
Amelioration of Nicotine-Induced Conditioned Place Preference Behaviors in Mice by an FABP3 Inhibitor
 ,
,  ,
,  , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
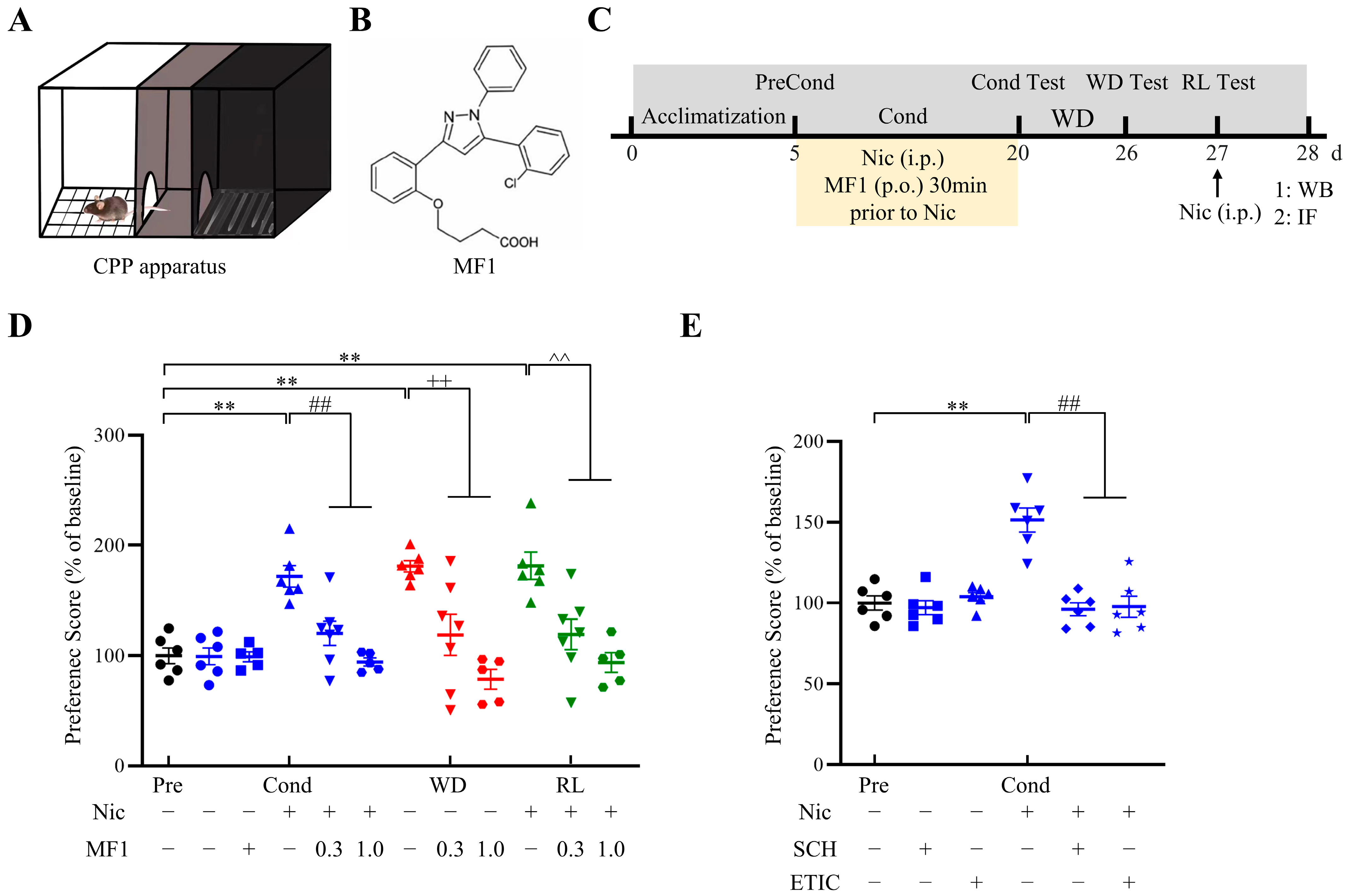
2.1. Nicotine-Induced CPP in Mice Was Significantly Inhibited by MF1 Treatment
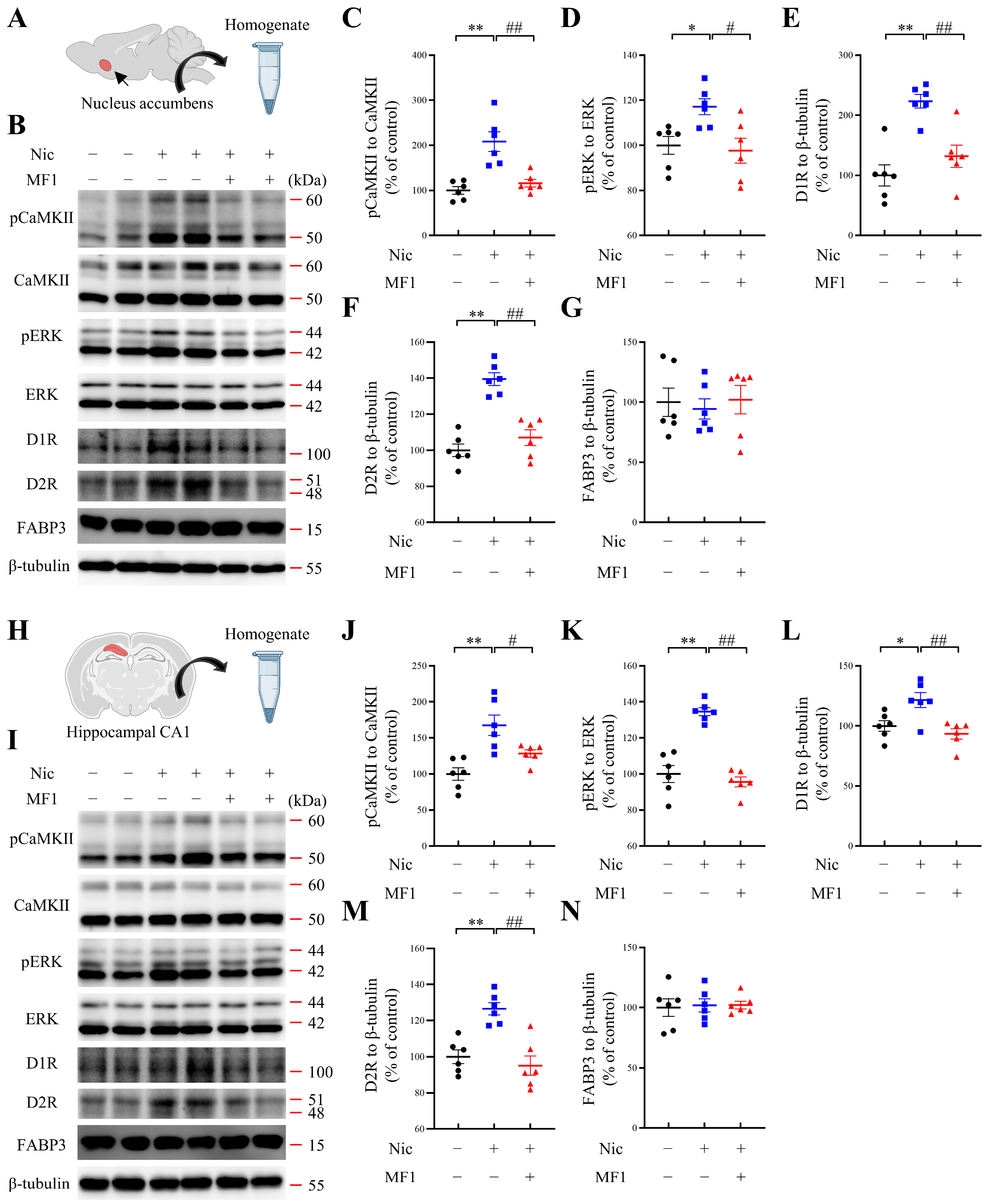
2.2. MF1 Ameliorates Nicotine-Induced Kinase Phosphorylation and Reduces DA Receptor Levels
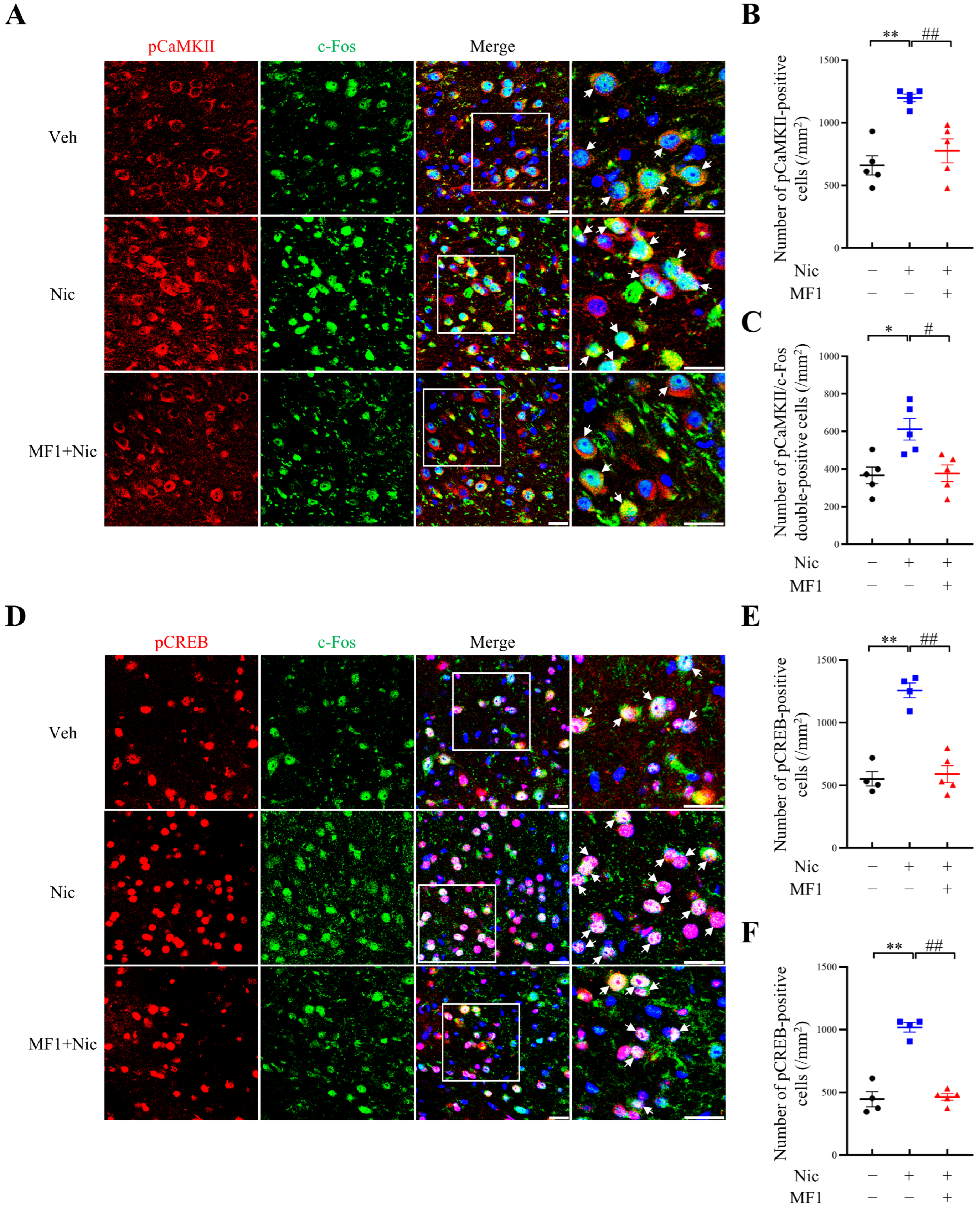
2.3. Inhibition of Nicotine-Induced Kinase Phosphorylation Is Associated with the Responsiveness of CREB/c-Fos Signals
2.4. MF1 Treatment Rescues QNP-Induced Increase in Kinase Phosphorylation in Cultured NAc Slices from WT Mice
2.5. Majority of GABAergic—But Not Cholinergic—Neurons Colocalize with FABP3 in the NAc
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Chemical Agents
4.3. CPP Apparatus
4.4. CPP Task
4.4.1. Acclimatization and Preconditioning Phase
4.4.2. Conditioning Phase
4.4.3. Nicotine Withdrawal and Relapse
4.5. Immunoblotting Analysis
4.6. Immunofluorescence
4.7. NAc Slices and Culture Preparations
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| aCSF | artificial cerebrospinal fluid |
| ANOVA | analysis of variance |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| ChAT | choline acetyltransferase |
| CMC | carboxymethylcellulose |
| CPP | conditioned place preference |
| CREB | cAMP-response element-binding protein |
| D1Rs | dopamine D1 receptors |
| D2Rs | dopamine D2 receptors |
| DA | dopamine |
| DAergic | dopaminergic |
| DAT | dopamine transporter |
| DMSO | dimethyl sulfoxide |
| ERK | extracellular signal-regulated kinase |
| FABP3 | fatty acid-binding protein 3 |
| FABP3−/− | fatty acid-binding protein 3 null |
| FABPs | Fatty acid-binding proteins |
| GABAergic | γ-aminobutyric acidergic |
| GAD | glutamate decarboxylase |
| GPCR | G protein-coupled receptor |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MSNs | medium spiny neurons |
| NAc | nucleus accumbens |
| nAChRs | nicotinic acetylcholine receptors |
| PBS | phosphate-buffered saline |
| QNP | quinpirole |
| SEM | standard error of the mean |
| TH | tyrosine hydroxylase |
| VTA | ventral tegmental area |
| WT | wild type |
References
- Roditis, M.; Lee, J.; Halpern-Felsher, B.L. Adolescent (Mis)Perceptions About Nicotine Addiction: Results from a Mixed-Methods Study. Health Educ. Behav. Off. Publ. Soc. Public Health Educ. 2016, 43, 156–164. [Google Scholar] [CrossRef]
- Benowitz, N.L. Clinical pharmacology of nicotine: Implications for understanding, preventing, and treating tobacco addiction. Clin. Pharmacol. Ther. 2008, 83, 531–541. [Google Scholar] [CrossRef]
