3.2.1. General Procedure for the Synthesis of Building Blocks BB1–BB4
All methyl ester carbamate building blocks (BB1, BB2b, BB2c, BB2d, BB2e, BB3f, BB3g and BB4h) were obtained from appropriate amino acids using the same two step procedure.
Step I: Thionylchloride (2 eq.) was slowly added to a cooled (−78 °C) suspension of amino acid (1 eq.) in CH3OH (20 mL/g) under an inert atmosphere. Then, the reaction mixture was warmed up to ambient temperature and stirred at this temperature overnight. Following this, the solvent was removed under reduced pressure and the solid residue was filtered and washed with diethyl ether. The product was taken to the next step without further purification.
Step II: The product (1 eq.,) from the previous step was dissolved in dry DCM (20 mL/g) at 0 °C, and DIPEA (3 eq.) was added. After stirring for 10 min, the solution was added dropwise into a flask containing solid N,N′-disuccinimidyl carbonate (DSC, 1.2 eq.). The reaction was stirred for 3 h under Ar at RT. Following this, the reaction mixture was washed with 1M KHSO
4 (3×) and brine (1×). In some cases, the products remained in the aqueous layer, so this layer was extracted with DCM until all the desired compound was in the organic layer. Combined organic layers were dried over Na
2SO
4. After evaporation under reduced pressure, the crude product was obtained as a colorless/yellowish oil. It was purified by dissolving it in small amount of DCM, followed by precipitation with diethyl ether and/or petroleum ether. The pure product was obtained as a white solid (except
BB4h, which was a yellowish oil used without further purification). Copies of
1H and
13C NMR spectra, as well as HRMS spectra of building blocks are given in the
Supporting Information, Figures S4–S23 and Figures S61–S70.
BB1: reagents in step I: 2-aminoisobutyric acid (2 g, 19.4 mmol), thionyl chloride (2.8 mL, 38.8 mmol) and methanol (40 mL). Reagents in step II: DSC (5.97 g, 23.3 mmol), DIPEA (10,1 mL, 58.2 mmol) and DCM (60 mL).
White solid, yield (after 2 steps): 56% (2.8g); 1H NMR (300 MHz, CDCl3) δ 6.18 (s, 1H), 3.82 (s, 3H), 2.84 (s, 4H), 1.64 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 173.74, 169.45, 149.21, 57.25, 52.71, 25.08, 23.98; HRMS ESI(+) calcd. for C10H15N2O6 259.09246 [M + H]+, found 259.09251.
BB2b: reagents in step I: Gly (293 mg, 3.9 mmol), thionyl chloride (0.57 mL, 7.8 mmol) and methanol (6 mL). Reagents in step II: DSC (1.17 g, 4.68 mmol), DIPEA (2.04 mL, 11.7 mmol) and DCM (6 mL).
White solid, yield (after 2 steps): 51% (0.46g); 1H NMR (300 MHz, CDCl3) δ 6.11 (s, 1H), 4.07 (d, J = 5.3 Hz, 2H), 3.81 (s, 3H), 2.85 (s, 4H); 13C NMR (75 MHz, CDCl3) δ 169.66, 169.08, 151.39, 52.72, 43.10, 25.47; HRMS ESI(+) calcd. for C8H11N2O6 231.06116 [M + H]+, found 231.06131.
BB2c: reagents in step I: L-Ala (1 g, 11.2 mmol), thionyl chloride (1.62 mL, 22.45 mmol) and methanol (20 mL). Reagents in step II: DSC (3.44 g, 13.44 mmol), DIPEA (7.0 mL, 40.32 mmol) and DCM (20 mL).
White solid, yield (after 2 steps): 51% (1.4 g); 1H NMR (300 MHz, CDCl3) δ 6.02 (d, J = 7.2 Hz, 1H), 4.40 (p, J = 7.2 Hz, 1H), 3.81 (s, 3H), 2.84 (s, 4H), 1.52 (d, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.79, 169.63, 150.48, 52.35, 50.09, 25.08, 17.70; HRMS ESI(+) calcd. for C9H13N2O6 245.07681 [M + H]+, found 245.07693.
BB2d: reagents in step I: L-Val (1 g, 8.54 mmol), thionyl chloride (1.24 mL, 17.08 mmol) and methanol (20 mL). Reagents in step II: DSC (2.63 g, 10.25 mmol), DIPEA (4.46 mL, 25.62 mmol) and DCM (20 mL).
White solid, yield (after 2 steps): 59% (1.38 g); 1H NMR (300 MHz, CDCl3) 1H NMR (300 MHz, CDCl3) δ 5.93 (d, J = 8.8 Hz, 1H), 4.29 (dd, J = 8.8, 4.7 Hz, 1H), 3.80 (s, 3H), 2.84 (s, 4H), 2.23 (m, 1H), 1.01 (d, J = 6.9 Hz, 1H), 0.96 (d, J = 6.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 171.17, 169.57, 151.34, 60.00, 52.54, 31.77, 25.47, 18.77, 17.45; HRMS ESI(+) calcd. for C11H17N2O6 273.10811 [M + H]+, found 273.10806.
BB2e: reagents in step I: L-Leu (2 g, 15.0 mmol), thionyl chloride (2.18 mL, 30 mmol) and methanol (20 mL). Reagents in step II: DSC (3.9 g, 15.24 mmol), DIPEA (6.6 mL, 38.1 mmol) and DCM (20 mL).
White solid, yield (after 2 steps): 66% (2.39 g); 1H NMR (300 MHz, CDCl3) δ 5.79 (d, J = 8.5 Hz, 1H), 4.41 (td, J = 8.4, 5.1 Hz, 1H), 3.80 (s, 3H), 2.84 (s, 4H), 1.82–1.57 (m, 3H), 0.98 (dd, J = 6.2, 3.7 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 171.82, 169.58, 150.84, 52.98, 52.17, 41.15, 25.07, 24.22, 22.30, 21.38; HRMS ESI(+) calcd. for C12H19N2O6 287.12376 [M + H]+, found 287.12383.
BB3f: reagents in step I: βhGly (2 g, 22.7 mmol), thionylchloride (3.3 mL, 45.3 mmol) and methanol (40 mL). Reagents in step II: DSC (6.98 mg, 27.24 mmol), DIPEA (11.86 mL, 68.1 mmol) and DCM (40 mL).
White solid, yield (after 2 steps): 62% (3.64 g); 1H NMR (300 MHz, CDCl3) δ 5.99 (s, 1H), 3.74 (s, 3H), 3.55 (q, J = 6.1 Hz, 2H), 2.84 (s, 4H), 2.63 (t, J = 6.0 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 172.46, 169.83, 151.41, 52.05, 37.42, 33.58, 25.47; HRMS ESI(+) calcd. for C9H13N2O6 245.07681 [M + H]+, found 245.07679.
BB3g: reagents in step I: (S)-3-Aminobutanoic acid (300 mg, 2.9 mmol), thionyl chloride (0.42 mL, 5.8 mmol) and methanol (7 mL). Reagents in step II: DSC (891 mg, 3.48 mmol), DIPEA (1.5 mL, 8.7 mmol) and DCM (10 mL).
White solid, yield: 53% (0.4 g); 1H NMR (300 MHz, CDCl3) δ 5.93 (d, J = 8.8 Hz, 1H), 4.13 (m, 1H), 3.74 (s, 3H), 2.84 (s, 4H), 2.64 (m, 2H), 1.35 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.09, 169.75, 150.24, 51.49, 44.97, 39.50, 25.08, 19.59; HRMS ESI(+) calcd. for C10H15N2O6 259.09246 [M + H]+, found 259.09259.
BB4h reagents in step I: γ-aminobutyric acid (1 g, 9.7 mmol), thionyl chloride (1.40 mL, 19.4 mmol) and methanol (20 mL). Reagents in step II: DSC (2.82 g, 11.64 mmol), DIPEA (5.0 mL, 29.1 mmol) and DCM (20 mL).
Yellowish oil, yield (after 2 steps): 64% (1.69 g); 1H NMR (300 MHz, CDCl3) δ 5.94 (s, 1H), 3.70 (s, 3H), 3.32 (q, J = 6.8 Hz, 2H), 2.83 (s, 4H), 2.42 (t, J = 7.2 Hz, 2H), 1.91 (p, J = 7.0 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 173.63, 170.05, 151.56, 51.85, 41.41, 31.11, 25.48, 24.46; HRMS ESI(+) calcd. for C10H15N2O6 259.09246 [M + H]+, found 259.09249.
Both methyl ester carbamate building blocks, BB4i and BB4j, were obtained starting from Boc-protected γ-amino acids. Boc deprotection procedure: protected γ-AA was treated with TFA (6 mL/g) and stirred at 0 °C for 1 h under Ar. When the deprotection reaction was complete, TFA was removed and the residue was co-evaporated with cyclohexane (3×) and diethyl ether (1×) and then reacted with thionyl chloride.
