
Berberine Induces Combined Cell Death in Gastrointestinal Cell Lines


Abstract
1. Introduction
2. Results
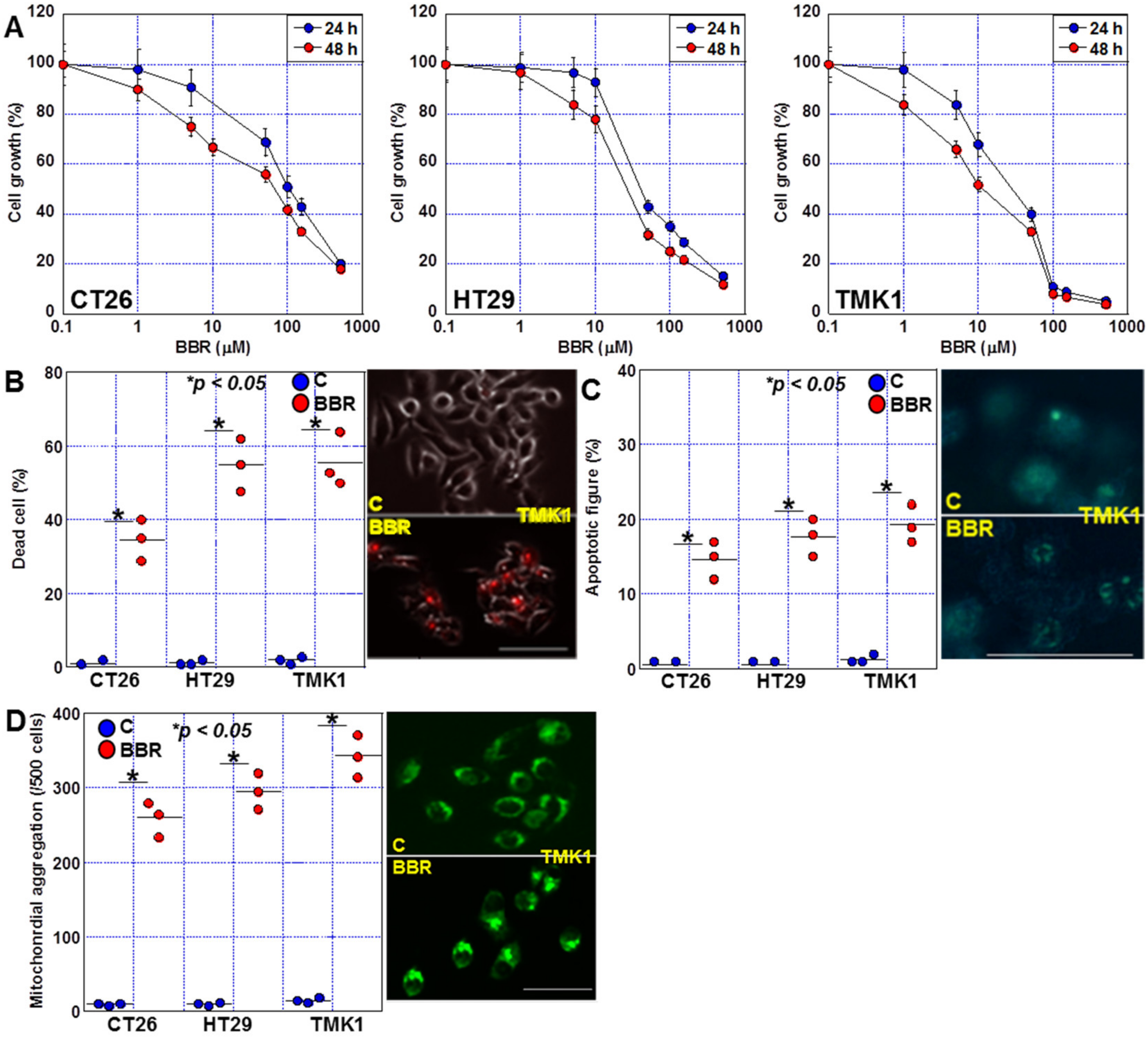
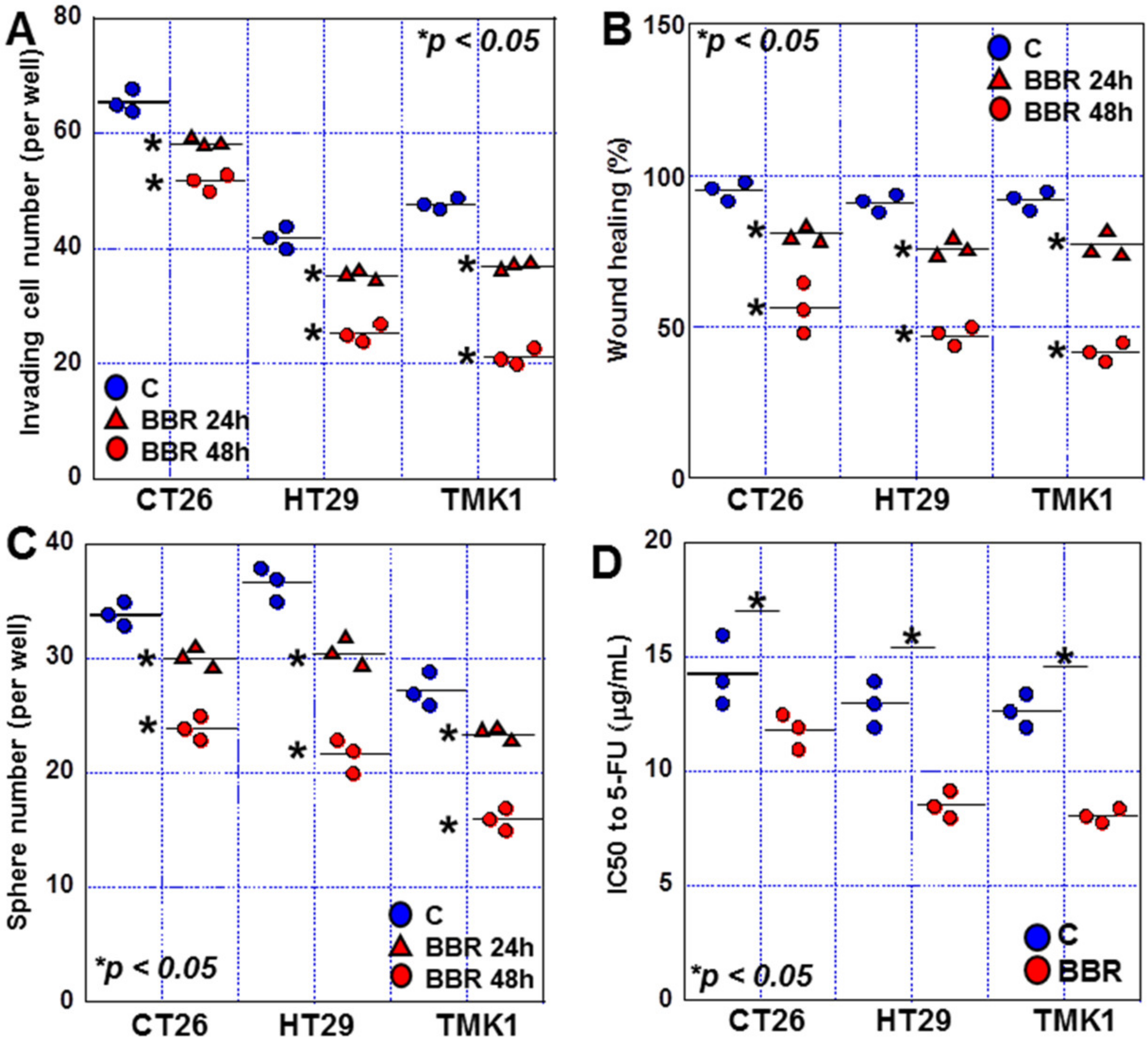
2.1. Effects of BBR on Proliferation, Invasion, and Sphere Formation in GIC Cells