- Changeux, J.P. The nicotinic acetylcholine receptor: The founding father of the pentameric ligand-gated ion channel superfamily. J. Biol. Chem. 2012, 287, 40207–40215. [Google Scholar] [CrossRef]
- Unwin, N. Structure and action of the nicotinic acetylcholine receptor explored by electron microscopy. FEBS Lett. 2003, 555, 91–95. [Google Scholar] [CrossRef]
- Dajas-Bailador, F.; Wonnacott, S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol. Sci. 2004, 25, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: From structure to pathology. Prog. Neurobiol. 2004, 74, 363–396. [Google Scholar] [CrossRef]
- Alijevic, O.; McHugh, D.; Rufener, L.; Mazurov, A.; Hoeng, J.; Peitsch, M. An electrophysiological characterization of naturally occurring tobacco alkaloids and their action on human alpha4beta2 and alpha7 nicotinic acetylcholine receptors. Phytochemistry 2020, 170, 112187. [Google Scholar] [CrossRef]
- Exley, R.; Maubourguet, N.; David, V.; Eddine, R.; Evrard, A.; Pons, S.; Marti, F.; Threlfell, S.; Cazala, P.; McIntosh, J.M.; et al. Distinct contributions of nicotinic acetylcholine receptor subunit alpha4 and subunit alpha6 to the reinforcing effects of nicotine. Proc. Natl. Acad. Sci. USA 2011, 108, 7577–7582. [Google Scholar] [CrossRef]
- McClure, J.B.; Swan, G.E.; Jack, L.; Catz, S.L.; Zbikowski, S.M.; McAfee, T.A.; Deprey, M.; Richards, J.; Javitz, H. Mood, side-effects and smoking outcomes among persons with and without probable lifetime depression taking varenicline. J. Gen. Intern. Med. 2009, 24, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.H.; Martin, R.M.; Knipe, D.W.; Higgins, J.P.; Gunnell, D. Risk of neuropsychiatric adverse events associated with varenicline: Systematic review and meta-analysis. BMJ (Clin. Res. Ed.) 2015, 350, h1109. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Koya, E.; Zhai, H.; Hope, B.T.; Shaham, Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006, 29, 695–703. [Google Scholar] [CrossRef]
- Gozen, O.; Balkan, B.; Yildirim, E.; Koylu, E.O.; Pogun, S. The epigenetic effect of nicotine on dopamine D1 receptor expression in rat prefrontal cortex. Synapse 2013, 67, 545–552. [Google Scholar] [CrossRef]
- Huang, W.; Ma, J.Z.; Payne, T.J.; Beuten, J.; Dupont, R.T.; Li, M.D. Significant association of DRD1 with nicotine dependence. Hum. Genet. 2008, 123, 133–140. [Google Scholar] [CrossRef]
- Yawata, S.; Yamaguchi, T.; Danjo, T.; Hikida, T.; Nakanishi, S. Pathway-specific control of reward learning and its flexibility via selective dopamine receptors in the nucleus accumbens. Proc. Natl. Acad. Sci. USA 2012, 109, 12764–12769. [Google Scholar] [CrossRef]
