

Checkpoint Kinase 1 Is a Key Signal Transducer of DNA Damage in the Early Mammalian Cleavage Embryo


{kind=link}
{kind=link}
Abstract
1. Introduction
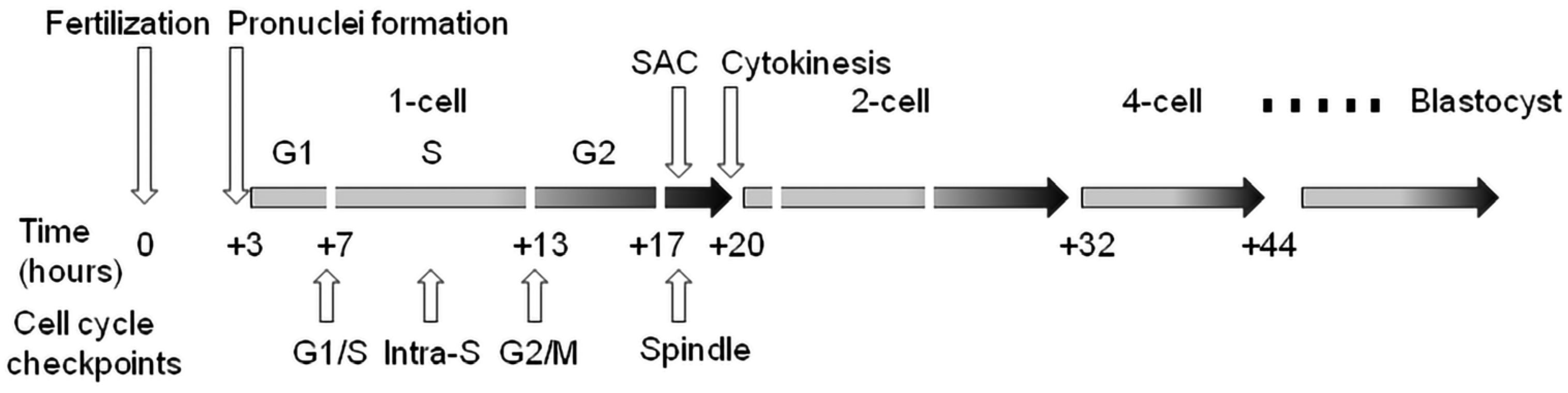
2. Cell Cycle Checkpoints
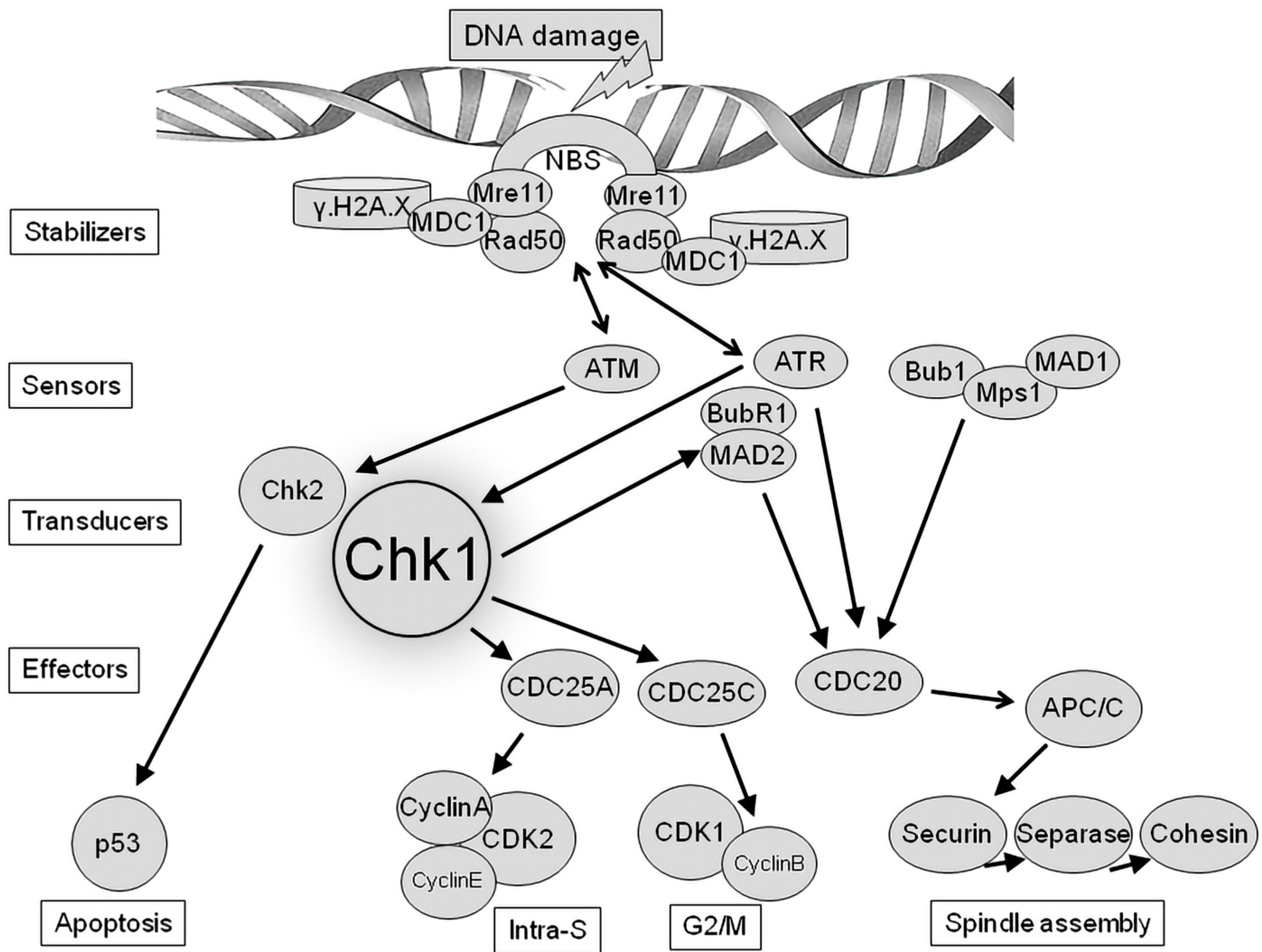
3. Chk1 as Regulator of DNA Damage Checkpoint
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Musson, R.; Gąsior, Ł.; Bisogno, S.; Ptak, G.E. DNA damage in preimplantation embryos and gametes: Specification, clinical relevance and repair strategies. Hum. Reprod. Update 2022, 28, 376–399. [Google Scholar] [CrossRef] [PubMed]
- Munisha, M.; Schimenti, J.C. Genome maintenance during embryogenesis. DNA Repair 2021, 106, 103195. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Hendrich, B.; Reik, W.; Dean, W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev. Biol. 2002, 241, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Pailas, A.; Niaka, K.; Zorzompokou, C.; Marangos, P. The DNA Damage Response in Fully Grown Mammalian Oocytes. Cells 2022, 11, 798. [Google Scholar] [CrossRef] [PubMed]
- Ladstatter, S.; Tachibana-Konwalski, K. A Surveillance mechanism ensure repair of DNA lesions during zygotic reprogramming. Cell 2016, 167, 1774–1787. [Google Scholar] [CrossRef]
- Kort, D.H.; Chia, G.; Treff, N.R.; Tanaka, A.J.; King, T.; Vensand, L.B. Human embryo commonly form abnormal nuclei during development: A mechanism of DNA damage, embryonic aneuploidy, and developmental arrest. Hum. Reprod. 2016, 31, 312–313. [Google Scholar] [CrossRef]
- Yurttas, P.; Morency, E.; Coonrod, S.A. Use of proteomics to identify highly abundant maternal factors that drive the egg-to-embryo transition. Reproduction 2010, 139, 809–823. [Google Scholar] [CrossRef]
- Hörmanseder, E.; Tischer, T.; Mayer, T.U. Modulation of cell cycle control during oocyte-to-embryo transitions. EMBO J. 2013, 32, 2191–2203. [Google Scholar] [CrossRef]
- Santos, M.A.; Teklenburg, G.; Macklon, N.S.; Van Opstal, D.; Schuring-Blom, G.H.; Krijtenburg, P.J.; de Vreeden-Elbertse, J.; Fauser, B.C.; Baart, E.B. The fate of the mosaic embryo: Chromosomal constitution and development of day 4, 5 and 8 human embryos. Hum. Reprod. 2010, 25, 1916–1926. [Google Scholar] [CrossRef]
- Mertzanidou, A.; Wilton, L.; Cheng, J.; Spits, C.; Vanneste, E.; Moreau, Y.; Vermeesch, J.R.; Sermon, K. Microarray analysis reveals abnormal chromosomal complements in over 70% of 14 normally developing human embryos. Hum. Reprod. 2013, 28, 256–264. [Google Scholar] [CrossRef]
- Mu, X.F.; Jin, X.L.; Farnham, M.M.J.; Li, Y.; O’Neil, C. DNA damage-sensing kinases mediate the mouse 2-cell embryo’s response to genotoxic stress. Biol. Reprod. 2011, 85, 524–535. [Google Scholar] [CrossRef]
- Pacchierotti, F.; Ranaldi, R.; Derijck, A.A.; Heijden, G.V.D.; Boer, P.D. In vivo repair of DNA damage induced by X-rays in the early stages of mouse fertilization, and the influence of maternal PARP1 ablation. Mutat. Res. 2011, 714, 44–52. [Google Scholar] [CrossRef]
- Ahmadi, A.; Ng, S.C. Fertilizing ability of DNA-damaged spermatozoa. J. Exp. Zool. 1999, 284, 696–704. [Google Scholar] [CrossRef]