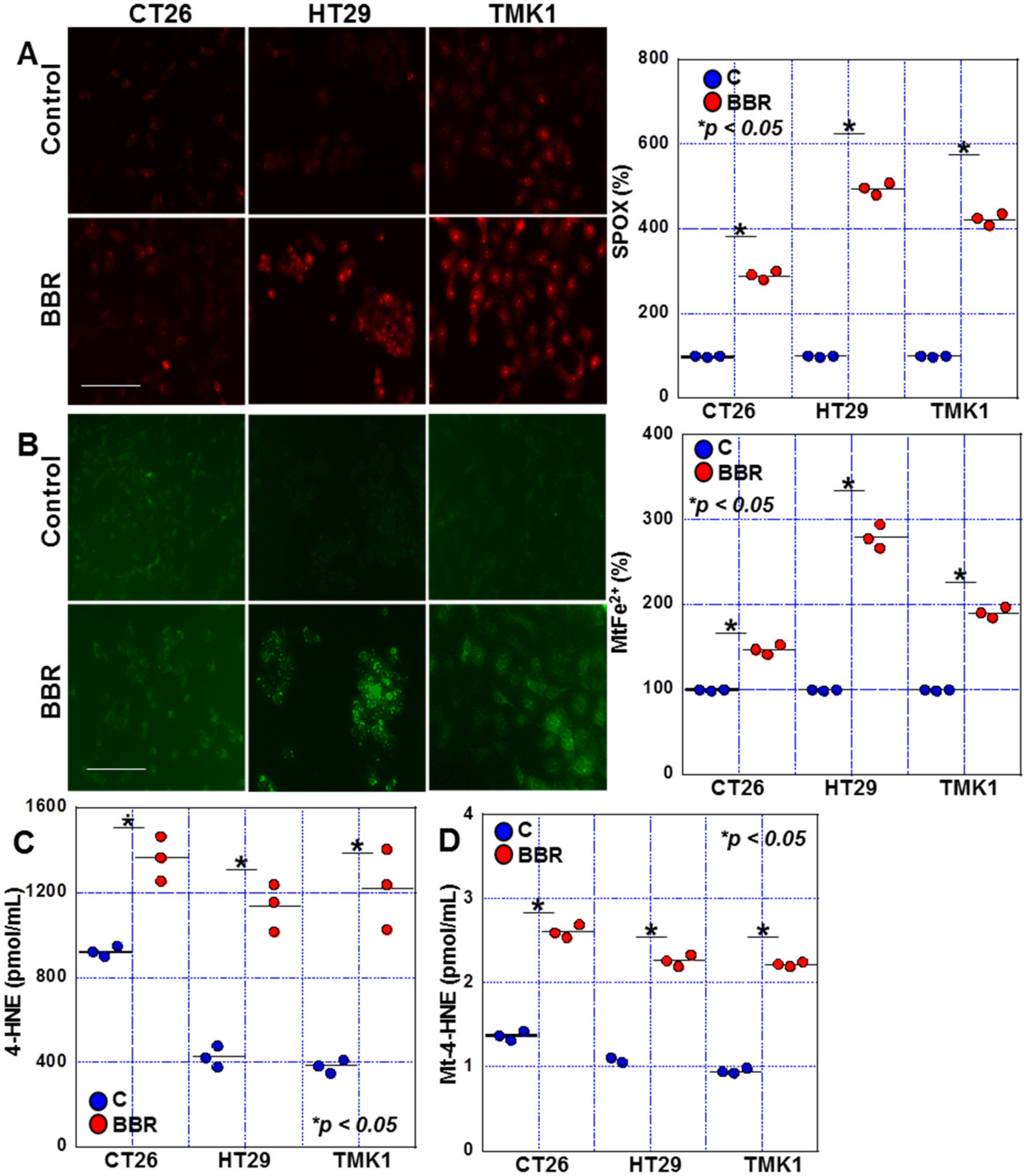
2.2. Effects of BBR on Mitochondrial ROS Production in GIC Cells
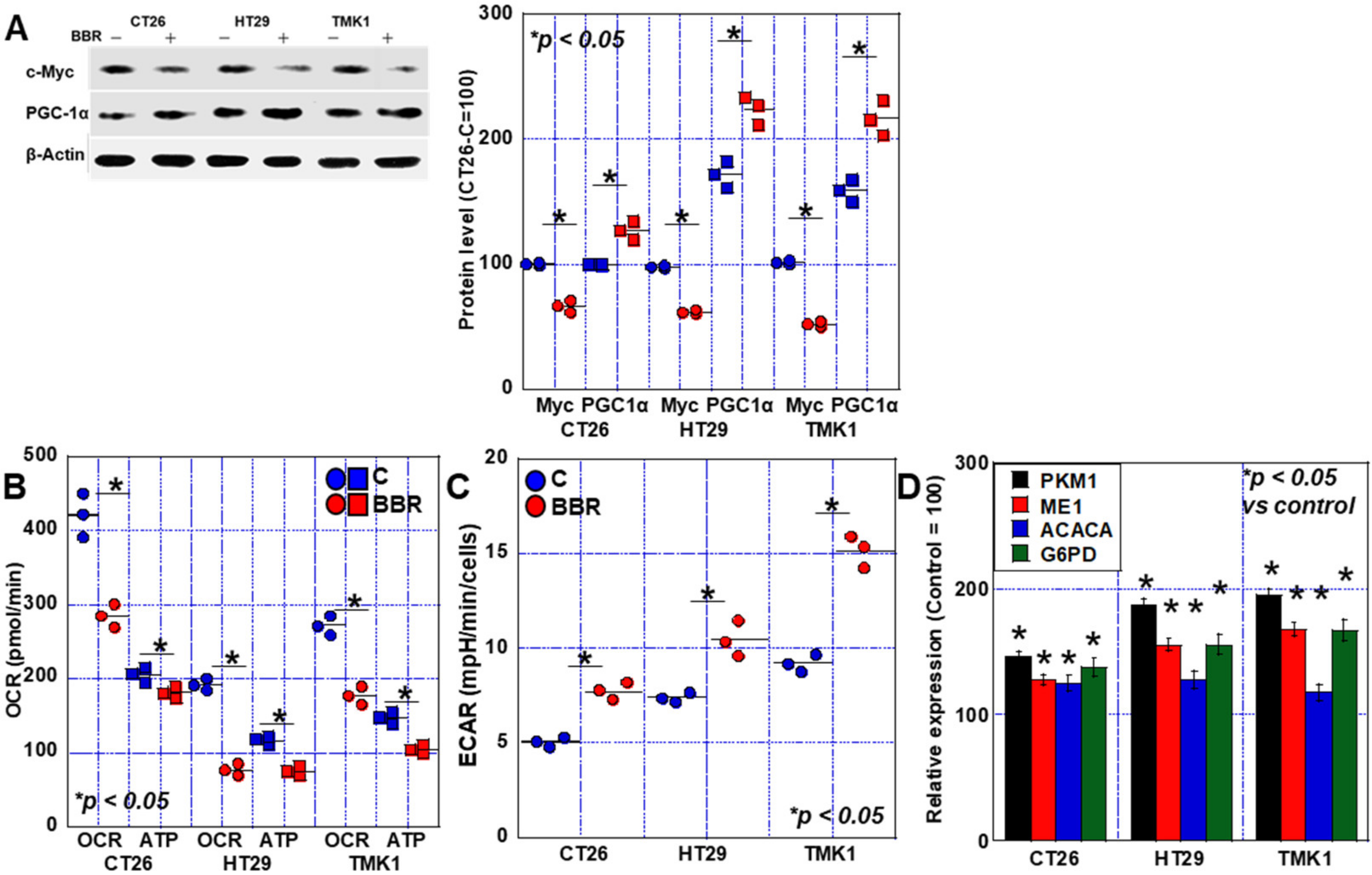
2.3. Alterations in the Energy Metabolism of GIC Cells Treated with BBR