- Wilar, G.; Shinoda, Y.; Sasaoka, T.; Fukunaga, K. Crucial Role of Dopamine D2 Receptor Signaling in Nicotine-Induced Conditioned Place Preference. Mol. Neurobiol. 2019, 56, 7911–7928. [Google Scholar] [CrossRef]
- Solis, O.; Garcia-Sanz, P.; Martin, A.B.; Granado, N.; Sanz-Magro, A.; Podlesniy, P.; Trullas, R.; Murer, M.G.; Maldonado, R.; Moratalla, R. Behavioral sensitization and cellular responses to psychostimulants are reduced in D2R knockout mice. Addict. Biol. 2021, 26, e12840. [Google Scholar] [CrossRef]
- Nakamura, T.; Sato, A.; Kitsukawa, T.; Momiyama, T.; Yamamori, T.; Sasaoka, T. Distinct motor impairments of dopamine D1 and D2 receptor knockout mice revealed by three types of motor behavior. Front. Integr. Neurosci. 2014, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Chausmer, A.L.; Elmer, G.I.; Rubinstein, M.; Low, M.J.; Grandy, D.K.; Katz, J.L. Cocaine-induced locomotor activity and cocaine discrimination in dopamine D2 receptor mutant mice. Psychopharmacology 2002, 163, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Tomasi, D.; Telang, F. Addiction: Beyond dopamine reward circuitry. Proc. Natl. Acad. Sci. USA 2011, 108, 15037–15042. [Google Scholar] [CrossRef] [PubMed]
- Guy, E.G.; Fletcher, P.J. Responding for a conditioned reinforcer, and its enhancement by nicotine, is blocked by dopamine receptor antagonists and a 5-HT(2C) receptor agonist but not by a 5-HT(2A) receptor antagonist. Pharmacol. Biochem. Behav. 2014, 125, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xie, C.; Wang, Q.; Liu, Z. Interactions of CaMKII with dopamine D2 receptors: Roles in levodopa-induced dyskinesia in 6-hydroxydopamine lesioned Parkinson’s rats. Sci. Rep. 2014, 4, 6811. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Shioda, N. Novel dopamine D2 receptor signaling through proteins interacting with the third cytoplasmic loop. Mol. Neurobiol. 2012, 45, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.J.; Muldoon, P.P.; Walters, C.; Damaj, M.I. Neuronal calcium/calmodulin-dependent protein kinase II mediates nicotine reward in the conditioned place preference test in mice. Behav. Pharmacol. 2016, 27, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Sohn, S.; Choe, E.S. Phosphorylation of GluA1-Ser831 by CaMKII Activation in the Caudate and Putamen Is Required for Behavioral Sensitization After Challenge Nicotine in Rats. Int. J. Neuropsychopharmacol. 2022, 25, 678–687. [Google Scholar] [CrossRef]
- Cates, H.M.; Thibault, M.; Pfau, M.; Heller, E.; Eagle, A.; Gajewski, P.; Bagot, R.; Colangelo, C.; Abbott, T.; Rudenko, G.; et al. Threonine 149 phosphorylation enhances DeltaFosB transcriptional activity to control psychomotor responses to cocaine. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 11461–11469. [Google Scholar] [CrossRef] [PubMed]
