
A Dual-Function “TRE-Lox” System for Genetic Deletion or Reversible, Titratable, and Near-Complete Downregulation of Cathepsin D





Abstract
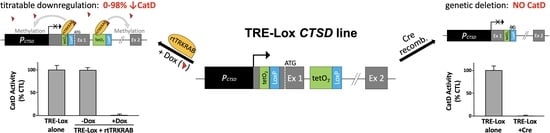
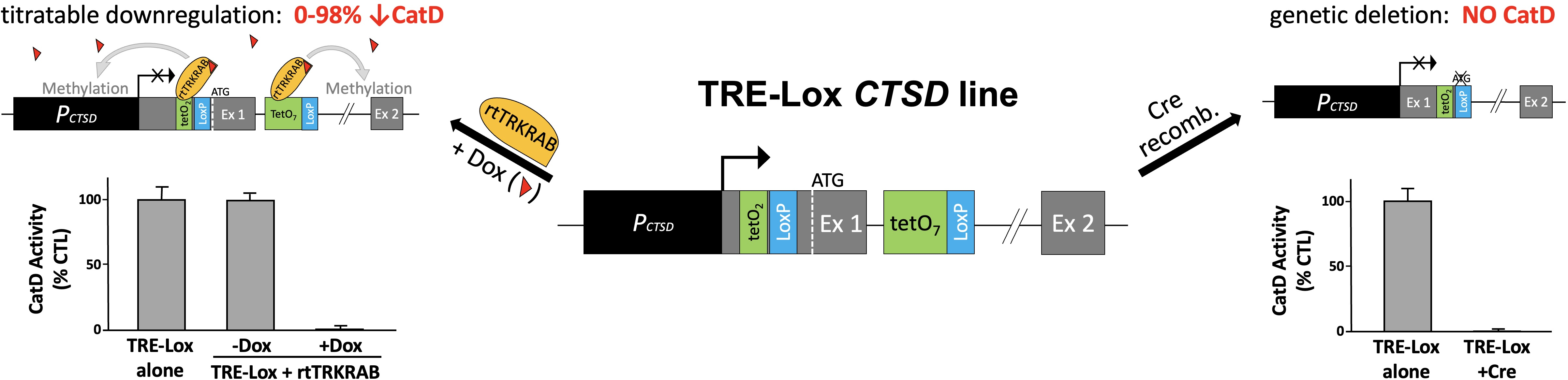{kind=link}
{kind=link}
{kind=link}
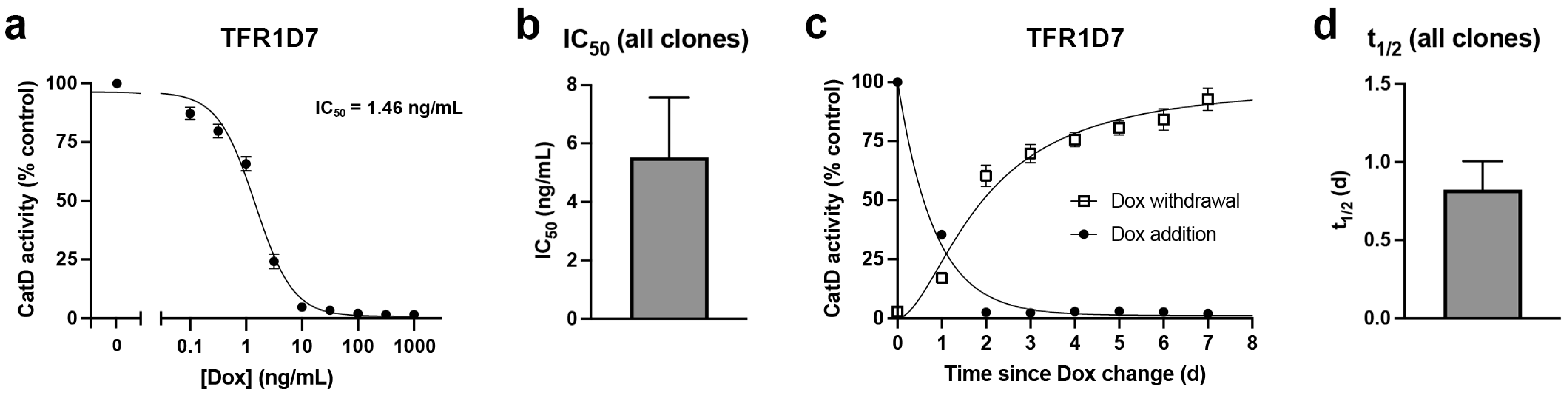{kind=link}
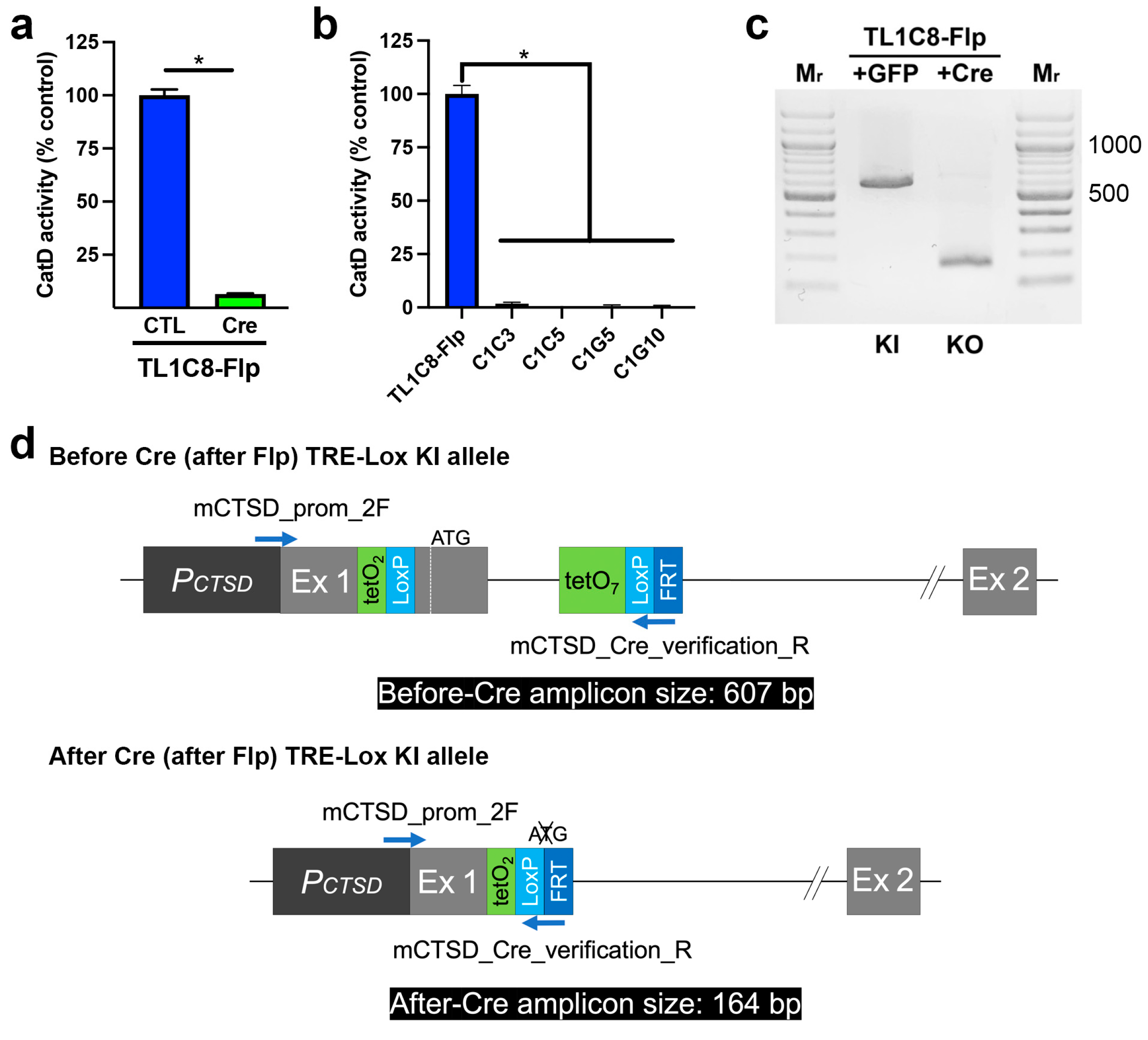{kind=link}
1. Introduction
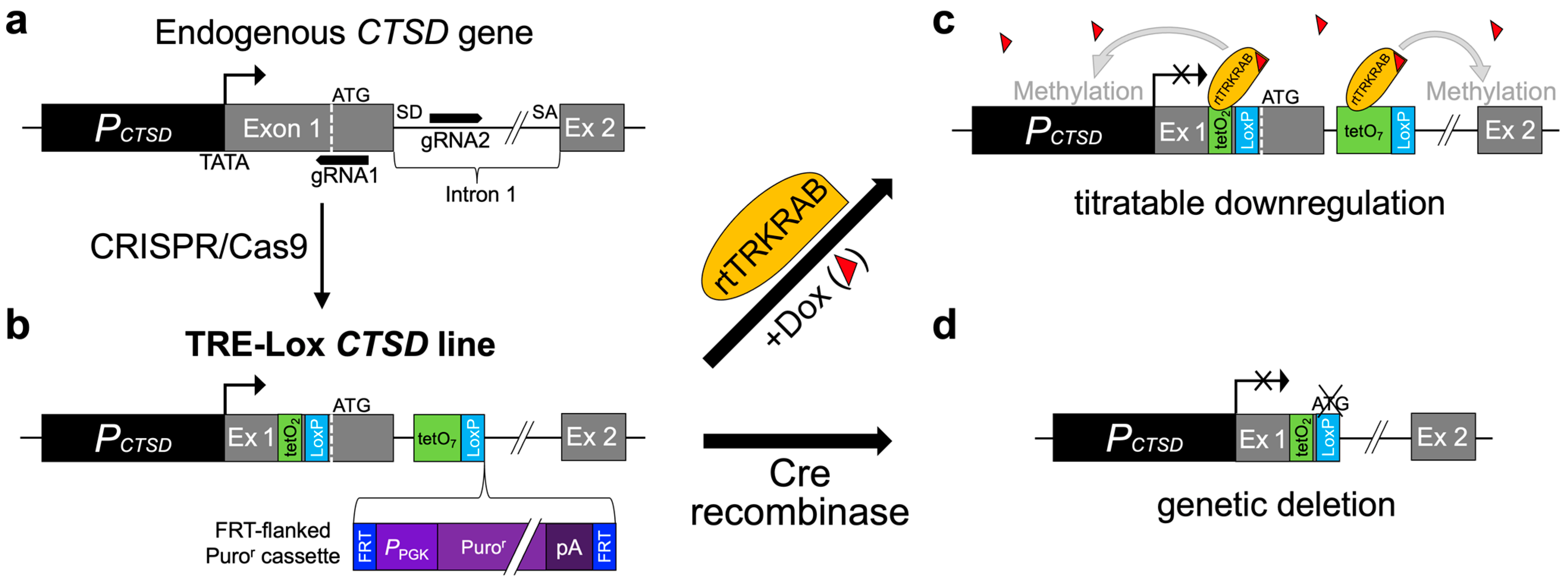
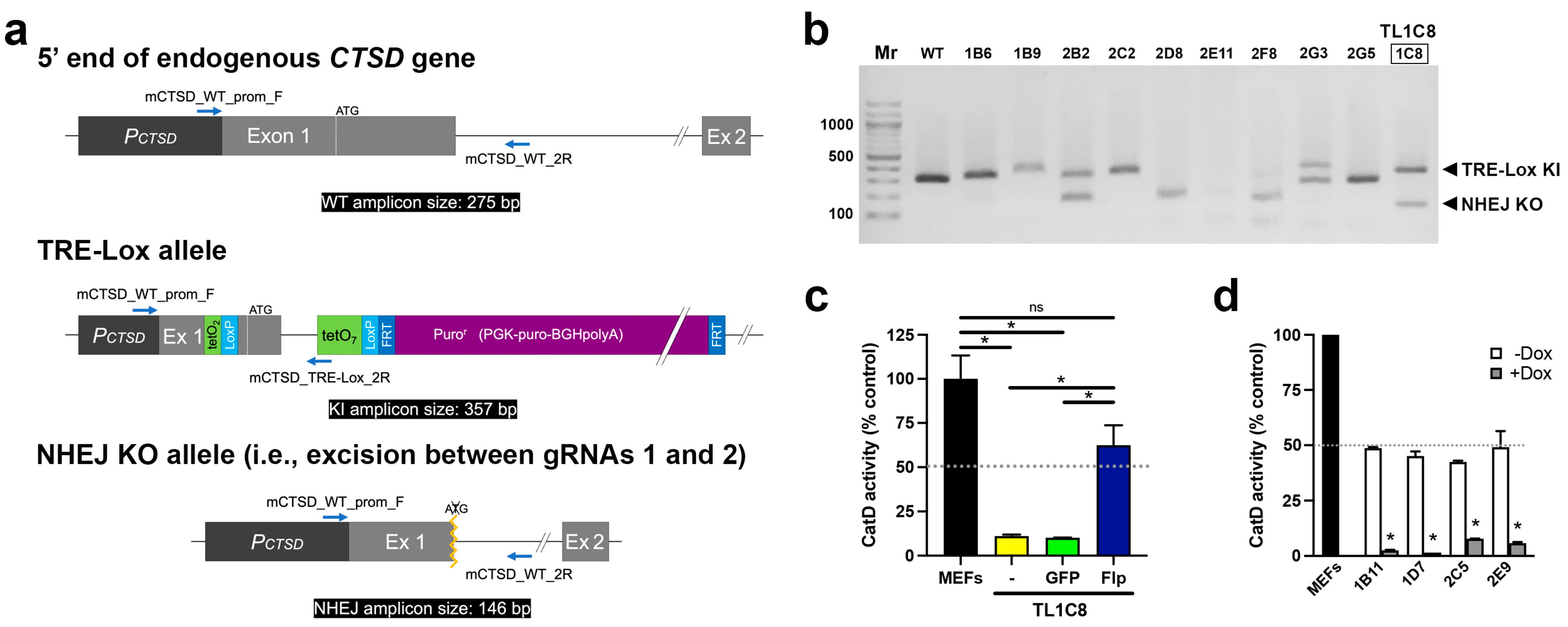
2. Results
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Targeted Modification of the CTSD Gene
4.3. Cloning of the rtTRKRABV9I and rtTR Expression Constructs and Generation of Stable Cell Lines Derived from Them
4.4. Cell Culture
4.5. CatD Activity Assays
4.6. Western Blotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orban, P.C.; Chui, D.; Marth, J.D. Tissue- and site-specific DNA recombination in transgenic mice. Proc. Natl. Acad. Sci. USA 1992, 89, 6861–6865. [Google Scholar] [CrossRef] [PubMed]
- Sauer, B. Functional expression of the cre-lox site-specific recombination system in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 1987, 7, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed]
- Gossen, M.; Freundlieb, S.; Bender, G.; Muller, G.; Hillen, W.; Bujard, H. Transcriptional activation by tetracyclines in mammalian cells. Science 1995, 268, 1766–1769. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Sen, G.L.; Blau, H.M. A brief history of RNAi: The silence of the genes. FASEB J. 2006, 20, 1293–1299. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D--many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Mijanovic, O.; Petushkova, A.I.; Brankovic, A.; Turk, B.; Solovieva, A.B.; Nikitkina, A.I.; Bolevich, S.; Timashev, P.S.; Parodi, A.; Zamyatnin, A.A., Jr. Cathepsin D-managing the delicate balance. Pharmaceutics 2021, 13, 837. [Google Scholar] [CrossRef] [PubMed]
- Vashishta, A.; Ohri, S.S.; Vetvicka, V. Pleiotropic effects of cathepsin D. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Suire, C.N.; Abdul-Hay, S.O.; Sahara, T.; Kang, D.; Brizuela, M.K.; Saftig, P.; Dickson, D.W.; Rosenberry, T.L.; Leissring, M.A. Cathepsin D regulates cerebral Abeta42/40 ratios via differential degradation of Abeta42 and Abeta40. Alzheimer’s Res. Ther. 2020, 12, 80. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Hetman, M.; Schmahl, W.; Weber, K.; Heine, L.; Mossmann, H.; Koster, A.; Hess, B.; Evers, M.; von Figura, K.; et al. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995, 14, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wani, W.Y.; Hottman, D.A.; Jeong, A.; Cao, D.; LeBlanc, K.J.; Saftig, P.; Zhang, J.; Li, L. Haplodeficiency of Cathepsin D does not affect cerebral amyloidosis and autophagy in APP/PS1 transgenic mice. J. Neurochem. 2017, 142, 297–304. [Google Scholar] [CrossRef]
- Yarmolinsky, M.; Hoess, R. The legacy of Nat Sternberg: The genesis of Cre-lox technology. Annu. Rev. Virol. 2015, 2, 25–40. [Google Scholar] [CrossRef]
- Deuschle, U.; Meyer, W.K.; Thiesen, H.J. Tetracycline-reversible silencing of eukaryotic promoters. Mol. Cell. Biol. 1995, 15, 1907–1914. [Google Scholar] [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef]