- Fatehi, A.N.; Bevers, M.M.; Schoevers, E.; Roelen, B.A.J.; Colenbrander, B.; Gadella, B.M. DNA damage in bovine sperm does not block fertilization and early embryonic development but induces apoptosis after the first cleavages. J. Androl. 2006, 27, 176–188. [Google Scholar] [CrossRef]
- Sedó, C.A.; Bilinski, M.; Lorenzi, D.; Uriondo, H.; Noblía, F.; Longobucco, V.; Lagar, E.V.; Nodar, F. Effect of sperm DNA fragmentation on embryo development: Clinical and biological aspects. J. Bras. Assist. Reprod. 2017, 21, 343–350. [Google Scholar] [CrossRef]
- Wossidlo, M.; Arand, J.; Sebastiano, V.; Leikhov, K.; Boiani, M.; Reihardt, R.; Scholler, H.; Walter, J. Dynamic link od DNA demethylation, DNA strand breaks and repair in mouse zygotes. EMBO J. 2010, 29, 1877–1888. [Google Scholar] [CrossRef]
- House, N.C.M.; Koch, M.R.; Freudenreich, C.H. Chromatin modification and DNA repair beyond double-strand breaks. Front. Genet. 2014, 5, 296. [Google Scholar] [CrossRef]
- Derijck, A.H.A.; van der Heijden, G.W.; Giele, M.; Philippens, M.E.P.; van Bavel, C.A.W.; de Boer, P. γH2AX signalling during sperm chromatin remodelling in the mouse zygote. DNA Repair 2006, 5, 959–971. [Google Scholar] [CrossRef]
- Carbone, M.C.; Tatone, C.; Delle Monache, S.; Marci, R.; Caserta, D.; Colonna, R.; Amicarelli, F. Antioxidant enzymatic defences in human follicular fluid: Characterization and age-dependent changes. Mol. Hum. Reprod. 2003, 9, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Hempstock, J.; Jauniaux, E. Oxygen, early embryonic metabolism and free radical-mediated embryopathies. Reprod. Biomed. Online 2003, 6, 84–96. [Google Scholar] [CrossRef]
- Luddi, A.; Capaldo, A.; Focarelli, R.; Gori, M.; Morgante, G.; Piomboni, P.; de Leo, V. Antioxidants reduce oxidative stress in follicular fluid of aged women undergoing IVF. Reprod. Biol. Endocrinol. 2016, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Fieder, M. Evidence for a maximum “shelf-life” of oocytes in mammals suggests that human menopause may be an implication of meiotic arrest. Sci. Rep. 2018, 8, 140099. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Riel, J.M.; Ward, M.A. Paternal DNA damage resulting from various sperm treatments persists after fertilization and is similar before and after DNA replication. J. Androl. 2012, 33, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Diez, C.; Gonzalez-Rojo, S.; Montfort, J.; Le Cam, A.; Bobe, J.; Robles, V.; Perez-Cerezales, S.; Herraez, M.P. Inhibition of zygotic DNA repair: Transcriptome analysis of the offspring in trout (Oncorhynchus mykiss). Reproduction 2015, 149, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Chen, X.; Wang, Z.; Wang, D. Is transcription in sperm stationary or dynamic? J. Reprod. Dev. 2017, 63, 439–443. [Google Scholar] [CrossRef]
- Martin, J.H.; Aitken, R.J.; Bromfield, E.G.; Nixon, B. DNA damage and repair in the female germline: Contributions to ART. Hum. Reprod. Update 2019, 25, 180–201. [Google Scholar] [CrossRef]