BB4i: reagents in step I: Boc-γ-Phe (250 mg, 0.85 mmol), TFA (1.5 mL), thionyl chloride (0.12 mL, 1.7 mmol) and methanol (5 mL). Reagents in step II: DSC (220 mg, 0.86 mmol), DIPEA (0.38 mL, 2.16 mmol) and DCM (5 mL).
White solid, yield (after 3 steps): 54% (0.16 g); 1H NMR (300 MHz, CDCl3) δ 7.42–7.18 (m, 5H), 5.31 (d, J = 7.0 Hz, 1H), 3.92 (m, 1H), 3.71 (s, 3H), 3.03–2.85 (m, 2H), 2.84 (s, 4H), 2.45 (t, J = 7.2 Hz, 2H), 2.04–1.72 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 173.54, 169.54, 150.78, 136.50, 129.09, 128.27, 126.40, 53.31, 51.49, 40.48, 30.23, 28.10, 25.08; HRMS ESI(+) calcd. for C17H21N2O6 349.13941 [M + H]+, found 349.13942.
BB4j: reagents in step I: Boc-γ-Leu (350 mg, 1.35 mmol), TFA (2 mL), thionylchloride (0.19 mL, 2.7 mmol) and methanol (7 mL). Reagents in step II: DSC (295 mg, 1.15 mmol), DIPEA (0.5 mL, 2.88 mmol) and DCM (7 mL).
Yellowish oil, yield (after 3 steps): 70% (295 mg); 1H NMR (300 MHz, CDCl3) δ 5.12 (d, J = 9.3 Hz, 1H), 3.81–3.73 (m, 1H), 3.72 (s, 3H), 2.83 (s, 4H), 2.45 (t, J = 7.4 Hz, 2H), 2.01–1.89 (m, 1H), 1.81–1.68 (m, 2H), 1.40–1.22 (m, 2H), 0.95 (dd, J = 6.6, 1.7 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 173.56, 169.79, 150.99, 51.37, 50.44, 43.94, 30.04, 29.93, 25.07, 24.32, 22.44, 21.71; HRMS ESI(+) calcd. for C14H23N2O6 315.15506 [M + H]+, found 315.15493.
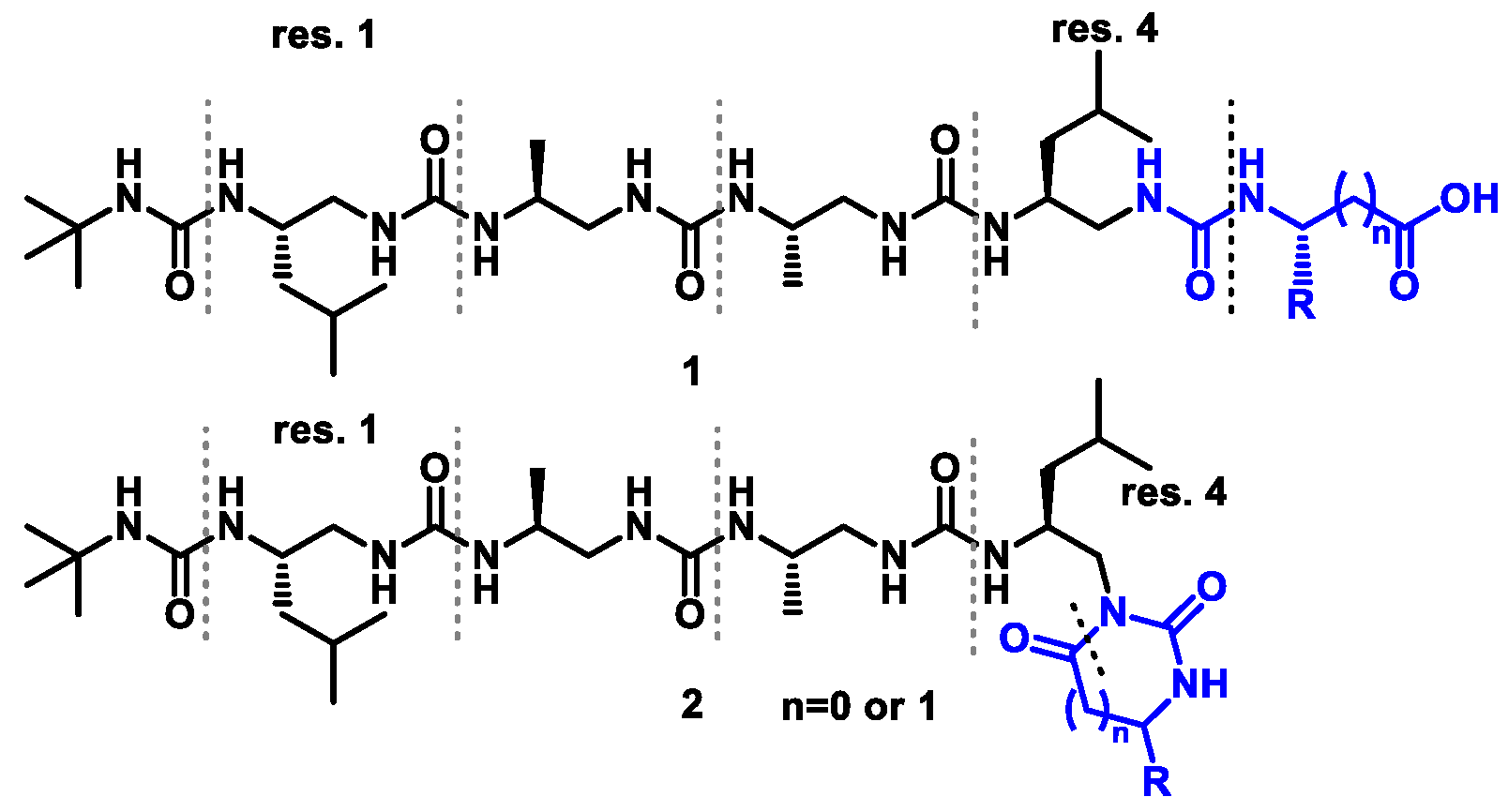
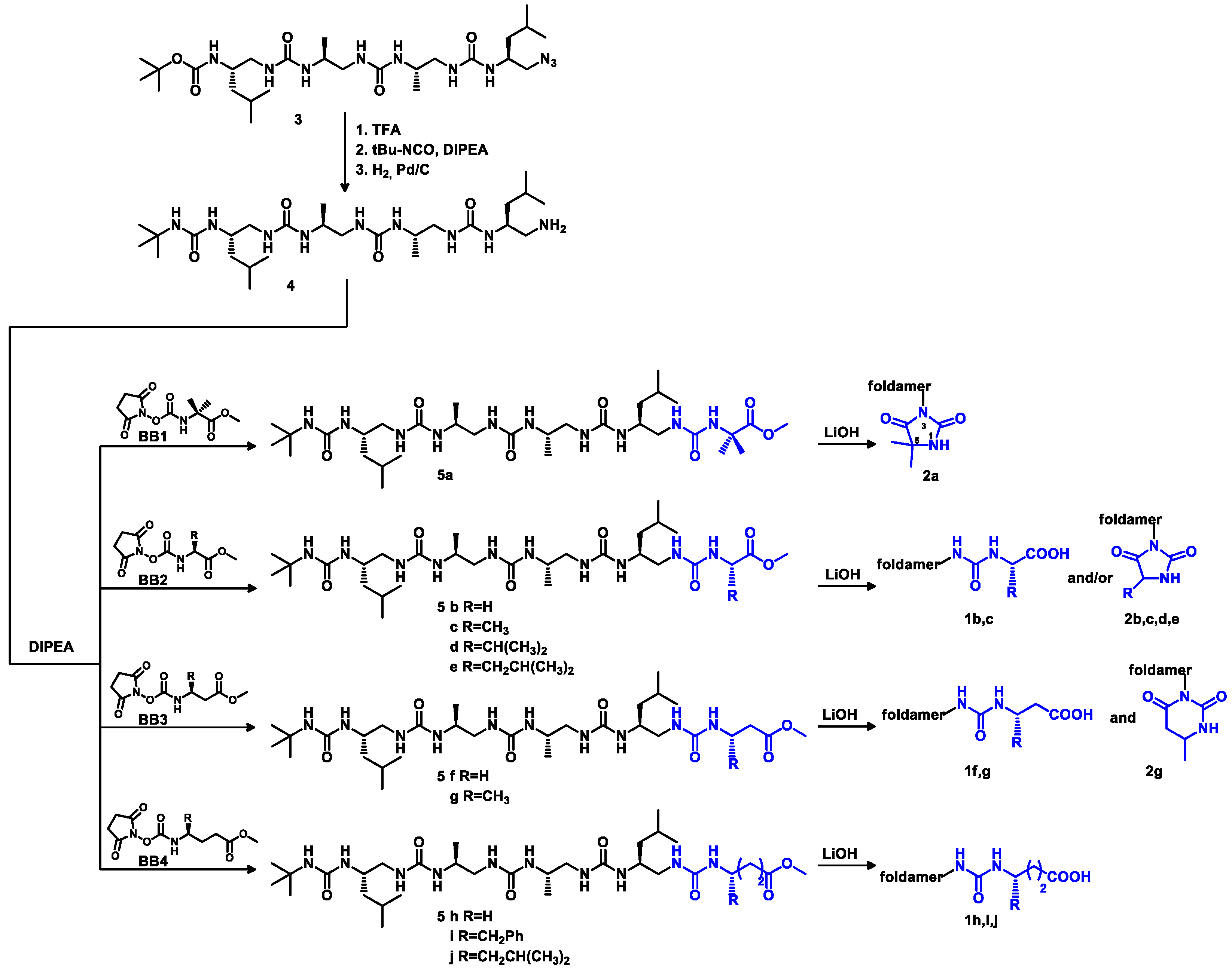
3.2.3. Synthesis of Oligourea Esters 5
Amine
4 (1 eq.) was dissolved in CH
3CN (40 mL/g) and DMF (10 mL/g) under Ar, and DIPEA (3 eq.) was added at basic pH. The mixture was left for 15 min, followed by a dropwise addition of appropriate building block
BB1–4 (1.2 eq.) in CH
3CN solution (7 mL/g). The reaction was left overnight at ambient temperature. When the reaction was complete, solvents were removed under vacuum and co-evaporated with toluene (3×) to remove DMF. The residue was redissolved in EtOAc, washed with 1M KHSO
4 (4×), saturated NaHCO
3 (1×), brine (1×) and water (1×) and then evaporated. The crude product was purified by silica gel chromatography (CH
2Cl
2:CH
3OH). The pure product was obtained as a white solid. Copies of
1H NMR spectra, as well as HRMS spectra of oligourea esters are given in the
Supporting Information, Figures S24–S33 and Figures S71–S80.