- Huntley, S.; Baggott, D.M.; Hamilton, A.T.; Tran-Gyamfi, M.; Yang, S.; Kim, J.; Gordon, L.; Branscomb, E.; Stubbs, L. A comprehensive catalog of human KRAB-associated zinc finger genes: Insights into the evolutionary history of a large family of transcriptional repressors. Genome Res. 2006, 16, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, P.; Georgiev, O.; Thiesen, H.J.; Hagmann, M.; Schaffner, W. Silencing of RNA polymerases II and III-dependent transcription by the KRAB protein domain of KOX1, a Kruppel-type zinc finger factor. Biol. Chem. 1997, 378, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Szulc, J.; Aebischer, P. Conditional gene expression and knockdown using lentivirus vectors encoding shRNA. Methods Mol. Biol. 2008, 434, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.M.; Guicherit, O.M.; Spicher, A.; Kringstein, A.M.; Fatyol, K.; Blakely, B.T.; Blau, H.M. Tetracycline-regulatable factors with distinct dimerization domains allow reversible growth inhibition by p16. Nat. Genet. 1998, 20, 389–393. [Google Scholar] [CrossRef]
- Groner, A.C.; Tschopp, P.; Challet, L.; Dietrich, J.E.; Verp, S.; Offner, S.; Barde, I.; Rodriguez, I.; Hiiragi, T.; Trono, D. The Kruppel-associated box repressor domain can induce reversible heterochromatization of a mouse locus in vivo. J. Biol. Chem. 2012, 287, 25361–25369. [Google Scholar] [CrossRef]
- Hetman, M.; Perschl, A.; Saftig, P.; Von Figura, K.; Peters, C. Mouse cathepsin D gene: Molecular organization, characterization of the promoter, and chromosomal localization. DNA Cell Biol. 1994, 13, 419–427. [Google Scholar] [CrossRef]