- Garcıa-Rodríguez, A.; Gosálvez, J.; Agarwal, A.; Roy, R.; Johnston, S. DNA damage and repair in human reproductive cells. Int. J. Mol. Sci. 2019, 20, 31. [Google Scholar] [CrossRef]
- Khokhlova, E.V.; Fesenko, Z.S.; Sopova, J.V.; Leonova, E.I. Features of DNA repair in the early stages of mammalian embryonic development. Genes 2020, 11, 1138. [Google Scholar] [CrossRef]
- Aitken, R.J. Role of sperm DNA damage in creating de-novo mutations in human offspring: The ‘post-meiotic oocyte collusion’ hypothesis. RBMO 2022, 45, 109–124. [Google Scholar] [CrossRef]
- Byrne, A.T.; Southgate, J.; Brison, D.R.; Leese, H.J. Analysis of apoptosis in the preimplantation bovine embryo using TUNEL. J. Reprod. Fertil. 1999, 117, 97–105. [Google Scholar] [CrossRef]
- Dumoulin, J.C.; Coonen, E.; Bras, M.; van Wissen, L.C.; Ignoul-Vanvuchelen, R.; Bergers- Jansen, J.M.; Derhaag, J.; Geraedts, J.P.; Evers, J. Comparison of in-vitro development of embryos originating from either conventional in-vitro fertilization or intracytoplasmic sperm injection. Hum. Reprod. 2000, 15, 402–409. [Google Scholar] [CrossRef]
- Shukla, V.; Høffding, M.K.; Hoffmann, E.R. Genome diversity and instability in human germ cells and preimplantation embryos. Semin. Cell Dev. Biol. 2021, 113, 132–147. [Google Scholar] [CrossRef]
- Girardi, L.; Serdarogullari, M.; Patassini, C.; Poli, M.; Fabiani, M.; Caroselli, S.; Coban, O.; Findikli, N.; Boynukalin, F.K.; Bahceci, M.; et al. Incidence, origin, and predictive model for the detection and clinical management of segmental aneuploidies in human embryos. Am. J. Hum. Genet. 2020, 106, 525–534. [Google Scholar] [CrossRef]
- Mayer, A.; Baran, V.; Sakakibara, Y.; Brzakova, A.; Ferencova, I.; Motlik, J.; Kitajima, T.S.; Schultz, R.M.; Solc, P. DNA damage response during mouse oocyte maturation. Cell Cycle 2016, 15, 546–558. [Google Scholar] [CrossRef]
- Baran, V.; Pisko, J. Cleavage of early mouse embryo with damaged DNA. Inter. J. Mol. Sci. 2022, 23, 3516. [Google Scholar] [CrossRef]
- Gawecka, J.E.; Marh, J.; Ortega, M.; Yamauchi, Y.; Ward, M.A.; Ward, W.S. Mouse zygotes respond sperm DNA damage by delaying paternal DNA replication and embryonic development. PLoS ONE 2013, 8, e56385. [Google Scholar] [CrossRef] [PubMed]
- Barton, T.S.; Robaire, B.; Hales, B.F. DNA damage recognition in rat zygote following chronic paternal cyclophospamide exposure. Toxicol. Sci. 2007, 100, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Colaco, S.; Sakkas, D. Paternal factors contributing to embryo quality. J. Assist. Reprod. Genet. 2018, 35, 1953–1968. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Y.; Ou-Yang, Y.C.; Wang, Z.W.; Wang, Z.B.; Jing, Z.Z.; Luo, S.H.; Hou, Y.; Liu, Y.H.; Schatten, H.; Sun, Q.Y. The effect of DNA double-strand breaks on mouse oocyte meiotic maturation. Cell Cycle 2013, 12, 1233–1241. [Google Scholar] [CrossRef]
- Marangos, P.; Carroll, J. Oocytes progress beyond prophase in the presence DNA damage. Curr. Biol. 2012, 22, 989–994. [Google Scholar] [CrossRef]
- Menezo, Y.; Dale, B.; Cohen, M. DNA damage and repair in human oocyte and embryos: A review. Zygote 2010, 18, 357–365. [Google Scholar] [CrossRef]