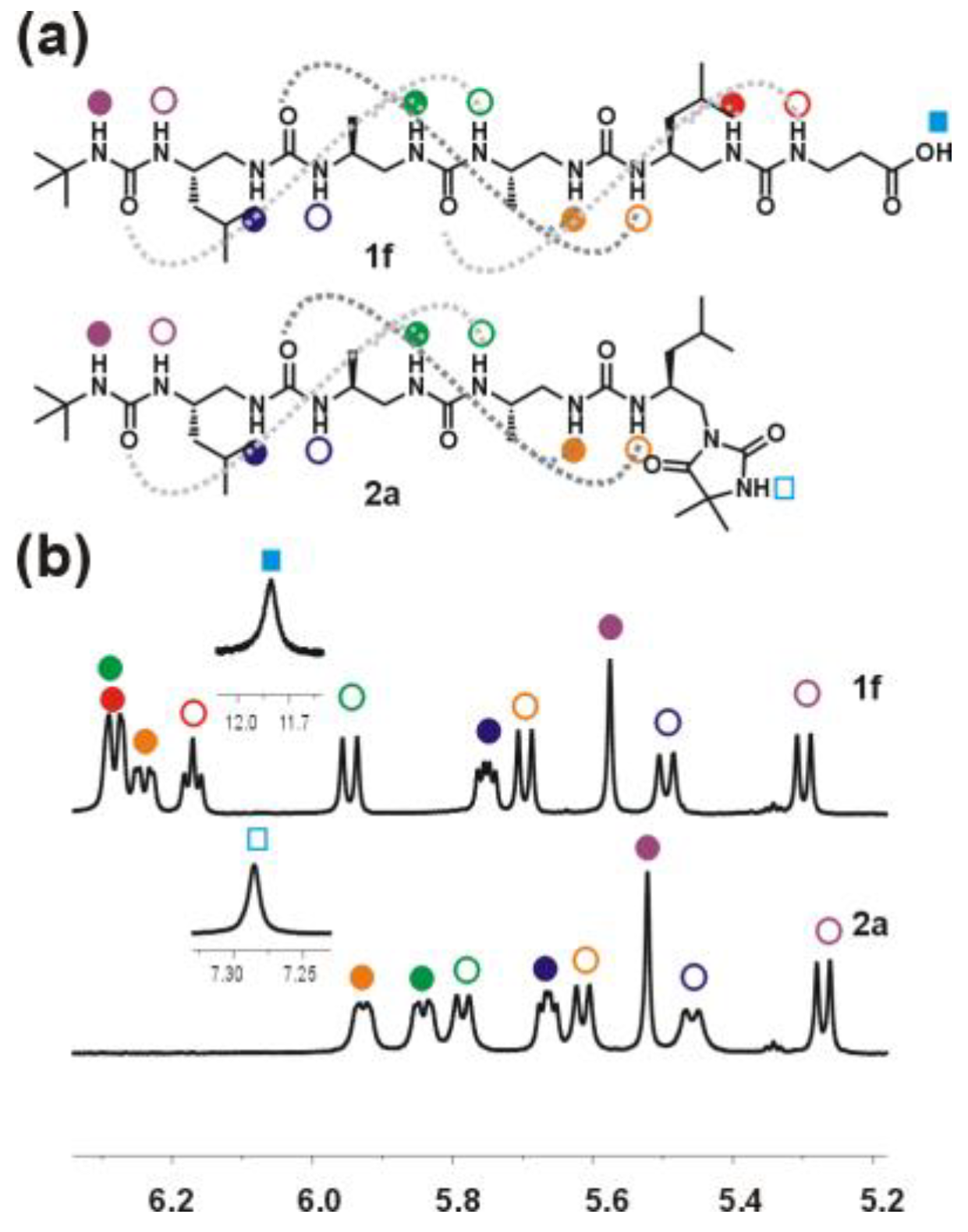
Oligourea ester 5a: Substrates: amine 4 (80 mg, 0.14 mmol), BB1 (44 mg, 0.17 mmol) and DIPEA (73 μL, 0.42 mmol). Purification: silica gel chromatography (CH2Cl2:MeOH (v/v) 95:5); yield: 97% (95mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 6.60 (s, 1H), 6.32 (m, 2H), 6.20 (d, J = 8.9 Hz, 1H), 6.04 (d, J = 10.5 Hz, 1H), 5.78 (dd, J = 8.3, 4.4 Hz, 1H), 5.71 (d, J = 9.7 Hz, 1H), 5.59 (s, 1H), 5.54 (d, J = 10.1 Hz, 1H), 5.31 (d, J = 9.5 Hz, 1H), 4.12–3.99 (m, 1H), 3.96–3.83 (m, 2H), 3.81–3.71 (m, 1H), 3.71–3.64 (m, 1H), 3.61 (s, 3H), 3.57–3.48 (m, 2H), 3.46–3.37 (m, 1H), 2.42–2.14 (m, 4H), 1.64 (m, 2H), 1.41 (s, 3H), 1.37 (s, 3H), 1.30 (s, 9H), 1.18 (q, J = 7.0 Hz, 4H), 1.02–0.83 (m, 18H); RP-HPLC: tR = 28.35 min; HRMS ESI(+) calcd. for C32H65N10O7 701.50322 [M + H]+, found 701.50324.
Oligourea ester 5b: Substrates: amine 4 (70 mg, 0.13 mmol), BB2b (35 mg, 0.15 mmol) and DIPEA (60 μL, 0.38 mmol). Purification: silica gel chromatography (CH2Cl2:MeOH (v/v) 95:5);yield: 59% (50 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 6.51 (d, J = 8.7 Hz, 1H), 6.44 (m, 1H), 6.32 (m, 2H), 6.00 (d, J = 10.5 Hz, 1H), 5.84–5.74 (m, 1H), 5.74 (d, J = 9.7 Hz, 1H), 5.59 (s, 1H), 5.53 (d, J = 10.2 Hz, 1H), 5.32 (d, J = 9.5 Hz, 1H), 4.13–3.72 (m, 6H), 3.66 (s, 3H), 3.65–3.35 (m, 4H), 2.47–2.11 (m, 4H), 1.65 (m, 2H), 1.30 (s, 9H), 1.24–1.12 (m, 4H), 1.04–0.68 (m, 18H); RP-HPLC: tR = 27.47 min; HRMS ESI(+) calcd. for C30H61N10O 673.47192 [M + H]+, found 673.47191.
Oligourea ester 5c: Substrates: amine 4 (200 mg, 0.36 mmol), BB2c (110 mg, 0.43 mmol) and DIPEA (188 μL, 1.08 mmol). Purification: silica gel chromatography (CH2Cl2:MeOH (v/v) 95:5), yield: 61% (150 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ 6.43 (d, J = 7.3 Hz, 1H), 6.41–6.22 (m, 3H), 6.00 (d, J = 10.5 Hz, 1H), 5.77 (m, 2H), 5.58 (s, 1H), 5.53 (d, J = 10.1 Hz, 1H), 5.31 (d, J = 9.5 Hz, 1H), 4.29 (p, J = 7.2 Hz, 1H), 4.05 (m, 1H), 3.96–3.71 (m, 3H), 3.67 (s, 3H), 3.55 (m, 3H), 3.41 (m, 1H), 2.41–2.05 (m, 4H), 1.64 (m, 2H), 1.35–1.27 (m, 12H), 1.23–1.12 (m, 4H), 0.99–0.94 (m, 6H), 0.94–0.87 (m, 12H); RP-HPLC: tR = 27.55 min; HRMS ESI(+) calcd. for C31H63N10O7 687.48757 [M + H]+, found 687.48776.