- Easton, A.C.; Lourdusamy, A.; Havranek, M.; Mizuno, K.; Solati, J.; Golub, Y.; Clarke, T.K.; Vallada, H.; Laranjeira, R.; Desrivieres, S.; et al. alphaCaMKII controls the establishment of cocaine’s reinforcing effects in mice and humans. Transl. Psychiatry 2014, 4, e457. [Google Scholar] [CrossRef]
- Shioda, N.; Takeuchi, Y.; Fukunaga, K. Advanced research on dopamine signaling to develop drugs for the treatment of mental disorders: Proteins interacting with the third cytoplasmic loop of dopamine D2 and D3 receptors. J. Pharmacol. Sci. 2010, 114, 25–31. [Google Scholar] [CrossRef]
- Liu, R.Z.; Li, X.; Godbout, R. A novel fatty acid-binding protein (FABP) gene resulting from tandem gene duplication in mammals: Transcription in rat retina and testis. Genomics 2008, 92, 436–445. [Google Scholar] [CrossRef]
- Shimamoto, C.; Ohnishi, T.; Maekawa, M.; Watanabe, A.; Ohba, H.; Arai, R.; Iwayama, Y.; Hisano, Y.; Toyota, T.; Toyoshima, M.; et al. Functional characterization of FABP3, 5 and 7 gene variants identified in schizophrenia and autism spectrum disorder and mouse behavioral studies. Hum. Mol. Genet. 2014, 23, 6495–6511. [Google Scholar] [CrossRef]
- Shioda, N.; Yamamoto, Y.; Watanabe, M.; Binas, B.; Owada, Y.; Fukunaga, K. Heart-type fatty acid binding protein regulates dopamine D2 receptor function in mouse brain. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 3146–3155. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Fukunaga, K. Differential subcellular localization of two dopamine D2 receptor isoforms in transfected NG108-15 cells. J. Neurochem. 2003, 85, 1064–1074. [Google Scholar] [CrossRef]
- Jia, W.; Wilar, G.; Kawahata, I.; Cheng, A.; Fukunaga, K. Impaired Acquisition of Nicotine-Induced Conditioned Place Preference in Fatty Acid-Binding Protein 3 Null Mice. Mol. Neurobiol. 2021, 58, 2030–2045. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Shinoda, Y.; Yamamoto, T.; Miyachi, H.; Fukunaga, K. Development of FABP3 ligands that inhibit arachidonic acid-induced alpha-synuclein oligomerization. Brain Res. 2019, 1707, 190–197. [Google Scholar] [CrossRef]
- Matsuo, K.; Cheng, A.; Yabuki, Y.; Takahata, I.; Miyachi, H.; Fukunaga, K. Inhibition of MPTP-induced alpha-synuclein oligomerization by fatty acid-binding protein 3 ligand in MPTP-treated mice. Neuropharmacology 2019, 150, 164–174. [Google Scholar] [CrossRef]
- Yabuki, Y.; Matsuo, K.; Kawahata, I.; Fukui, N.; Mizobata, T.; Kawata, Y.; Owada, Y.; Shioda, N.; Fukunaga, K. Fatty Acid Binding Protein 3 Enhances the Spreading and Toxicity of alpha-Synuclein in Mouse Brain. Int. J. Mol. Sci. 2020, 21, 2230. [Google Scholar] [CrossRef] [PubMed]