- Bazrgar, M.; Gourabi, H.; Yazdi, P.E.; Vazirinasab, H.; Fakhri, M.; Hassani, F.; Valojerdi, M.R. DNA repair signalling pathway genes are overexpressed in poor-quality pre-implantation human embryos with complex aneuploidy. Eur. J. Obs. Gynecol. Reprod. Biol. 2014, 175, 152–156. [Google Scholar] [CrossRef]
- Palmer, N.; Kaldis, P. Regulation of the embryonic cell cycle during mammalian preimplantation development. Curr. Top. Dev. Biol. 2016, 120, 2–53. [Google Scholar] [CrossRef]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef]
- Wang, W.H.; Sun, Q.Y. Meiotic spindle, spindle checkpoint and embryonic aneuploidy. Front. Biosci. 2016, 11, 620–636. [Google Scholar] [CrossRef]
- Ford, E.; Currie, C.E.; Taylor, D.M.; Erent, M.; Marston, A.L.; Hartshorne, G.M.; McAinsh, A.D. The First Mitotic Division of the Human Embryo Is Highly Error-Prone. bioRxiv 2020, 1–13. [Google Scholar] [CrossRef]
- Cavazza, T.; Takeda, Y.; Politi, A.Z.; Aushev, M.; Aldag, P.; Baker, C.; Choudhary, M.; Bucevičius, J.; Lukinavičius, G.; Elder, K.; et al. Parental Genome Unification Is Highly Error-Prone in Mammalian Embryos. Cell 2021, 184, 2860–2877.e22. [Google Scholar] [CrossRef]
- Palmerola, K.L.; Amrane, S.; Angeles, A.D.L.; Xu, S.; Wang, N.; Pinho, J.; Zuccaro, M.V.; Taglialatela, A.; Massey, D.J.; Turocy, J.; et al. Replication Stress Impairs Chromosome Segregation and Preimplantation Development in Human Embryos. Cell 2022, 185, 2988–3007.e20. [Google Scholar] [CrossRef]
- Svoboda, P. Mammalian zygotic genome activation. Semin. Cell Dev. Biol. 2018, 84, 18–126. [Google Scholar] [CrossRef]
- Yukawa, M.; Oda, S.; Mitani, H.; Nagata, M.; Aoki, F. Deficiency in response to DNA double-strand breaks in mouse early preimplantation embryos. Biochem. Biophys. Res. Commun. 2007, 358, 578–584. [Google Scholar] [CrossRef]
- Marchetti, F.; Bishop, J.B.; Lowe, X.; Generoso, W.M.; Hozier, J.; Wyrobek, A.J. Etoposide induces heritable chromosomal aberration and aneuploidy during male meiosis in mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 3953–3957. [Google Scholar] [CrossRef] [PubMed]
- Carson, S.A.; Kallen, A.N. Diagnosis and Management of Infertility: A Review. JAMA 2021, 326, 65–76. [Google Scholar] [CrossRef]
- Gruhn, J.R.; Hoffman, E.R. Errors of the Egg: The Establishment and Progression of Human Aneuploidy Research in the Maternal Germline. Annu. Rev. Genet. 2022, 56, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Middelkamp, S.; van Tol, H.T.A.; Spierings, D.C.J.; Boymans, S.; Guryev, V.; Roelen, B.J.A.; Lansdorp, P.M.; Cuppen, E.; Kuijk, E.W. Sperm DNA damage causes genomic instability in early embryonic development. Sci. Adv. 2020, 6, eaaz7602. [Google Scholar] [CrossRef] [PubMed]
