caroni, P. Thrombospondin-4, an extracellular matrix protein expressed in the developing and adult nervous system promotes neurite outgrowth. J. Cell Biol. 1995, 131, 1083–1094. [Google Scholar] [CrossRef]
- Vanhoutte, D.; Schips, T.G.; Kwong, J.Q.; Davis, J.; Tjondrokoesoemo, A.; Brody, M.J.; Sargent, M.A.; Kanisicak, O.; Yi, H.; Gao, Q.Q.; et al. Thrombospondin expression in myofibers stabilizes muscle membranes. eLife 2016, 5, e17589. [Google Scholar] [CrossRef]
- Amenta, A.R.; Creely, H.E.; Mercado, M.L.T.; Hagiwara, H.; McKechnie, B.A.; Lechner, B.E.; Rossi, S.G.; Wang, Q.; Owens, R.T.; Marrero, E.; et al. Biglycan is an extracellular MuSK binding protein important for synapse stability. J. Neurosci. 2012, 32, 2324–2334. [Google Scholar] [CrossRef] [PubMed]
- Janin, A.; N’Guyen, K.; Habib, G.; Dauphin, C.; Chanavat, V.; Bouvagnet, P.; Eschalier, R.; Streichenberger, N.; Chevalier, P.; Millat, G. Truncating mutations on myofibrillar myopathies causing genes as prevalent molecular explanations on patients with dilated cardiomyopathy. Clin. Genet. 2017, 92, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.-Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.T.; Wolfe, D. Tissue processing and hematoxylin and eosin staining. Methods Mol. Biol. 2014, 1180, 31–43. [Google Scholar] [CrossRef]
- Gangfuß, A.; Hentschel, A.; Heil, L.; Gonzalez, M.; Schönecker, A.; Depienne, C.; Nishimura, A.; Zengeler, D.; Kohlschmidt, N.; Sickmann, A.; et al. Proteomic and morphological insights and clinical presentation of two young patients with novel mutations of BVES (POPDC1). Mol. Genet. Metab. 2022, 136, 226–237. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Mutation | Onset | Pattern of Muscle Weakness | Muscle Atrophy | Creatin-Kinase (CK) Level |
|---|---|---|---|---|---|
| p1 | Heterozygous c.320C>T; p.(Ser107Leu) Published in [12] | Congenital | Pronounced paresis of distal leg muscles | Calf atrophy, pes cavus | 600–650 U/L |
| p2 | Heterozygous c.1195C>T; p.(Arg399Cys) | Early adulthood | Severe paresis pronounced in proximal and distal leg muscles, mild paresis of proximal and distal arm muscles, MRI: adductor muscles spared | Proximal and distal leg muscles | 200–300 U/L |
| p3 (son of p4) | Heterozygous c.2189G>A; p.(Arg730His) | Early childhood | Distal paresis of leg muscles | Distal leg muscles | 200–600 U/L |
| p4 (father of p3) | Heterozygous c.2189G>A; p.(Arg730His) | Early childhood | Distal paresis of leg muscles | Distal leg muscles (right >>left) | 600–650 U/L |
| p5 (mother of p6) | BICD2: heterozygous c.1904G>T; p.(Arg635Leu), FLNC: heterozygous c.2272G>A; p.(Val758Met) | Early childhood | Pronounced paresis of distal leg muscles, slight proximal paresis of arms and legs | Distal leg muscles and shoulder muscles | 150–200 U/L |
| p6 (daughter of p5) | BICD2: heterozygous c.1904G>T; (p.Arg635Leu), FLNC: heterozygous c.2272G>A; (p.Val758Met) | Early childhood | Pronounced paresis of distal leg muscles, slight proximal paresis of arms and legs, hyperlordosis | Distal leg muscles | 50–100 U/L |
| p7 | BICD2: heterozygous c.2452A>G; p.(Lys818Glu), COL6A1: heterozygous c.1694G>A; p.(Arg565Gln) | Late adulthood | Proximal paresis of the lower limb muscles | Thigh muscles | 200–300 U/L |
| Patient | Biopsied Muscle Age at Biopsy | Light Microscopy | Fiber Size | Oxidative Enzyme Reaction | IF/WB Analysis |
|---|---|---|---|---|---|