- Placzek, A.N.; Zhang, T.A.; Dani, J.A. Nicotinic mechanisms influencing synaptic plasticity in the hippocampus. Acta Pharmacol. Sin. 2009, 30, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Kawahata, I.; Cheng, A.; Fukunaga, K. The Role of CaMKII and ERK Signaling in Addiction. Int. J. Mol. Sci. 2021, 22, 3189. [Google Scholar] [CrossRef]
- Greer, P.L.; Greenberg, M.E. From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron 2008, 59, 846–860. [Google Scholar] [CrossRef]
- Volkow, N.D.; Morales, M. The Brain on Drugs: From Reward to Addiction. Cell 2015, 162, 712–725. [Google Scholar] [CrossRef]
- Sofuoglu, M.; Mooney, M. Cholinergic functioning in stimulant addiction: Implications for medications development. CNS Drugs 2009, 23, 939–952. [Google Scholar] [CrossRef]
- Filip, M.; Zaniewska, M.; Frankowska, M.; Wydra, K.; Fuxe, K. The importance of the adenosine A2A receptor-dopamine D2 receptor interaction in drug addiction. Curr. Med. Chem. 2012, 19, 317–355. [Google Scholar] [CrossRef] [PubMed]
- Linker, K.E.; Gad, M.; Tawadrous, P.; Cano, M.; Green, K.N.; Wood, M.A.; Leslie, F.M. Microglial activation increases cocaine self-administration following adolescent nicotine exposure. Nat. Commun. 2020, 11, 306. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A.; De Biasi, M. Cellular mechanisms of nicotine addiction. Pharmacol. Biochem. Behav. 2001, 70, 439–446. [Google Scholar] [CrossRef]
- Zhang, R.; Manza, P.; Tomasi, D.; Kim, S.W.; Shokri-Kojori, E.; Demiral, S.B.; Kroll, D.S.; Feldman, D.E.; McPherson, K.L.; Biesecker, C.L.; et al. Dopamine D1 and D2 receptors are distinctly associated with rest-activity rhythms and drug reward. J. Clin. Investig. 2021, 131, e149722. [Google Scholar] [CrossRef] [PubMed]
- Rosa, H.Z.; Barcelos, R.C.S.; Segat, H.J.; Roversi, K.; Dias, V.T.; Milanesi, L.H.; Burger, M.E. Physical exercise modifies behavioral and molecular parameters related to opioid addiction regardless of training time. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2020, 32, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Robison, L.S.; Swenson, S.; Hamilton, J.; Thanos, P.K. Exercise Reduces Dopamine D1R and Increases D2R in Rats: Implications for Addiction. Med. Sci. Sport. Exerc. 2018, 50, 1596–1602. [Google Scholar] [CrossRef]
- Jobson, C.L.M.; Renard, J.; Szkudlarek, H.; Rosen, L.G.; Pereira, B.; Wright, D.J.; Rushlow, W.; Laviolette, S.R. Adolescent Nicotine Exposure Induces Dysregulation of Mesocorticolimbic Activity States and Depressive and Anxiety-like Prefrontal Cortical Molecular Phenotypes Persisting into Adulthood. Cereb. Cortex 2019, 29, 3140–3153. [Google Scholar] [CrossRef]