- Ribas-Maynou, J.; Novo, S.; Torres, T.; Salas-Huetos, A.; Rovira, S.; Antich, M.; Yeste, M. Sperm DNA integrity does play a crucial role for embryo development after ICSI, notably when good-quality oocytes from young donors are used. Biol. Res. 2022, 55, 41. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Poli, M.; Rienzi, L.; Girardi, L.; Patassini, C.; Fabiani, M.; Cimadomo, D.; Benini, F.; Farcomeni, A.; Cuzzi, J.; et al. Mosaic Human Preimplantation Embryos and Their Developmental Potential in a Prospective, Non-Selection Clinical Trial. Am. J. Hum. Genet. 2021, 108, 2238–2247. [Google Scholar] [CrossRef]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol. Cell. Oncol. 2015, 2, e1012976. [Google Scholar] [CrossRef]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149 (Suppl. SC), 124–138. [Google Scholar] [CrossRef]
- Simoneau, A.; Zou, L. An extending ATR–CHK1 circuitry: The replication stress response and beyond. Curr. Opin. Genet. Dev. 2021, 71, 92–98. [Google Scholar] [CrossRef]
- Muralidharan, S.V.; Nilsson, L.M.; Lindberg, M.F.; Nilsson, J.A. Small Molecule Inhibitors and a Kinase-Dead Expressing Mouse Model Demonstrate That the Kinase Activity of Chk1 Is Essential for Mouse Embryos and Cancer Cells. Life Sci. Alliance 2020, 3, e202000671. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Takai, H.; Tominaga, K.; Motoyama, N.; Minamishima, Y.A.; Nagahama, H.; Tsukiyama, T.; Ikeda, K.; Nakayama, K.; Nakanishi, M.; Nakayama, K.-I. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev. 2000, 14, 1439–1447. [Google Scholar] [CrossRef]
- Takai, H.; Naka, K.; Okada, Y.; Watanabe, M.; Harada, N.; Saito, S.; Anderson, C.W.; Appella, E.; Nakanishi, M.; Suzuki, H.; et al. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 2002, 21, 5195–5205. [Google Scholar] [CrossRef]
- Sidi, S.; Sanda, T.; Kennedy, R.D.; Hagen, A.T.; Jette, C.A.; Hoffmans, R.; Pascual, J.; Imamura, S.; Kishi, S.; Amatruda, J.F.; et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 2008, 133, 864–877. [Google Scholar] [CrossRef]
- Smits, V.A.J.; Gillespie, D.A. DNA damage control: Regulation and functions of checkpoint kinase 1. FEBS J. 2015, 282, 3681–3692. [Google Scholar] [CrossRef]
- Li, J.; Cui, P.; Sun, Q.; Du, Z.; Chen, Z.; Li, Z.; Liu, C.; Cao, Y.; Yang, Z.; Liu, R.; et al. PSPC1 regulates CHK1 phosphorylation through phase separation and participates in mouse oocyte maturation. Acta Biochim. Biophys. Sin. 2021, 53, 1527–1537. [Google Scholar] [CrossRef]
- Michelena, J.; Gatti, M.; Teloni, F.; Imhof, R.; Altmeyer, M. Basal CHK1 activity safeguards its stability to maintain intrinsic S-phase checkpoint functions. J. Cell Biol. 2019, 218, 2865–2875. [Google Scholar] [CrossRef]
- Chen, L.; Chao, S.B.; Wang, Z.B.; Qi, S.T.; Zhu, X.L.; Yang, S.W.; Yang, C.R.; Zhang, Q.H.; Ouyang, Y.C.; Hou, Y.; et al. Checkpoint kinase 1 is essential for meiotic cell cycle regulation in mouse oocytes. Cell Cycle 2012, 11, 1948–1955. [Google Scholar] [CrossRef][Green Version]
- Dai, X.X.; Duan, X.; Liu, H.L.; Cui, X.S.; Kim, N.H.; Sun, S.C. Chk2 regulates cell cycle progression during mouse oocyte maturation and early embryo development. Mol. Cell 2014, 37, 126. [Google Scholar] [CrossRef]
- Stringer, J.M.; Winship, A.; Liew, S.H.; Hutt, K. The capacity of oocytes for DNA repair. Cell. Mol. Life Sci. 2018, 75, 2777–2792. [Google Scholar] [CrossRef]