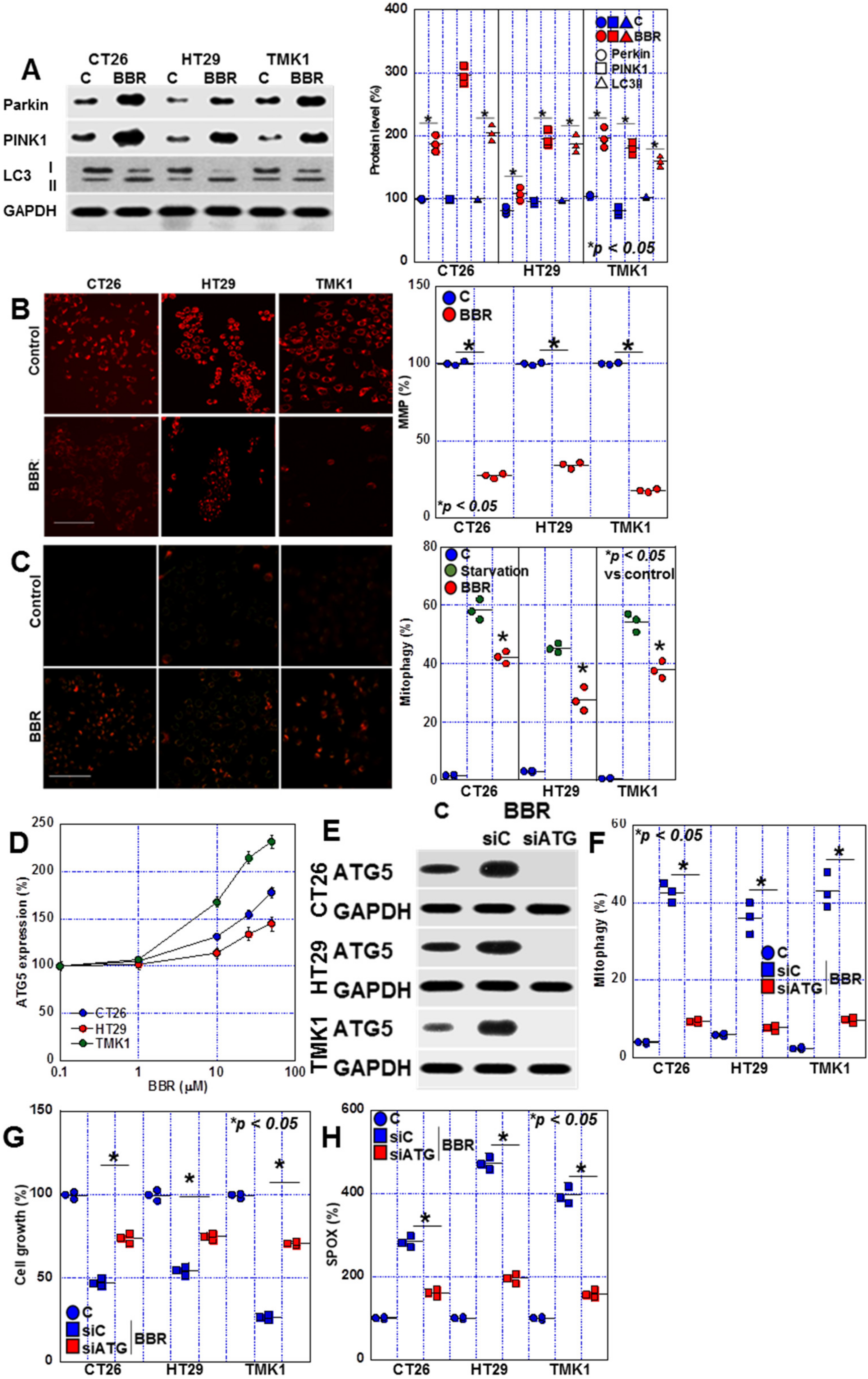
2.4. Changes in Mitophagy in BBR-Treated GIC Cells
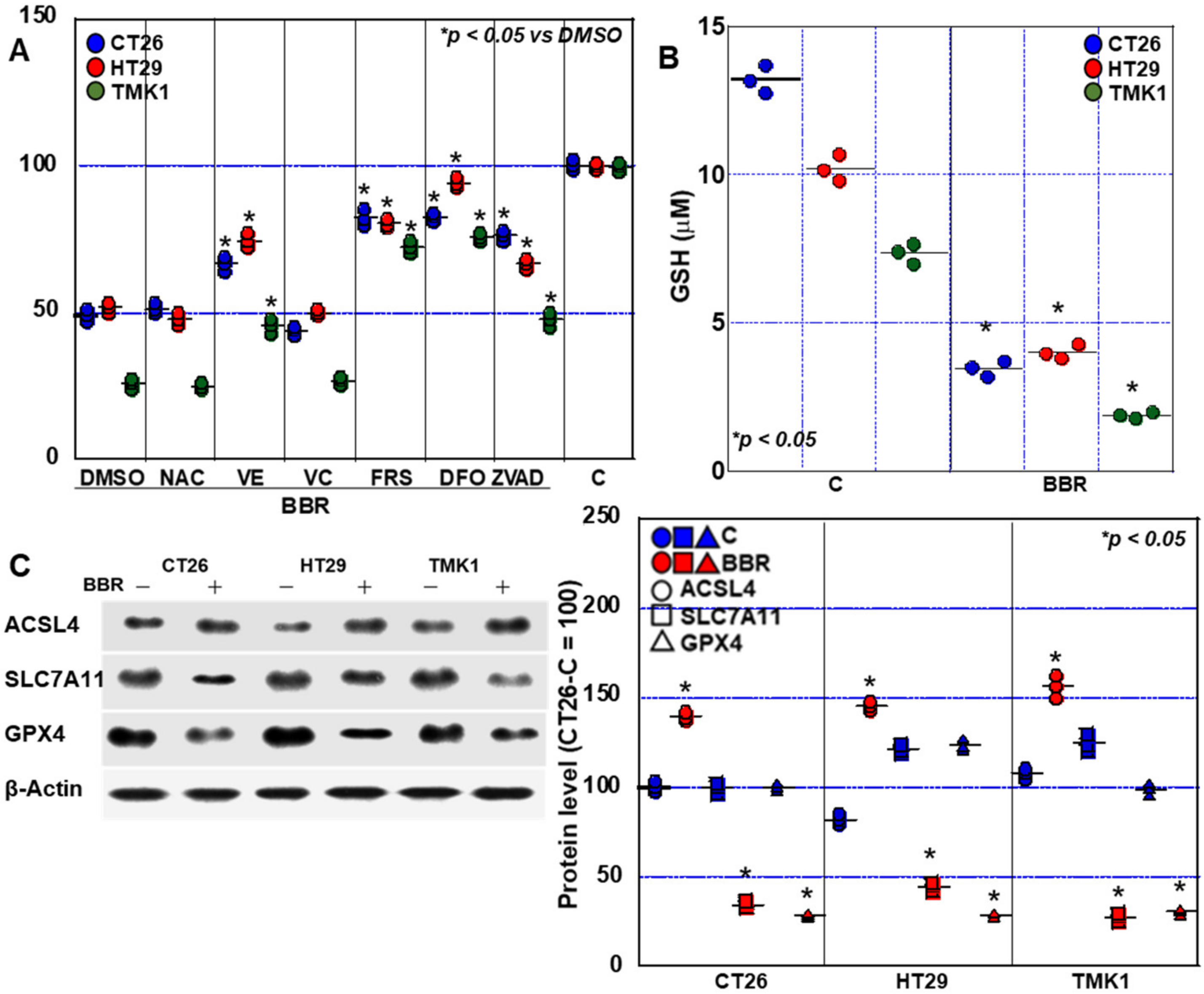
2.5. Induction of Cell Death by BBR in GIC Cells
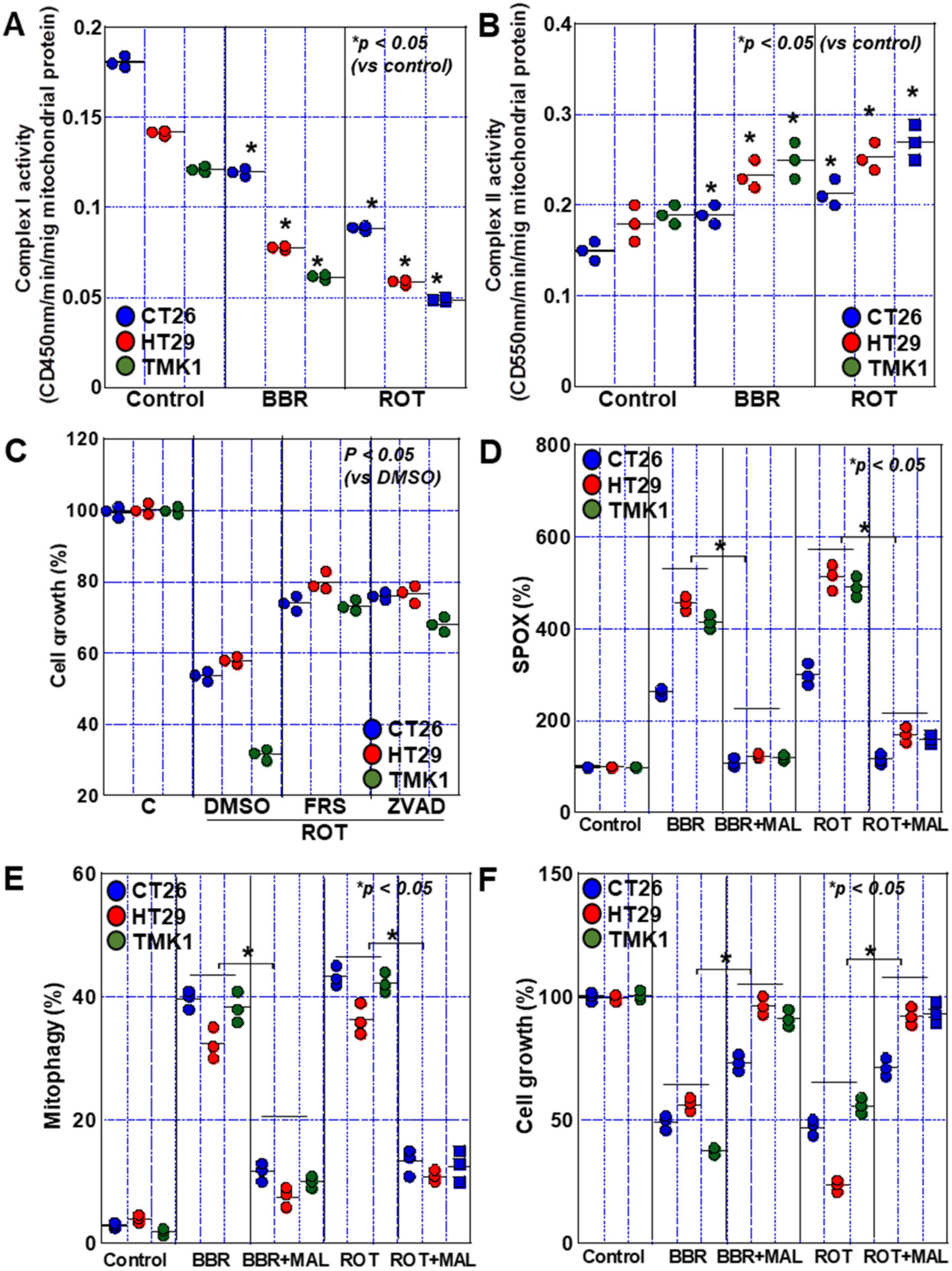
2.6. Complex I Inhibition by BBR