- Zhou, X.; Vink, M.; Klaver, B.; Berkhout, B.; Das, A.T. Optimization of the Tet-On system for regulated gene expression through viral evolution. Gene Ther. 2006, 13, 1382–1390. [Google Scholar] [CrossRef]
- Wiznerowicz, M.; Jakobsson, J.; Szulc, J.; Liao, S.; Quazzola, A.; Beermann, F.; Aebischer, P.; Trono, D. The Kruppel-associated box repressor domain can trigger de novo promoter methylation during mouse early embryogenesis. J. Biol. Chem. 2007, 282, 34535–34541. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Zheng, Q.; Cai, X.; Tan, M.H.; Schaffert, S.; Arnold, C.P.; Gong, X.; Chen, C.Z.; Huang, S. Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques 2014, 57, 115–124. [Google Scholar] [CrossRef]
- Labun, K.; Krause, M.; Torres Cleuren, Y.; Valen, E. CRISPR genome editing made easy through the CHOPCHOP Website. Curr. Protoc. 2021, 1, e46. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Nishikawa, A.; Kume, S.; Chayama, K.; Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 2014, 4, 5400. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Cepko, C.L. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. USA 2007, 104, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Tenenbaum, L.; Berkhout, B. Tet-On systems for doxycycline-inducible gene expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Urlinger, S.; Baron, U.; Thellmann, M.; Hasan, M.T.; Bujard, H.; Hillen, W. Exploring the sequence space for tetracycline-dependent transcriptional activators: Novel mutations yield expanded range and sensitivity. Proc. Natl. Acad. Sci. USA 2000, 97, 7963–7968. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.G.; Zhou, H.; Xu, Z. Multiple shRNAs expressed by an inducible pol II promoter can knock down the expression of multiple target genes. Biotechniques 2006, 41, 64–68. [Google Scholar] [CrossRef]
- Szulc, J.; Wiznerowicz, M.; Sauvain, M.O.; Trono, D.; Aebischer, P. A versatile tool for conditional gene expression and knockdown. Nat. Methods 2006, 3, 109–116. [Google Scholar] [CrossRef]
- Campa, C.C.; Weisbach, N.R.; Santinha, A.J.; Incarnato, D.; Platt, R.J. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 2019, 16, 887–893. [Google Scholar] [CrossRef]
- Magnusson, J.P.; Rios, A.R.; Wu, L.; Qi, L.S. Enhanced Cas12a multi-gene regulation using a CRISPR array separator. Elife 2021, 10, e66406. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N. Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006320. [Google Scholar] [CrossRef]
- Elegheert, J.; Behiels, E.; Bishop, B.; Scott, S.; Woolley, R.E.; Griffiths, S.C.; Byrne, E.F.X.; Chang, V.T.; Stuart, D.I.; Jones, E.Y.; et al. Lentiviral transduction of mammalian cells for fast, scalable and high-level production of soluble and membrane proteins. Nat. Protoc. 2018, 13, 2991–3017. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Caccianini, L.; Fudenberg, G.; So, K.; Kameswaran, V.; Nagle, A.; Uebersohn, A.; Hajj, B.; Saux, A.L.; Coulon, A.; et al. Molecular basis of CTCF binding polarity in genome folding. Nat. Commun. 2020, 11, 5612. [Google Scholar] [CrossRef] [PubMed]