- Zhang, M.; Kothari, P.; Mullins, M.; Lampson, M.A. Regulation of zygotic genome activation and DNA damage checkpoint acquisition at the mid-blastula transition. Cell Cycle 2014, 13, 3828–3838. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Marchal, A.; Huang, Y.; Guillot-Ferriols, M.T.; Ferrer-RodaI, M.; Guixe, A.; Garcia-Caldes, M.; Roig, I. The DNA damage response is required for oocyte cyst breakdown and follicle formation in mice. PLOS Genet. 2020, 16, e1009067. [Google Scholar] [CrossRef] [PubMed]
- Remillard-Labrosse, G.; Dean, N.L.; Allais, A.; Mihajlovic, A.I.; Jin, S.G.; Son, W.Y.; Chung, J.T.; Pansera, M.; Henderson, S.; Mahfoudh, A.; et al. Human oocytes harboring damaged DNA can complete meiosis I. Fertil. Steril. 2020, 113, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, V.D.; Bolcun-Filas, E.; Kogo, H.; Kurahashi, H.; Schimenti, J.C. The DNA damage checkpoint eliminates mouse oocytes with chromosome synapsis failure. Mol. Cell 2017, 67, 1026–1036. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Rinaldi, V.D.; Bloom, J.C.; Schimenti, J.C. Oocyte Elimination Through DNA Damage Signaling from CHK1/CHK2 to p53 and p63. Genetics 2020, 215, 373–378. [Google Scholar] [CrossRef]
- Gillespie, D.A. When more is less: Heritable gain-of-function chk1 mutations impair human fertility. FEBS J. 2022, 1–6. [Google Scholar] [CrossRef]
- Crncec, A.; Hochegger, H. Triggering mitosis. FEBS Lett. 2019, 593, 2868–2888. [Google Scholar] [CrossRef]
- Ju, J.Q.; Li, X.H.; Pan, M.H.; Xu, Y.; Sun, M.H.; Xu, Y.; Sun, S.C. CHK1 monitors spindle assembly checkpoint and DNA damage repair during the first cleavage of mouse early embryos. Cell Prolif. 2020, 53, e12895. [Google Scholar] [CrossRef]
- Iyer, D.R.; Rhind, N. The Intra-S Checkpoint Responses to DNA Damage. Genes 2017, 8, 74. [Google Scholar] [CrossRef]
- Kermi, E.; Lo Furno, D. Maiorano. Regulation of DNA replication in early embryonic cleavages. Genes 2017, 8, 42. [Google Scholar] [CrossRef]
- Gómez-González, B.; Aguilera, A. Transcription-mediated replication hindrance: A major driver of genome instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef]
- Gnan, S.; Liu, Y.; Spagnuolo, M.; Chen, C.L. The Impact of Transcription-mediated Replication Stress on Genome Instability and Human Disease. Genome Instab. Dis. 2020, 1, 207–234. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Niida, H.; Nakanishi, M. DNA damage checkpoints in mammals. Mutagenesis 2006, 21, 3–9. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- Goto, H.; Natsume, T.; Kanemaki, M.T.; Kaito, A.; Wang, S.; Gabazza, E.C.; Inagaki, M.; Mizoguchi, A. Chk1-mediated Cdc25A degradation as a critical mechanism for normal cell cycle progression. J. Cell Sci. 2019, 132, jcs223123. [Google Scholar] [CrossRef]
- Sur, S.; Agrawal, D.K. Phosphatases and kinases regulating CDC25 activity in the cell cycle: Clinical implications of CDC25 overexpression and potential treatment strategies. Mol. Cell. Biochem. 2016, 416, 33–46. [Google Scholar] [CrossRef]