Oligourea ester 5d: Substrates: amine 4 (96 mg, 0.17 mmol), BB2d (57 mg, 0.21 mmol) and DIPEA (90 μL, 0.51 mmol). Purification: silica gel chromatography (CH2Cl2:MeOH (v/v) 96:4), yield: 54% (65 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 6.52 (d, J = 9.1 Hz, 1H), 6.40–6.24 (m, 3H), 6.02 (d, J = 10.5 Hz, 1H), 5.81 (dd, J = 8.2, 4.4 Hz, 1H), 5.73 (d, J = 9.8 Hz, 1H), 5.61 (s, 1H), 5.58 (d, J = 10.1 Hz, 1H), 5.34 (d, J = 9.5 Hz, 1H), 4.19 (dd, J = 8.5, 5.2 Hz, 1H), 4.06 (m, 1H), 3.96–3.71 (m, 3H), 3.68 (s, 3H), 3.60 (m, 3H), 3.49–3.35 (m, 1H), 2.46–2.05 (m, 4H), 1.78–1.51 (m, 2H), 1.30 (s, 9H), 1.18 (q, J = 6.8 Hz, 4H), 1.01–0.82 (m, 24H); RP-HPLC: tR = 29.34 min; HRMS ESI(+) calcd. for C33H67N10O7 715.51887 [M + H]+, found 715.51910.
Oligourea ester 5e: Substrates: amine 4 (65 mg, 0.12 mmol), BB2e (41 mg, 0.144 mmol) and DIPEA (63 μL, 0.36 mmol). Purification: silica gel chromatography (CH2Cl2:MeOH (v/v) 96:4), yield: 65% (57 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 6.45 (d, J = 8.9 Hz, 1H), 6.36–6.21 (m, 3H), 6.01 (d, J = 10.5 Hz, 1H), 5.89–5.79 (m, 1H), 5.75 (d, J = 9.9 Hz, 1H), 5.60 (s, 1H), 5.54 (d, J = 10.1 Hz, 1H), 5.33 (d, J = 9.5 Hz, 1H), 4.30 (td, J = 8.3, 6.2 Hz, 1H), 4.12–3.99 (m, 1H), 3.99–3.70 (m, 3H), 3.67 (s, 3H), 3.64–3.49 (m, 3H), 3.48–3.34 (m, 1H), 2.45–2.15 (m, 4H), 1.77–1.59 (m, 3H), 1.55–1.46 (m, 2H), 1.30 (s, 9H), 1.23–1.09 (m, 4H), 1.01–0.82 (m, 24H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ: 52.71, 52.17, 48.31, 48.00, 47.90, 47.74, 46.89, 46.87, 45.67, 45.28, 44.07, 42.67, 42.46, 30.24, 25.89, 25.50, 23.59, 23.39, 23.28, 22.52, 22.45, 21.97, 18.98, 18.40; RP-HPLC: tR = 30.49 min; HRMS ESI(+) calcd. for C34H69N10O7 729.53452 [M + H]+, found 729.53485.
Oligourea ester 5f: Substrates: amine 4 (70 mg, 0.13 mmol), BB3f (39 mg, 0.16 mmol) and DIPEA (67 μL, 0.39 mmol). Purification: silica gel chromatography (CH2Cl2:MeOH (v/v) 95:5), yield: 66% (59 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ 6.37–6.13 (m, 4H), 5.97 (d, J = 10.5 Hz, 1H), 5.77 (dd, J = 8.3, 4.4 Hz, 1H), 5.69 (d, J = 9.7 Hz, 1H), 5.59 (s, 1H), 5.51 (d, J = 10.1 Hz, 1H), 5.31 (d, J = 9.6 Hz, 1H), 4.05–3.70 (m, 4H), 3.62 (s, 3H), 3.59–3.43 (m, 4H), 3.34 (q, J = 6.3 Hz, 2H), 2.46–2.30 (m, 4H), 2.26–2.11 (m, 2H), 1.65 (m, 2H), 1.30 (s, 9H), 1.25–1.11 (m, 4H), 1.09–0.75 (m, 18H); RP-HPLC: tR = 27.92 min; HRMS ESI(+) calcd. for C31H63N10O7 687.48757 [M + H]+, found 687.48774.
Oligourea ester 5g: Substrates: amine 4 (110 mg, 0.20 mmol), BB3g (62 mg, 0.24 mmol) and DIPEA (105 μL, 0.60 mmol). Purification: silica gel chromatography (CH2Cl2:MeOH (v/v) 95:5), yield: 70% (100 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 6.33 (dd, J = 10.0, 3.1 Hz, 1H), 6.27 (dd, J = 9.9, 3.3 Hz, 1H), 6.19 (d, J = 8.7 Hz, 1H), 6.06 (d, J = 8.3 Hz, 1H), 5.99 (d, J = 10.4 Hz, 1H), 5.78 (dd, J = 8.3, 4.4 Hz, 1H), 5.71 (d, J = 9.7 Hz, 1H), 5.60 (s, 1H), 5.53 (d, J = 10.2 Hz, 1H), 5.32 (d, J = 9.5 Hz, 1H), 4.13–3.97 (m, 2H), 3.96–3.81 (m, 2H), 3.81–3.70 (m, 1H), 3.63 (s, 3H), 3.61–3.47 (m, 3H), 3.47–3.35 (m, 1H), 2.65–2.61 (m, 1H), 2.47–2.28 (m, 3H), 2.28–2.14 (m, 2H), 1.73–1.56 (m, 2H), 1.30 (s, 9H), 1.22–1.15 (m, 4H), 1.13 (d, J = 6.6 Hz, 3H), 1.00–0.86 (m, 18H); RP-HPLC: tR = 27.84 min; HRMS ESI(+) calcd. for C32H65N10O7 701.50322 [M + H]+, found 701.50334.
Oligourea ester 5h: Substrates: amine 4 (85 mg, 0.15 mmol), BB4h (46 mg, 0.18 mmol) and DIPEA (78 μL, 0.45 mmol). Purification: silica gel chromatography (CH2Cl2:MeOH (v/v) 95:5), yield: 86% (90 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 6.30 (dd, J = 14.7, 10.2 Hz, 2H), 6.18–6.07 (m, 2H), 6.00 (d, J = 10.4 Hz, 1H), 5.78 (dd, J = 8.2, 4.4 Hz, 1H), 5.68 (d, J = 9.7 Hz, 1H), 5.60 (s, 1H), 5.53 (d, J = 10.1 Hz, 1H), 5.32 (d, J = 9.6 Hz, 1H), 4.06–3.69 (m, 4H), 3.62 (s, 3H), 3.60–3.32 (m, 4H), 3.11 (p, J = 6.4 Hz, 2H), 2.47–2.29 (m, 4H), 2.21 (m, 2H), 1.68 (m, 4H), 1.30 (s, 9H), 1.27–1.10 (m, 4H), 1.03–0.80 (m, 18H); RP-HPLC: tR = 27.63 min; HRMS ESI(+) calcd. for C32H65N10O7 701.50322 [M + H]+, found 701.50304.
Oligourea ester 5i: Substrates: amine 4 (60 mg, 0.11 mmol), BB4i (35 mg, 0.13 mmol) and DIPEA (52 μL, 0.33 mmol). Purification: silica gel chromatography (CH2Cl2:MeOH (v/v) 95:5), yield: 78% (68 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ 7.43–7.10 (m, 5H), 6.36–6.22 (m, 2H), 6.18 (d, J = 9.4 Hz, 1H), 5.97 (dd, J = 12.5, 9.6 Hz, 2H), 5.77 (dd, J = 8.3, 4.4 Hz, 1H), 5.61 (d, J = 10.8 Hz, 1H), 5.60 (s, 1H), 5.52 (d, J = 10.4 Hz, 1H), 5.31 (d, J = 9.6 Hz, 1H), 4.05–3.77 (m, 4H), 3.80–3.66 (m, 1H), 3.60 (s, 3H), 3.61–3.34 (m, 4H), 2.72 (dd, J = 6.9, 5.1 Hz, 2H), 2.60–2.50 (m, 1H), 2.42–2.25 (m, 3H), 2.24–2.11 (m, 2H), 1.77–1.60 (m, 2H), 1.59–1.44 (m, 2H), 1.31 (s, 9H), 1.23–1.09 (m, 4H), 0.97–0.85 (m, 18H); RP-HPLC: tR = 30.59 min; HRMS ESI(+) calcd. for C39H71N10O7 791.55017 [M + H]+, found 791.55042.