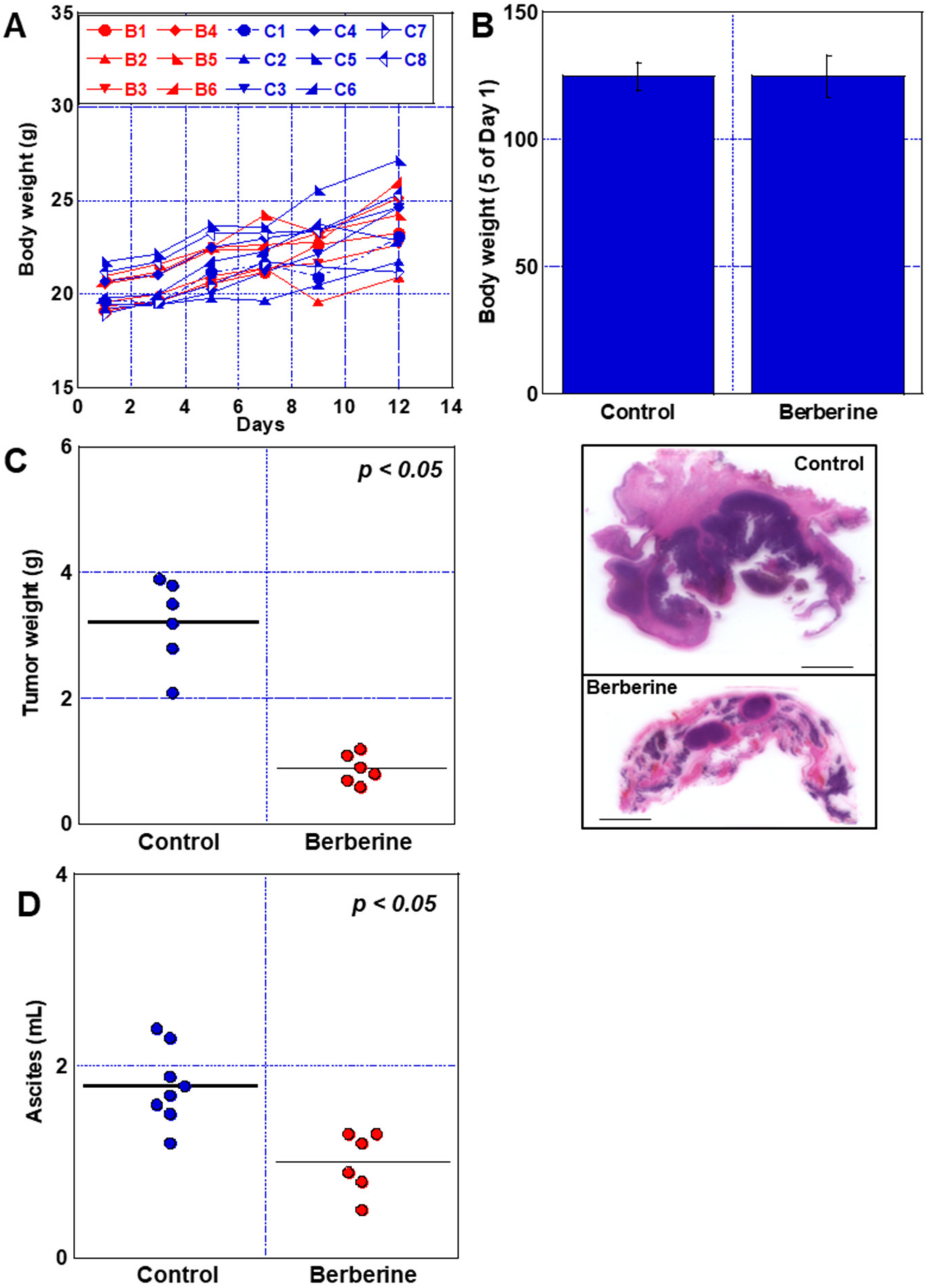
2.7. Effect of BBR in Mouse Peritoneal Dissemination Model Using CT26 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Cell Growth
4.3. Chamber Invasion Assay
4.4. Wound Healing Assay
4.5. Sphere Assay
4.6. Mitochondrial Imaging
4.7. Enzyme-Linked Immunosorbent Assay (ELISA) and Fluorometric Assay
4.8. Extracellular Flux Analysis
4.9. Glycolytic Stress Test
4.10. qRT-PCR for RNA
4.11. Animals
4.12. Western Blot Analysis
4.13. Small Interfering RNA
4.14. Activity of Mitochondrial Complex
4.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wakao, F. Cancer Statistics in Japan 2021; Foundation for Promotion of Cancer Research: Tokyo, Japan, 2021; p. 146. [Google Scholar]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Japanese Association of Clinical Cancer Centers. Survival Studies in Japanese Association of Clinical Cancer Centers. Available online: https://www.zengankyo.ncc.go.jp/etc/seizonritsu/seizonritsu2013.html (accessed on 25 October 2022).