| p1 | Vastus lateralis 32 years | Lipofibromatosis, fiber size variation with some angular atrophic fibers, type-1-fiber predominance, grouping of both fiber types, fiber splittings, central nuclei, few necrotic and regenerating fibers, few nuclear clumps | Type 1 fibers: 9–186 µm Type 2 fibers: 4–125 µm | No abnormalities | Reduced: dystrophin 2, dysferlin, alpha-dystroglycan Normal: dystrophin 1 and 3, alpha-sarcoglycan, gamma-sarcoglycan, laminin alpha 2 (80 kD + 300 kD), dysferlin, calpain 2C4 |
| p2 | Gastrocnemius, caput mediale 40 years | Mild lipofibromatosis, fiber necrosis and regenerating fibers, central nuclei, vacuolar changes | Type 1 fibers: 16–161 µm Type 2 fibers: 4–136 µm | Few fibers with irregular oxidative enzyme reaction | Aggregates of: desmin myotilin |
| p3 | Quadriceps femoris 19 months | Fiber size variation, few angular atrophic fibers, very few regenerating fibers, very few necrosis | Type 1 fibers: 7.5–17.5 µm Type 2 fibers: 7.5–25 µm | No abnormalities | Reduced: dystrophin 1–3 alpha-dystroglycan Normal: beta-dystroglycan alpha-, beta-, gamma-, and delta-sarcoglycan laminin alpha 2 (80 kD + 300 kD), dysferlin, collagen VI, utrophin, neonatal myosin, calpain 3, dysferlin |
| p5 | Tibialis anterior 55 years | Central nuclei, few fiber splittings, fiber size variation, type1-fiber-predominance | Type 1 fibers: 9–106 µm Type 2 fibers: 19–90 µm | Many fibers with irregular oxidative enzyme reaction | Reduced: dysferlin (normal in WB analysis) Normal dystrophin 2, alpha-sarcoglycan, gamma-sarcoglycan, alpha-dystroglycan, laminin alpha 2 (80 kD + 300 kD), myotilin, caveolin 3, calpain 2C4 |
| p7 | Vastus medialis 67 years | Mild lipofibromatosis, slight fiber size variation | Type 1 fibers: 13–116 µm Type 2 fibers: 20–107 µm | No abnormalities | Reduced: dysferlin Normal: Dystrophin 1, 2, and 3, alpha- and gamma-sarcoglycan, alpha-dystroglycan, calpain 2C4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unger, A.; Roos, A.; Gangfuß, A.; Hentschel, A.; Gläser, D.; Krause, K.; Doering, K.; Schara-Schmidt, U.; Hoffjan, S.; Vorgerd, M.; et al. Microscopic and Biochemical Hallmarks of BICD2-Associated Muscle Pathology toward the Evaluation of Novel Variants. Int. J. Mol. Sci. 2023, 24, 6808. https://doi.org/10.3390/ijms24076808
Unger A, Roos A, Gangfuß A, Hentschel A, Gläser D, Krause K, Doering K, Schara-Schmidt U, Hoffjan S, Vorgerd M, et al. Microscopic and Biochemical Hallmarks of BICD2-Associated Muscle Pathology toward the Evaluation of Novel Variants. International Journal of Molecular Sciences. 2023; 24(7):6808. https://doi.org/10.3390/ijms24076808
Chicago/Turabian StyleUnger, Andreas, Andreas Roos, Andrea Gangfuß, Andreas Hentschel, Dieter Gläser, Karsten Krause, Kristina Doering, Ulrike Schara-Schmidt, Sabine Hoffjan, Matthias Vorgerd, and et al. 2023. "Microscopic and Biochemical Hallmarks of BICD2-Associated Muscle Pathology toward the Evaluation of Novel Variants" International Journal of Molecular Sciences 24, no. 7: 6808. https://doi.org/10.3390/ijms24076808
APA StyleUnger, A., Roos, A., Gangfuß, A., Hentschel, A., Gläser, D., Krause, K., Doering, K., Schara-Schmidt, U., Hoffjan, S., Vorgerd, M., & Güttsches, A.-K. (2023). Microscopic and Biochemical Hallmarks of BICD2-Associated Muscle Pathology toward the Evaluation of Novel Variants. International Journal of Molecular Sciences, 24(7), 6808. https://doi.org/10.3390/ijms24076808