Oligourea ester 5j: Substrates: amine 4 (70 mg, 0.125 mmol), BB4j (47 mg, 0.15 mmol) and DIPEA (65 μL, 0.38 mmol). Purification: silica gel chromatography (CH2Cl2:MeOH (v/v) 96:4), yield: 57% (54 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ 6.38–6.21 (m, 2H), 6.16 (d, J = 9.2 Hz, 1H), 5.99 (d, J = 10.4 Hz, 1H), 5.78 (dd, J = 8.3, 4.4 Hz, 1H), 5.69 (d, J = 9.3 Hz, 1H), 5.65 (d, J = 9.8 Hz, 1H), 5.58 (s, 1H), 5.53 (d, J = 10.2 Hz, 1H), 5.30 (d, J = 9.5 Hz, 1H), 4.04–3.68 (m, 5H), 3.63 (s, 3H), 3.62–3.47 (m, 3H), 3.47–3.37 (m, 1H), 2.49–2.26 (m, 4H), 2.28–2.12 (m, 2H), 1.74–1.61 (m, 3H), 1.58–1.39 (m, 2H), 1.30 (s, 9H), 1.27–1.10 (m, 6H), 1.07–0.79 (m, 24H); RP-HPLC: tR = 30.72 min; HRMS ESI(+) calcd. for C36H73N10O7 757.56582 [M + H]+, found 757.56642.
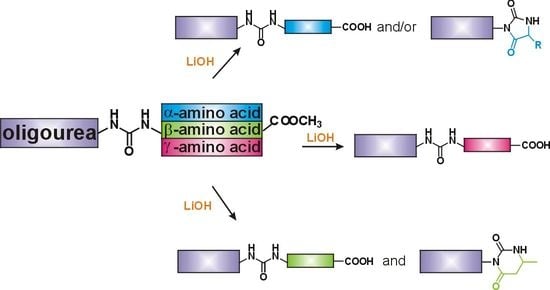
3.2.4. Hydrolysis of Oligourea Ester 5
The corresponding ester
5 (1 eq.) was dissolved in MeOH (7 mM). LiOH × H
2O (10 eq) was dissolved in water (the ratio of MeOH to H
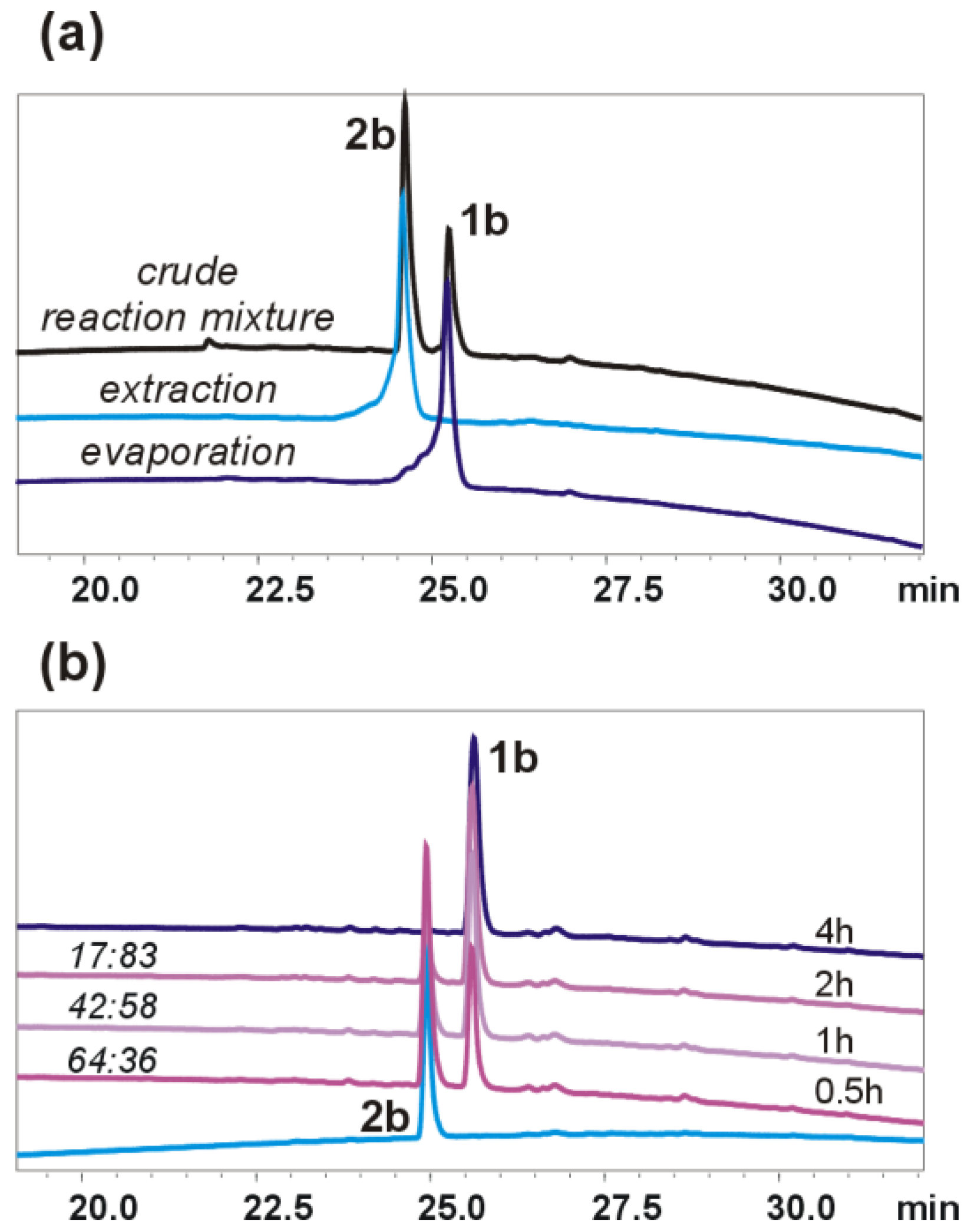
2O was 4:1) and added to the ester solution. The reaction mixture was stirred for 2 h at RT. The progress of the reaction was monitored by HPLC (for
5g, the reaction mixture was stirred overnight). The work-up conditions depended on the obtained product/products. To isolate oligourea acid
1, the solvents were completely and slowly removed under vacuum (water bath at 45 °C). The residue was redissolved in CH
2Cl
2 and distilled water. The water layer was extracted with CH
2Cl
2 (2×) with the addition of brine because of the formation of an emulsion. The organic layers were removed. HCl (1M) was added to water layer until an acidic pH and it was extracted with CH
2Cl
2 until all the product was completely extracted into the organic layer. The solvent was evaporated, and the crude product was dissolved in CH
3CN, quickly passed through a syringe filter (PA 0.2μm) and lyophilized from CH
3CN with addition of water. The product of type
1 was obtained as a white solid. To isolate oligourea hydantoin/dihydrouracil
2, the reaction mixture was diluted with CH
2Cl
2 and distilled water. The layers were separated, and the water layer was extracted with CH
2Cl
2 with the addition of brine (emulsion formation) until all the cyclic product was completely extracted into the organic layer. The solvent was evaporated, and the crude product was dissolved in CH
3CN, quickly passed through a syringe filter (PA 0.2μm) and lyophilized from CH
3CN with the addition of water. The product of type
2 was obtained as a white solid. Copies of
1H NMR spectra, as well as HRMS spectra of oligourea acids and oligourea hydantoins/dihydrouracils are given in the
Supporting Information, Figures S34–S46 and Figures S81–S93.
Oligourea acid 1b: Substrates: 5b (20 mg, 0.03 mmol) and LiOH × H2O (12.6 mg, 0.30 mmol). Yield: 61% (12 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 6.54 (d, J = 8.9 Hz, 1H), 6.44 (t, J = 5.8 Hz, 1H), 6.40–6.30 (m, 2H), 6.03 (d, J = 10.5 Hz, 1H), 5.87–5.76 (m, 2H), 5.64 (s, 1H), 5.56 (d, J = 10.1 Hz, 1H), 5.44–5.33 (m, 1H), 4.10–3.98 (m, 1H), 3.98–3.83 (m, 3H), 3.81 (dd, J = 5.8, 3.4 Hz, 2H), 3.66–3.48 (m, 3H), 3.48–3.36 (m, 1H), 2.39–2.27 (m, 1H), 2.27–2.16 (m, 2H), 2.16–2.02 (m, 1H), 1.72–1.54 (m, 2H), 1.30 (s, 9H), 1.23–1.12 (m, 4H), 1.00–0.86 (m, 18H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ: 48.07, 48.06, 47.88, 47.84, 46.90, 46.82, 46.08, 45.43, 43.58, 42.78, 42.71, 29.82, 25.89, 25.70, 23.51, 23.41, 22.48, 18.91, 18.51, 14.42; RP-HPLC: tR = 25.52 min; HRMS ESI(+) calcd. for C29H59N10O7 [M + H]+ 659.4568, found 659.4566.