- Saldivar, J.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- Branigan, T.B.; Kozono, D.; Schade, A.E.; Deraska, P.; Rivas, H.G.; Sambel, L.; Reavis, H.D.; Shapiro, G.I.; D’Andrea, A.D.; DeCaprio, J.A. MMB-FOXM1-Driven Premature Mitosis Is Required for CHK1 Inhibitor Sensitivity. Cell Rep. 2021, 34, 108808. [Google Scholar] [CrossRef]
- Lam, M.H.; Liu, Q.; Elledge, S.J.; Rosen, J.M. Chk1 Is Haploinsufficient for MultipleFunctions Critical to Tumor Suppression. Cancer Cell 2004, 6, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Zonderland, G.; Vanzo, R.; Amitash, S.; Martín-Doncel, E.; Coscia, F.; Mund, A.; Lerdrup, M.; Benada, J.; Boos, D.; Toledo, L. The TRESLIN-MTBP Complex Couples Completion of DNA Replication with S/G2 Transition. Mol. Cell 2022, 82, 3350–3365.e7. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, C.; Karanika, K.; Fernández-Casañas, M.; Herbert, A.; Olukoga, T.; Özgürses, M.E.; Chan, K.L. DNA replication is highly resilient and persistent under the challenge of mild replication stress. Cell Rep. 2022, 39, 110701. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Shi, R.; Bian, J.; Li, Y.; Wang, P.; Wang, H.; Liao, J.; Zhu, W.G.; Xu, X. PARP1 and CHK1 coordinate PLK1 enzymatic activity during the DNA damage response to promote homologous recombination-mediated repair. Nucleic Acids Res. 2021, 49, 7554–7570. [Google Scholar] [CrossRef] [PubMed]
- Lebrec, V.; Poteau, M.; Morretton, J.P.; Gavet, O. Chk1 dynamics in G2 phase upon replication stress predict daughter cell outcome. Dev. Cell 2022, 57, 638–653. [Google Scholar] [CrossRef]
- Solc, P.; Schultz, R.M.; Motlik, J. Prophase I arrest and progression to metaphase I in mouse oocytes: Comparison of resumption of meiosis and recovery from G2-arrest in somatic cells. Mol. Hum. Reprod. 2010, 16, 654–664. [Google Scholar] [CrossRef]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, T.; Wu, K.; Hou, Z.; Zhao, S.; Zhang, C.; Gao, Y.; Gao, M.; Chen, Z.J. Dominant mutations in CHK1 cause pronuclear fusion failure and zygote arrest that can be rescued by CHK1 inhibitor. Cell Res. 2021, 31, 814–817. [Google Scholar] [CrossRef]
- Chen, B.; Guo, J.; Wang, T.; Lee, Q.; Ming, J.; Ding, F.; Li, H.; Zhang, Z.; Li, L.; Cao, Y.; et al. Maternal heterozygous mutation in CHEK1 leads to mitotic arrest in human zygotes. Protein Cell 2022, 13, 148–154. [Google Scholar] [CrossRef]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef]
- Wei, Y.; Multi, S.; Yang, C.R.; Ma, J.; Zhang, Q.H.; Wang, Z.B.; Li, M.; Wei, L.; Ge, Z.-J.; Zhang, C.-H.; et al. Spindle assembly checkpoint regulates mitotic cell cycle progression during preimplantation embryo development. PLoS ONE 2011, 6, e21557. [Google Scholar] [CrossRef]
- Tang, J.; Erikson, R.L.; Liu, X. Checkpoint kinase 1 (Chk1) is required for mitotic progression through negative regulation of polo-like kinase 1 (Plk1). Proc. Natl. Acad. Sci. USA 2006, 103, 11964–11969. [Google Scholar] [CrossRef]
- Peddibhotla, S.; Lam, M.H.; Gonzalez-Rimbau, M.; Rosen, J.M. The DNA-Damage Effector Checkpoint Kinase 1 Is Essential for Chromosome Segregation and Cytokinesis. Proc. Natl. Acad. Sci. USA 2009, 106, 5159–5164. [Google Scholar] [CrossRef]