- Hamada, M.; Higashi, H.; Nairn, A.C.; Greengard, P.; Nishi, A. Differential regulation of dopamine D1 and D2 signaling by nicotine in neostriatal neurons. J. Neurochem. 2004, 90, 1094–1103. [Google Scholar] [CrossRef]
- Perreault, M.L.; Hasbi, A.; Alijaniaram, M.; Fan, T.; Varghese, G.; Fletcher, P.J.; Seeman, P.; O’Dowd, B.F.; George, S.R. The dopamine D1-D2 receptor heteromer localizes in dynorphin/enkephalin neurons: Increased high affinity state following amphetamine and in schizophrenia. J. Biol. Chem. 2010, 285, 36625–36634. [Google Scholar] [CrossRef]
- Perreault, M.L.; Hasbi, A.; O’Dowd, B.F.; George, S.R. The dopamine d1-d2 receptor heteromer in striatal medium spiny neurons: Evidence for a third distinct neuronal pathway in Basal Ganglia. Front. Neuroanat. 2011, 5, 31. [Google Scholar] [CrossRef]
- Hasbi, A.; Fan, T.; Alijaniaram, M.; Nguyen, T.; Perreault, M.L.; O’Dowd, B.F.; George, S.R. Calcium signaling cascade links dopamine D1-D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc. Natl. Acad. Sci. USA 2009, 106, 21377–21382. [Google Scholar] [CrossRef]
- Rashid, A.J.; So, C.H.; Kong, M.M.; Furtak, T.; El-Ghundi, M.; Cheng, R.; O’Dowd, B.F.; George, S.R. D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc. Natl. Acad. Sci. USA 2007, 104, 654–659. [Google Scholar] [CrossRef]
- Song, B.; Lai, B.; Zheng, Z.; Zhang, Y.; Luo, J.; Wang, C.; Chen, Y.; Woodgett, J.R.; Li, M. Inhibitory phosphorylation of GSK-3 by CaMKII couples depolarization to neuronal survival. J. Biol. Chem. 2010, 285, 41122–41134. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Xue, Y.X.; Ding, Z.B.; Xue, L.F.; Xu, C.M.; Lu, L. Glycogen synthase kinase 3beta in the basolateral amygdala is critical for the reconsolidation of cocaine reward memory. J. Neurochem. 2011, 118, 113–125. [Google Scholar] [CrossRef]
- Xu, C.M.; Wang, J.; Wu, P.; Xue, Y.X.; Zhu, W.L.; Li, Q.Q.; Zhai, H.F.; Shi, J.; Lu, L. Glycogen synthase kinase 3beta in the nucleus accumbens core is critical for methamphetamine-induced behavioral sensitization. J. Neurochem. 2011, 118, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Fukunaga, K.; Miyamoto, E. Activation of nuclear Ca2+/calmodulin-dependent protein kinase II and brain-derived neurotrophic factor gene expression by stimulation of dopamine D2 receptor in transfected NG108-15 cells. J. Neurochem. 2002, 82, 316–328. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Miyamoto, E.; Fukunaga, K. Activation of the rat dopamine D2 receptor promoter by mitogen-activated protein kinase and Ca2+/calmodulin-dependent protein kinase II pathways. J. Neurochem. 2002, 83, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, I.; Sekimori, T.; Wang, H.; Wang, Y.; Sasaoka, T.; Bousset, L.; Melki, R.; Mizobata, T.; Kawata, Y.; Fukunaga, K. Dopamine D2 Long Receptors Are Critical for Caveolae-Mediated alpha-Synuclein Uptake in Cultured Dopaminergic Neurons. Biomedicines 2021, 9, 49. [Google Scholar] [CrossRef]