Oligourea acid 1c: Substrates: 5c (9 mg, 0.013 mmol) and LiOH × H2O (5.5 mg, 0.13 mmol). Yield: 92% (8 mg), NMR peaks were doubled because of the epimerization of the stereogenic center of the amino acid residue at the C-terminus and the formation of a mixture of diastereomeric products. 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 12.08 (bs, 1H), 6.52 (d, J = 6.8 Hz, 1H), 6.49–6.37 (m, 2H), 6.36–6.28 (m, 1H), 6.09–5.96 (m, 1H), 5.84–5.77 (m, 1H), 5.73 (d, J = 9.7 Hz, 1H), 5.63–5.58 (m, 1H), 5.57–5.50 (m, 1H), 5.36–5.30 (m, 1H), 4.24–4.15 (m, 1H), 4.08–3.98 (m, 1H), 3.96–3.82 (m, 2H), 3.79–3.69 (m, 1H), 3.57–3.48 (m, 3H), 3.48–3.36 (m, 1H), 2.45–2.30 (m, 2H), 2.29–2.15 (m, 2H), 1.72–1.48 (m, 2H), 1.33–1.27 (m, 12H), 1.23–1.13 (m, 4H), 0.98–0.94 (m, 6H), 0.93–0.87 (m, 12H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ 49.61, 47.91, 47.90, 47.89, 47.86, 46.94, 46.83, 46.07, 45.43, 43.49, 42.68, 29.83, 25.81, 25.59, 23.52, 23.41, 22.48, 18.72, 18.64, 14.38; RP-HPLC: tR = 26.23 min; HRMS ESI(+) calcd. for C30H61N10O7 [M + H]+ 673.4725, found 673.4722.
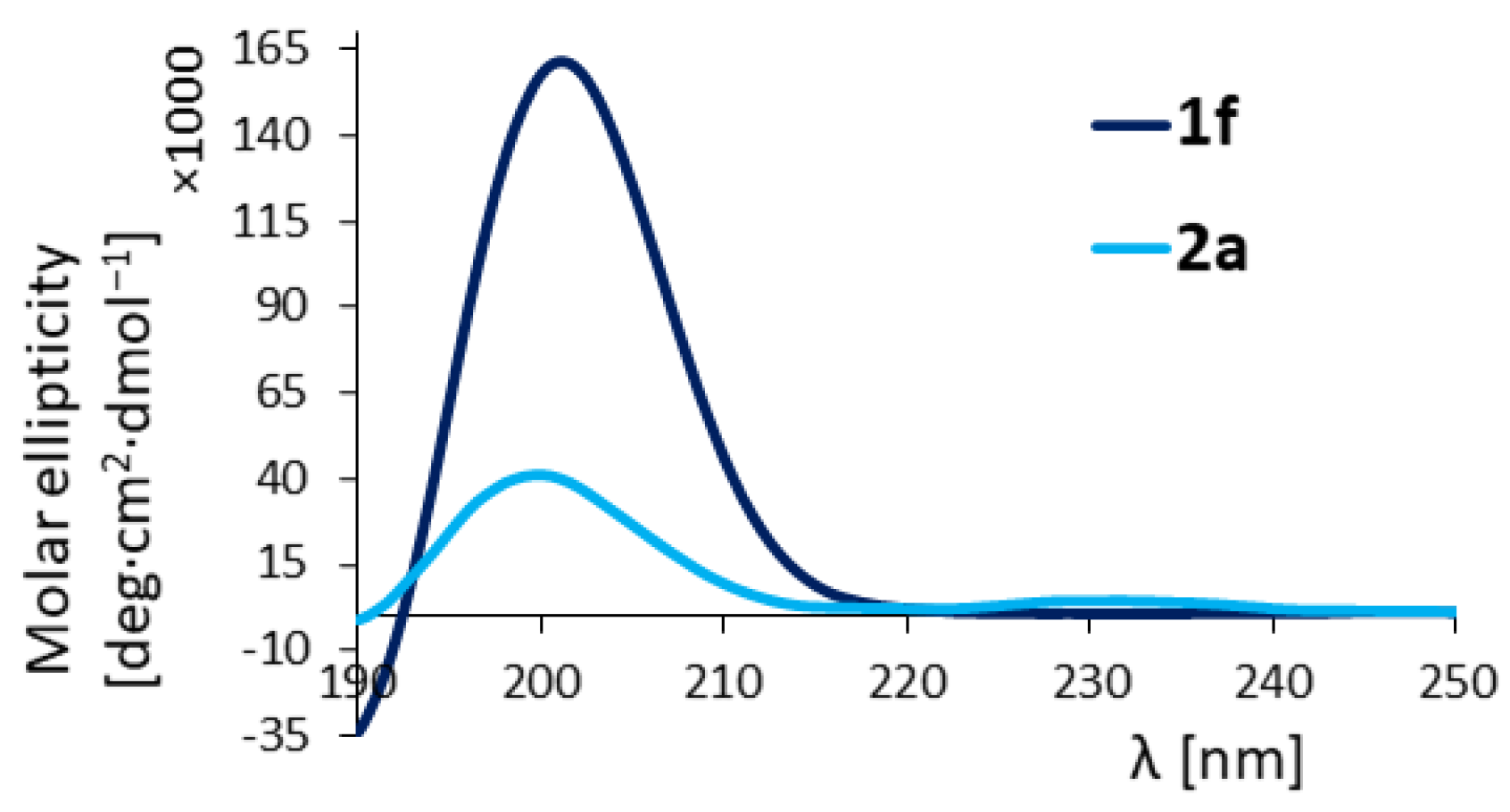
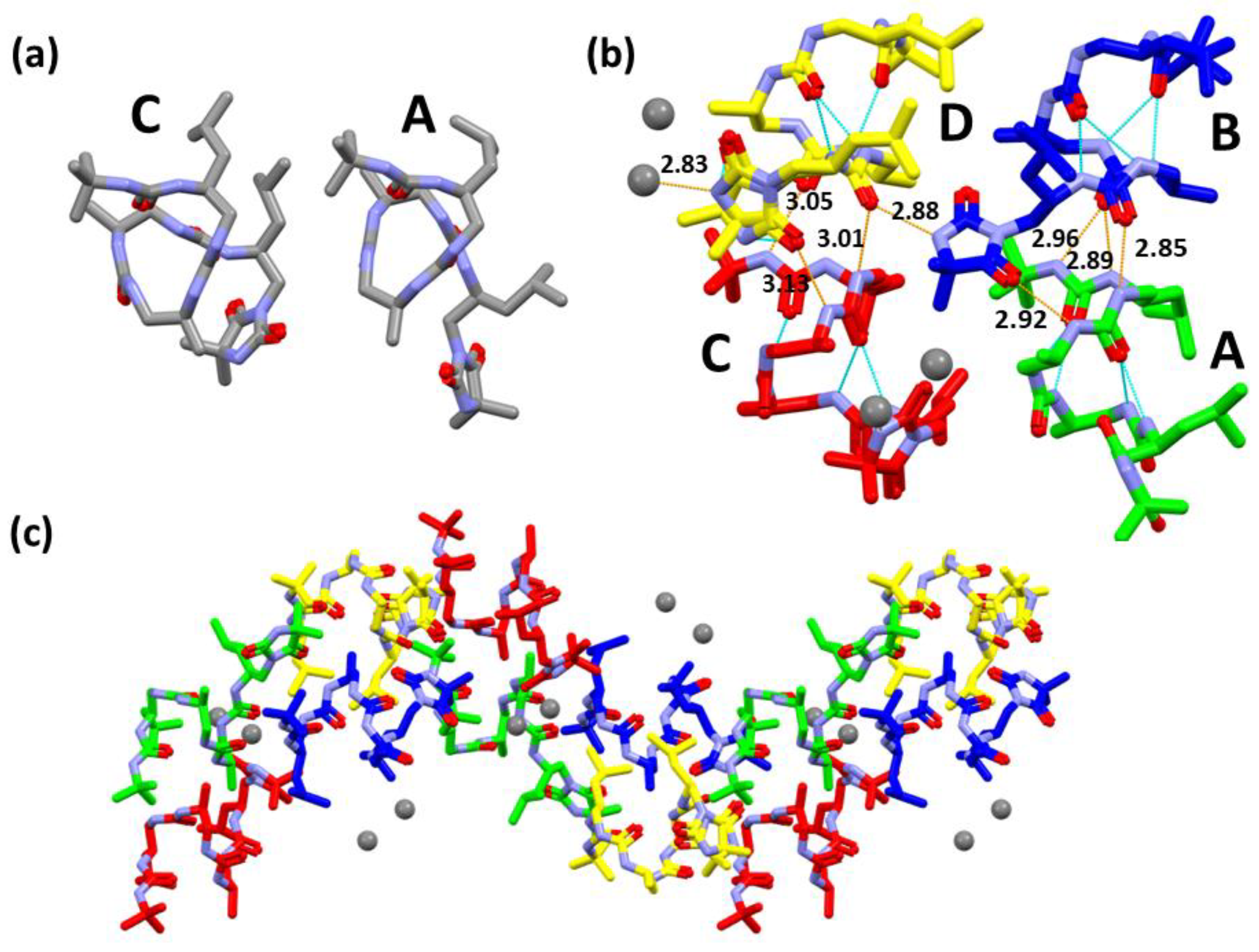
Oligourea acid 1f: Substrates: 5f (40 mg, 0.06 mmol), LiOH × H2O (25.2 mg, 0.6 mmol). Yield: 64% (25 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 11.80 (bs, 1H), 6.37–6.24 (m, 3H), 6.22–6.17 (m, 1H), 5.98 (d, J = 10.5 Hz, 1H), 5.78 (dd, J = 8.2, 4.3 Hz, 1H), 5.72 (d, J = 9.6 Hz, 1H), 5.60 (s, 1H), 5.53 (d, J = 10.1 Hz, 1H), 5.33 (d, J = 9.6 Hz, 1H), 4.10–3.69 (m, 4H), 3.64–3.38 (m, 4H), 3.37–3.27 (m, 2H), 2.41 (t, J = 6.3 Hz, 2H), 2.37–2.06 (m, 4H), 1.72–1.57 (m, 2H), 1.30 (s, 9H), 1.23–1.11 (m, 4H), 1.00–0.80 (m, 18H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ: 48.29, 47.97, 47.96, 47.78, 45.30, 46.88, 46.87, 45.76, 43.90, 42.71, 36.45, 36.39, 29.86, 25.82, 25.55, 23.55, 23.44, 22.51, 22.49, 19.00, 18.51; RP-HPLC: tR = 25.27 min; HRMS ESI(+) calcd. for C30H61N10O7 [M + H]+ 673.4725, found 673.4723.