- Carrassa, L.; Sanchez, Y.; Erba, E.; Damia, G. U2OS cells lacking Chk1 undergo aberrant mitosis and fail to activate the spindle checkpoint. J. Cell. Mol. Med. 2009, 13, 1565–1576. [Google Scholar] [CrossRef]
- Fishler, T.; Li, Y.Y.; Wang, R.H.; Kim, H.S.; Sengupta, K.; Vassilopoulos, A.; Lahusen, T.; Xu, X.; Lee, M.H.; Liu, Q.; et al. Genetic Instability and Mammary Tumor Formation in Mice Carrying Mammary-Specific Disruption of Chk1 and P53. Oncogene 2010, 29, 4007–4017. [Google Scholar] [CrossRef]
- Zachos, G.; Black, E.J.; Walker, M.; Scott, M.T.; Vagnarelli, P.; Earnshaw, W.C.; Gillespie, D.A.F. Chk1 is required for spindle checkpoint function. Dev. Cell 2007, 12, 247–260. [Google Scholar] [CrossRef]
- Chila, R.; Celenza, C.; Lupi, M.; Damia, G.; Carrassa, L. Chk1-Mad2 interaction: A crosslink between the DNA damage checkpoint and the mitotic spindle checkpoint. Cell Cycle 2013, 12, 1083–1090. [Google Scholar] [CrossRef]
- Yang, X.; Xu, W.; Hu, Z.; Zhang, Y.; Xu, N. Chk1 is required for the metaphase-anaphase transition via regulating the expression and localization of Cdc20 and Mad2. Life Sci. 2014, 106, 12–18. [Google Scholar] [CrossRef]
- Petsalaki, E.; Zachos, G. Chk1 and Mps1 jointly regulate correction of merotelic kinetochore attachments. J. Cell Sci. 2013, 126 Pt 5, 1235–1246. [Google Scholar] [CrossRef]
- Liu, X.M.; Chen, F.; Wang, L.; Zhang, F.; Huo, L.J. Checkpoint kinases are required for oocyte meiotic progression by the maintenance of normal spindle structure and chromosome condensation. Exp. Cell Res. 2021, 405, 112657. [Google Scholar] [CrossRef]
- Jacobs, K.; deVelde, H.; Paepe, C.D.; Serson, K.; Spits, C. Mitotic spindle disruption in human preimplantation embryos activates the spindle assembly checkpoint but not apoptosis until day 5 of development. Molec. Hum. Reprod. 2017, 23, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tang, M.; Chen, Z.; Nie, L.; Li, S.; Xiong, Y.; Szymonowicz, K.A.; Park, J.-M.; Zhang, H.; Feng, X.; et al. Genetic vulnerabilities upon inhibition of DNA damage response. Nucleic Acids Res. 2021, 49, 8214–8231. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baran, V.; Mayer, A. Checkpoint Kinase 1 Is a Key Signal Transducer of DNA Damage in the Early Mammalian Cleavage Embryo. Int. J. Mol. Sci. 2023, 24, 6778. https://doi.org/10.3390/ijms24076778
Baran V, Mayer A. Checkpoint Kinase 1 Is a Key Signal Transducer of DNA Damage in the Early Mammalian Cleavage Embryo. International Journal of Molecular Sciences. 2023; 24(7):6778. https://doi.org/10.3390/ijms24076778
Chicago/Turabian StyleBaran, Vladimír, and Alexandra Mayer. 2023. "Checkpoint Kinase 1 Is a Key Signal Transducer of DNA Damage in the Early Mammalian Cleavage Embryo" International Journal of Molecular Sciences 24, no. 7: 6778. https://doi.org/10.3390/ijms24076778
APA StyleBaran, V., & Mayer, A. (2023). Checkpoint Kinase 1 Is a Key Signal Transducer of DNA Damage in the Early Mammalian Cleavage Embryo. International Journal of Molecular Sciences, 24(7), 6778. https://doi.org/10.3390/ijms24076778