- Cheng, A.; Jia, W.; Kawahata, I.; Fukunaga, K. Impact of Fatty Acid-Binding Proteins in alpha-Synuclein-Induced Mitochondrial Injury in Synucleinopathy. Biomedicines 2021, 9, 560. [Google Scholar] [CrossRef]
- Sharma, M.; Celver, J.; Octeau, J.C.; Kovoor, A. Plasma membrane compartmentalization of D2 dopamine receptors. J. Biol. Chem. 2013, 288, 12554–12568. [Google Scholar] [CrossRef]
- He, D.; Lasek, A.W. Anaplastic Lymphoma Kinase Regulates Internalization of the Dopamine D2 Receptor. Mol. Pharmacol. 2020, 97, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.I.; Min, C.; Jung, K.S.; Cheong, S.Y.; Zheng, M.; Cheong, S.J.; Oak, M.H.; Cheong, J.H.; Lee, B.K.; Kim, K.M. The N-terminal region of the dopamine D2 receptor, a rhodopsin-like GPCR, regulates correct integration into the plasma membrane and endocytic routes. Br. J. Pharmacol. 2012, 166, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Reith, M.E.A.; Liu-Chen, L.Y.; Kortagere, S. Biased signaling agonist of dopamine D3 receptor induces receptor internalization independent of beta-arrestin recruitment. Pharmacol. Res. 2019, 143, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Shioda, N.; Yabuki, Y.; Wang, Y.; Uchigashima, M.; Hikida, T.; Sasaoka, T.; Mori, H.; Watanabe, M.; Sasahara, M.; Fukunaga, K. Endocytosis following dopamine D2 receptor activation is critical for neuronal activity and dendritic spine formation via Rabex-5/PDGFRbeta signaling in striatopallidal medium spiny neurons. Mol. Psychiatry 2017, 22, 1205–1222. [Google Scholar] [CrossRef]
- Beniyama, Y.; Matsuno, K.; Miyachi, H. Structure-guided design, synthesis and in vitro evaluation of a series of pyrazole-based fatty acid binding protein (FABP) 3 ligands. Bioorg. Med. Chem. Lett. 2013, 23, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Carboni, E.; Vacca, C. Conditioned place preference. A simple method for investigating reinforcing properties in laboratory animals. Methods Mol. Med. 2003, 79, 481–498. [Google Scholar]
- Jackson, K.J.; McLaughlin, J.P.; Carroll, F.I.; Damaj, M.I. Effects of the kappa opioid receptor antagonist, norbinaltorphimine, on stress and drug-induced reinstatement of nicotine-conditioned place preference in mice. Psychopharmacology 2013, 226, 763–768. [Google Scholar] [CrossRef]
- Cheng, A.; Jia, W.; Kawahata, I.; Fukunaga, K. A novel fatty acid-binding protein 5 and 7 inhibitor ameliorates oligodendrocyte injury in multiple sclerosis mouse models. eBioMedicine 2021, 72, 103582. [Google Scholar] [CrossRef]