Oligourea acid 1g: Substrates: 5g (45 mg, 0.064 mmol) and LiOH × H2O (26.8 mg, 0.64 mmol). Yield: 73% (32 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 11.87 (bs, 1H), 6.42–6.27 (m, 2H), 6.26–6.19 (m, 1H), 6.08 (d, J = 8.3 Hz, 1H), 6.02 (d, J = 10.5 Hz, 1H), 5.84–5.78 (m, 1H), 5.75 (d, J = 9.7 Hz, 1H), 5.65 (s, 1H), 5.56 (d, J = 10.2 Hz, 1H), 5.41–5.34 (m, 1H), 4.10–3.96 (m, 2H), 3.95–3.82 (m, 2H), 3.81–3.69 (m, 1H), 3.62–3.49 (m, 3H), 3.42 (ddd, J = 13.8, 8.4, 3.4 Hz, 1H), 2.47–2.13 (m, 6H), 1.73–1.55 (m, 2H), 1.30 (s, 9H), 1.23–1.15 (m, 4H), 1.14 (d, J = 6.7 Hz, 3H), 0.99–0.85 (m, 18H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ: 48.14, 48.08, 47.75, 47.74, 46.84, 46.81, 45.98, 45.39, 43.60, 43.43, 42.94, 42.71, 29.85, 25.88, 25.54, 23.56, 23.40, 22.53, 22.50, 21.45, 18.96, 18.52; RP-HPLC: tR = 26.23 min; HRMS ESI(+) calcd. for C31H63N10O7 [M + H]+ 687.4881, found 687.4875.
Oligourea acid 1h: Substrates: 5h (70 mg, 0.10 mmol) and LiOH × H2O (42 mg, 1.0 mmol). Yield: 84% (58 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 6.47–6.22 (m, 3H), 6.11–6.05 (m, 1H), 6.02 (d, J = 10.4 Hz, 1H), 5.81 (dd, J = 8.2, 4.4 Hz, 1H), 5.75 (d, J = 9.8 Hz, 1H), 5.60 (s, 1H), 5.57 (d, J = 10.3 Hz, 1H), 5.33 (d, J = 9.5 Hz, 1H), 4.08–3.96 (m, 1H), 3.97–3.69 (m, 3H), 3.67–3.47 (m, 3H), 3.47–3.35 (m, 1H), 3.32–3.15 (m, 1H), 3.12–2.99 (m, 1H), 2.46–2.35 (m, 2H), 2.31 (t, J = 7.4 Hz, 2H), 2.27–2.14 (m, 2H), 1.80–1.57 (m, 4H), 1.30 (s, 9H), 1.26–1.11 (m, 4H), 1.02–0.80 (m, 18H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ: 48.55, 48.02, 47.83, 47.78, 46.84, 46.76, 45.98, 45.49, 43.70, 42.66, 39.52, 32.37, 29.85, 27.33, 25.89, 25.58, 23.54, 23.41, 22.79, 22.47, 18.95, 18.48; RP-HPLC: tR = 25.63 min; HRMS ESI(+) calcd. for C31H63N10O7 [M + H]+ 687.4881, found 687.4878.
Oligourea acid 1i: Substrates: 5i (50 mg, 0.063 mmol) and LiOH × H2O (26.4 mg, 0.63 mmol). Yield: 69% (34 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 11.96 (bs, 1H), 7.35–7.13 (m, 5H), 6.56–6.46 (m, 1H), 6.38–6.29 (m, 2H), 6.04 (d, J = 10.5 Hz, 1H), 5.88–5.81 (m, 2H), 5.79 (d, J = 9.9 Hz, 1H), 5.63 (s, 1H), 5.60 (d, J = 10.3 Hz, 1H), 5.36 (d, J = 9.5 Hz, 1H), 4.12–3.67 (m, 5H), 3.65–3.47 (m, 3H), 3.47–3.35 (m, 1H), 2.78–2.65 (m, 2H), 2.46–2.31 (m, 3H), 2.32–2.15 (m, 3H), 1.93–1.79 (m, 1H), 1.75–1.56 (m, 2H), 1.54–1.39 (m, 1H), 1.31 (s, 9H), 1.23–1.09 (m, 4H), 1.01–0.80 (m, 18H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ 130.29, 129.04, 126.90, 51.31, 50.89, 49.01, 47.99, 47.95, 47.79, 46.84, 46.67, 46.13, 45.67, 43.43, 42.98, 42.66, 32.47, 32.05, 29.86, 25.87, 25.61, 23.52, 23.42, 22.91, 22.46, 18.84, 18.49. RP-HPLC: tR = 29.48 min; HRMS ESI(+) calcd. for C38H69N10O7 [M + H]+ 777.5351, found 777.5349.
Oligourea acid 1j: Substrates: 5j (45 mg, 0.059 mmol) and LiOH × H2O (24.8 mg, 0.59 mmol). Yield: 75% (33 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 12.00 (s, 1H), 6.57–6.48 (m, 1H), 6.40–6.28 (m, 2H), 6.03 (d, J = 10.4 Hz, 1H), 5.88–5.77 (m, 2H), 5.66–5.54 (m, 3H), 5.31 (d, J = 9.5 Hz, 1H), 4.11–3.96 (m, 1H), 3.96–3.83 (m, 2H), 3.82–3.71 (m, 2H), 3.70–3.47 (m, 3H), 3.47–3.34 (m, 1H), 2.42–2.23 (m, 4H), 1.91–1.75 (m, 2H), 1.73–1.58 (m, 3H), 1.47–1.36 (m, 2H), 1.30 (s, 9H), 1.25–1.08 (m, 6H), 1.03–0.67 (m, 24H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ: 48.99, 47.96, 47.93, 47.80, 47.54, 46.87, 46.76, 46.37, 46.23, 45.68, 43.43, 42.66, 33.45, 32.08, 29.83, 25.82, 25.74, 23.51, 22.47, 22.44, 18.44, 18.85; RP-HPLC: tR = 29.49 min; HRMS ESI(+) calcd. for C35H71N10O7 [M + H]+ 743.5507, found 743.5504.
Oligourea hydantoin 2a: Substrates: 5a (40 mg, 0.057 mmol) and LiOH × H2O (23.9 mg, 0.57 mmol). Yield: 87% (35 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 7.34 (s, 1H), 6.01–5.94 (m, 1H), 5.89 (dd, J = 9.6, 3.3 Hz, 1H), 5.83 (d, J = 8.9 Hz, 1H), 5.72 (dd, J = 7.8, 4.6 Hz, 1H), 5.66 (d, J = 9.2 Hz, 1H), 5.59 (s, 1H), 5.52 (d, J = 9.4 Hz, 1H), 5.35 (d, J = 9.0 Hz, 1H), 4.15–3.97 (m, 1H), 3.92–3.69 (m, 3H), 3.61–3.37 (m, 2H), 3.34 (dd, J = 6.9, 1.9 Hz, 2H), 3.32–3.20 (m, 1H), 2.69–2.63 (m, 1H), 2.59–2.51 (m, 1H), 2.41 (q, J = 10.9 Hz, 1H), 1.77–1.54 (m, 2H), 1.34 (d, J = 1.5 Hz, 6H), 1.28 (s, 9H), 1.25–1.10 (m, 4H), 1.02 (d, J = 6.9 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H), 0.95–0.82 (m, 12H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ: 47.89, 47.35, 46.89, 46.48, 46.44, 46.36, 46.14, 43.90, 42.99, 42.90, 29.86, 25.78, 25.47, 25.16, 24.97, 23.64, 23.26, 22.51, 22.18, 19.67, 18.99; RP-HPLC: tR = 25.28 min; HRMS ESI(+) calcd. for C31H61N10O6 [M + H]+ 669.4776, found 669.4769.