- Chtarto, A.; Humbert-Claude, M.; Bockstael, O.; Das, A.T.; Boutry, S.; Breger, L.S.; Klaver, B.; Melas, C.; Barroso-Chinea, P.; Gonzalez-Hernandez, T.; et al. A regulatable AAV vector mediating GDNF biological effects at clinically-approved sub-antimicrobial doxycycline doses. Mol. Ther. Methods Clin. Dev. 2016, 5, 16027. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terron, H.M.; Maranan, D.S.; Burgard, L.A.; LaFerla, F.M.; Lane, S.; Leissring, M.A. A Dual-Function “TRE-Lox” System for Genetic Deletion or Reversible, Titratable, and Near-Complete Downregulation of Cathepsin D. Int. J. Mol. Sci. 2023, 24, 6745. https://doi.org/10.3390/ijms24076745
Terron HM, Maranan DS, Burgard LA, LaFerla FM, Lane S, Leissring MA. A Dual-Function “TRE-Lox” System for Genetic Deletion or Reversible, Titratable, and Near-Complete Downregulation of Cathepsin D. International Journal of Molecular Sciences. 2023; 24(7):6745. https://doi.org/10.3390/ijms24076745
Chicago/Turabian StyleTerron, Heather M., Derek S. Maranan, Luke A. Burgard, Frank M. LaFerla, Shelley Lane, and Malcolm A. Leissring. 2023. "A Dual-Function “TRE-Lox” System for Genetic Deletion or Reversible, Titratable, and Near-Complete Downregulation of Cathepsin D" International Journal of Molecular Sciences 24, no. 7: 6745. https://doi.org/10.3390/ijms24076745
APA StyleTerron, H. M., Maranan, D. S., Burgard, L. A., LaFerla, F. M., Lane, S., & Leissring, M. A. (2023). A Dual-Function “TRE-Lox” System for Genetic Deletion or Reversible, Titratable, and Near-Complete Downregulation of Cathepsin D. International Journal of Molecular Sciences, 24(7), 6745. https://doi.org/10.3390/ijms24076745