- Guo, Q.; Kawahata, I.; Cheng, A.; Wang, H.; Jia, W.; Yoshino, H.; Fukunaga, K. Fatty acid-binding proteins 3 and 5 are involved in the initiation of mitochondrial damage in ischemic neurons. Redox Biol. 2023, 59, 102547. [Google Scholar] [CrossRef]
- Fukunaga, K.; Goto, S.; Miyamoto, E. Immunohistochemical localization of Ca2+/calmodulin-dependent protein kinase II in rat brain and various tissues. J. Neurochem. 1988, 51, 1070–1078. [Google Scholar] [CrossRef]
- Sun, M.; Shinoda, Y.; Fukunaga, K. KY-226 Protects Blood-brain Barrier Function Through the Akt/FoxO1 Signaling Pathway in Brain Ischemia. Neuroscience 2019, 399, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Wang, Y.F.; Shinoda, Y.; Kawahata, I.; Yamamoto, T.; Jia, W.B.; Yamamoto, H.; Mizobata, T.; Kawata, Y.; Fukunaga, K. Fatty acid-binding protein 7 triggers alpha-synuclein oligomerization in glial cells and oligodendrocytes associated with oxidative stress. Acta Pharmacol. Sin. 2022, 43, 552–562. [Google Scholar] [CrossRef]
- Kasahara, J.; Fukunaga, K.; Miyamoto, E. Activation of calcium/calmodulin-dependent protein kinase IV in long term potentiation in the rat hippocampal CA1 region. J. Biol. Chem. 2001, 276, 24044–24050. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, S.; Shioda, N.; Yamamoto, Y.; Tagashira, H.; Fukunaga, K. The T-type voltage-gated calcium channel as a molecular target of the novel cognitive enhancer ST101: Enhancement of long-term potentiation and CaMKII autophosphorylation in rat cortical slices. J. Neurochem. 2012, 121, 44–53. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, W.; Kawahata, I.; Cheng, A.; Sasaki, T.; Sasaoka, T.; Fukunaga, K. Amelioration of Nicotine-Induced Conditioned Place Preference Behaviors in Mice by an FABP3 Inhibitor. Int. J. Mol. Sci. 2023, 24, 6644. https://doi.org/10.3390/ijms24076644
Jia W, Kawahata I, Cheng A, Sasaki T, Sasaoka T, Fukunaga K. Amelioration of Nicotine-Induced Conditioned Place Preference Behaviors in Mice by an FABP3 Inhibitor. International Journal of Molecular Sciences. 2023; 24(7):6644. https://doi.org/10.3390/ijms24076644
Chicago/Turabian StyleJia, Wenbin, Ichiro Kawahata, An Cheng, Takuya Sasaki, Toshikuni Sasaoka, and Kohji Fukunaga. 2023. "Amelioration of Nicotine-Induced Conditioned Place Preference Behaviors in Mice by an FABP3 Inhibitor" International Journal of Molecular Sciences 24, no. 7: 6644. https://doi.org/10.3390/ijms24076644
APA StyleJia, W., Kawahata, I., Cheng, A., Sasaki, T., Sasaoka, T., & Fukunaga, K. (2023). Amelioration of Nicotine-Induced Conditioned Place Preference Behaviors in Mice by an FABP3 Inhibitor. International Journal of Molecular Sciences, 24(7), 6644. https://doi.org/10.3390/ijms24076644