Oligourea hydantoin 2b: Substrates: 5b (15 mg, 0.022 mmol) and LiOH × H2O (9.2 mg, 0.22 mmol). Yield: 55% (8 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 7.13 (s, 1H), 6.01–5.93 (m, 1H), 5.88–5.83 (m, 1H), 5.81 (d, J = 8.7 Hz, 1H), 5.73–5.66 (m, 1H), 5.63 (d, J = 9.2 Hz, 1H), 5.54 (s, 1H), 5.48 (d, J = 10.3 Hz, 1H), 5.27 (d, J = 9.3 Hz, 1H), 4.11–4.00 (m, 1H), 3.94–3.86 (m, 1H), 3.83 (dd, J = 3.5, 1.2 Hz, 2H), 3.80–3.67 (m, 1H), 3.61–3.43 (m, 3H), 3.37 (d, J = 6.8 Hz, 2H), 3.35–3.25 (m, 1H), 2.53 (d, J = 4.8 Hz, 1H), 2.45–2.27 (m, 1H), 2.16–2.00 (m, 1H), 1.77–1.51 (m, 2H), 1.28 (s, 9H), 1.23 (t, J = 7.4 Hz, 2H), 1.19–1.12 (m, 2H), 1.02 (d, J = 6.9 Hz, 3H), 0.99 (d, J = 6.7 Hz, 3H), 0.93–0.83 (m, 12H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ: 47.84, 47.24, 47.01, 46.85, 46.53, 46.44, 46.29, 46.06, 43.79, 42.92, 42.83, 29.81, 25.75, 25.49, 23.60, 22.28, 22.13, 19.59, 19.00, 14.39; RP-HPLC: tR = 24.88 min; HRMS ESI(+) calcd. for C29H57N10O6 [M + H]+ 641.4463, found 641.4456.
Oligourea hydantoin 2c: Substrates: 5c (30 mg, 0.044 mmol) and LiOH × H2O (18.5 mg, 0.44 mmol). Yield: 65% (19 mg); NMR peaks were doubled because of the epimerization of the stereogenic center of the amino acid residue at the C-terminus and the formation of a mixture of diastereomeric products, 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 7.27 (s, 1H), 6.03–5.92 (m, 1H), 5.91–5.78 (m, 2H), 5.73–5.67 (m, 1H), 5.65 (d, J = 10.8 Hz, 1H), 5.55 (s, 1H), 5.52–5.45 (m, 1H), 5.30 (d, J = 9.4 Hz, 1H), 4.13–3.93 (m, 1H), 3.94–3.81 (m, 3H), 3.80–3.70 (m, 1H), 3.65–3.41 (m, 2H), 3.41–3.30 (m, 2H), 3.32–3.26 (m, 1H), 2.76–2.64 (m, 1H), 2.45–2.03 (m, 2H), 1.71–1.52 (m, 2H), 1.28 (s, 9H), 1.26–1.13 (m, 4H), δ 1.12 (d, J = 6.2 Hz, 3H), 1.02 (d, J = 6.9 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H), 0.95–0.82 (m, 12H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ: 53.36, 47.87, 47.31, 46.84, 46.47, 46.31, 46.08, 43.84, 42.91, 42.89, 29.83, 25.76, 25.48, 23.62, 22.35, 22.29, 19.65, 19.02, 17.69; RP-HPLC: tR = 25.28 min; HRMS ESI(+) calcd. for C30H59N10O6 [M + H]+ 655.4619, found 655.4614
Oligourea hydantoin 2d: Substrates: 5d (30 mg, 0.042 mmol) and LiOH × H2O (17.6 mg, 0.42 mmol). Yield: 87% (25 mg); NMR peaks were doubled because of the epimerization of the stereogenic center of the amino acid residue at the C-terminus and the formation of a mixture of diastereomeric products, 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ: 7.32 (d, J = 7.3 Hz, 1H), 6.03–5.94 (m, 1H), 5.90–5.77 (m, 2H), 5.71–5.61 (m, 2H), 5.56–5.49 (m, 1H), 5.50–5.43 (m, 1H), 5.25 (d, J = 9.3 Hz, 1H), 4.06 (m, 1H), 3.94–3.83 (m, 3H), 3.81–3.66 (m, 1H), 3.59–3.46 (m, 2H), 3.36 (d, J = 7.1 Hz, 2H), 3.34–3.30 (m, 1H), 2.57–2.53 (m, 1H), 2.41–2.30 (m, 1H), 2.16–2.07 (m, 1H), 1.71–1.50 (m, 3H), 1.29 (s, 9H), 1.24–1.10 (m, 4H), 1.04–0.96 (m, 6H), 0.93–0.82 (m, 18H); 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ 62.90, 47.81, 47.32, 46.87, 46.47, 46.42, 46.35, 46.11, 43.62, 42.97, 42.90, 30.85, 29.84, 25.80, 25.42, 23.70, 22.87, 22.54, 22.43, 19.61, 19.07, 16.65, 14.45; RP-HPLC: tR = 26.19 min; HRMS ESI(+) calcd. for C32H63N10O6 [M + H]+ 683.4932, found 683.4923
Oligourea hydantoin 2e: Substrates: 5e (40 mg, 0.055 mmol) and LiOH × H2O (23.0 mg, 0.55 mmol). Yield: 89% (34 mg) NMR peaks were doubled because of the epimerization of the stereogenic center of the amino acid residue at the C-terminus and the formation of a mixture of diastereomeric products, 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ 7.45 (d, J = 11.1 Hz, 1H), 6.05–5.94 (m, 1H), 5.93–5.78 (m, 2H), 5.70 (dd, J = 7.7, 4.5 Hz, 1H), 5.63 (dd, J = 9.3, 4.5 Hz, 1H), 5.55 (d, J = 2.2 Hz, 1H), 5.54–5.43 (m, 1H), 5.30 (d, J = 9.3 Hz, 1H), 4.14–3.98 (m, 1H), 3.97–3.81 (m, 2H), 3.82–3.70 (m, 1H), 3.59–3.43 (m, 3H), 3.40–3.16 (m, 2H), 3.32–3.22 (m, 1H), 2.56–2.52 (m, 2H), 2.45–2.30 (m, 1H), 1.90–1.76 (m, 1H), 1.77–1.47 (m, 2H), 1.28 (s, 9H), 1.27–1.10 (m, 6H), 1.09–0.77 (m, 24H); 13C NMR as HSQC (125 MHz, CD3CN) δ 56.35, 47.93, 47.33, 46.95, 46.60, 46.50, 46.40, 46.28, 43.90, 42.88, 42.78, 42.25, 29.85, 25.81, 25.58, 25.53, 25.52, 23.60, 23.58, 22.57, 22.11, 21.74, 21.60, 19.61, 18.95; RP-HPLC: tR = 27.09 min; HRMS ESI(+) calcd. for C33H65N10O6 [M + H]+ 697.5088, found 697.5085
Oligourea dihydrouracil 2g: Substrates: 5g (35 mg, 0.050 mmol) and LiOH × H2O (21.0 mg, 0.50 mmol). Yield: 42% (4 mg); 1H NMR (300 MHz, 10% DMSO-d6 in CD3CN) δ 6.75 (s, 1H), 6.06–5.98 (m, 1H), 5.90–5.78 (m, 2H), 5.71–5.62 (m, 1H), 5.56–5.48 (m, 2H), 5.43 (d, J = 9.5 Hz, 1H), 5.24 (d, J = 9.4 Hz, 1H), 4.15–4.00 (m, 1H), 3.99–3.82 (m, 2H), 3.82–3.64 (m, 2H), 3.61 (d, J = 5.6 Hz, 2H), 3.59–3.40 (m, 2H), 3.40–3.29 (m, 1H), 2.78–2.66 (m, 1H), 2.46–2.39 (m, 1H), 2.39–2.17 (m, 2H), 2.15–2.03 (m, 1H), 1.76–1.55 (m, 2H), 1.29 (s, 9H), 1.18 (d, J = 6.4 Hz, 3H), 1.20–1.11 (m, 4H), 1.02 (d, J = 6.9 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H), 0.95–0.77 (m, 12H). 13C NMR as HSQC (125 MHz, 10% DMSO-d6 in CD3CN) δ: 47.80, 47.46, 46.91, 46.67, 46.61, 46.21, 46.17, 44.37, 43.46, 43.07, 42.90, 39.89, 29.83, 25.78, 25.61, 23.98, 23.62, 22.49, 22.35, 20.65, 19.81, 19.03. RP-HPLC: tR = 25.90 min; HRMS ESI(+) calcd. for C31H61N10O6 [M + H]+ 669.4775, found 669.4772










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}