- Shabbir, U.; Rubab, M.; Daliri, E.B.; Chelliah, R.; Javed, A.; Oh, D.H. Curcumin, Quercetin, Catechins and Metabolic Diseases: The Role of Gut Microbiota. Nutrients 2021, 13, 206. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Goel, A. Curcumin and colorectal cancer: An update and current perspective on this natural medicine. Semin. Cancer Biol. 2022, 80, 73–86. [Google Scholar] [CrossRef]
- Hojo, Y.; Kishi, S.; Mori, S.; Fujiwara-Tani, R.; Sasaki, T.; Fujii, K.; Nishiguchi, Y.; Nakashima, C.; Luo, Y.; Shinohara, H.; et al. Sunitinib and Pterostilbene Combination Treatment Exerts Antitumor Effects in Gastric Cancer via Suppression of PDZD8. Int. J. Mol. Sci. 2022, 23, 4002. [Google Scholar] [CrossRef]
- Mori, S.; Kishi, S.; Honoki, K.; Fujiwara-Tani, R.; Moriguchi, T.; Sasaki, T.; Fujii, K.; Tsukamoto, S.; Fujii, H.; Kido, A.; et al. Anti-Stem Cell Property of Pterostilbene in Gastrointestinal Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9347. [Google Scholar] [CrossRef] [PubMed]
- Imenshahidi, M.; Hosseinzadeh, H. Berberine and barberry (Berberis vulgaris): A clinical review. Phytother. Res. PTR 2019, 33, 504–523. [Google Scholar] [CrossRef]
- Gong, J.; Hu, M.; Huang, Z.; Fang, K.; Wang, D.; Chen, Q.; Li, J.; Yang, D.; Zou, X.; Xu, L.; et al. Berberine Attenuates Intestinal Mucosal Barrier Dysfunction in Type 2 Diabetic Rats. Front. Pharmacol. 2017, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Fan, X.; Yuan, B.; Takagi, N.; Liu, S.; Han, X.; Ren, J.; Liu, J. Berberine inhibits NLRP3 Inflammasome pathway in human triple-negative breast cancer MDA-MB-231 cell. BMC Complement. Altern. Med. 2019, 19, 216. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.P.; Wang, Z.F.; Huang, X.L.; Tan, M.X.; Luo, Z.H.; Wang, S.L.; Zou, B.Q.; Liang, H. Two telomerase-targeting Pt(ii) complexes of jatrorrhizine and berberine derivatives induce apoptosis in human bladder tumor cells. Dalton Trans. 2019, 48, 15247–15254. [Google Scholar] [CrossRef]
- Luo, Y.; Tian, G.; Zhuang, Z.; Chen, J.; You, N.; Zhuo, L.; Liang, B.; Song, Y.; Zang, S.; Liu, J.; et al. Berberine prevents non-alcoholic steatohepatitis-derived hepatocellular carcinoma by inhibiting inflammation and angiogenesis in mice. Am. J. Transl. Res. 2019, 11, 2668–2682. [Google Scholar]
- Liu, Y.; Hua, W.; Li, Y.; Xian, X.; Zhao, Z.; Liu, C.; Zou, J.; Li, J.; Fang, X.; Zhu, Y. Berberine suppresses colon cancer cell proliferation by inhibiting the SCAP/SREBP-1 signaling pathway-mediated lipogenesis. Biochem. Pharmacol. 2020, 174, 113776. [Google Scholar] [CrossRef]
- Samadi, P.; Sarvarian, P.; Gholipour, E.; Asenjan, K.S.; Aghebati-Maleki, L.; Motavalli, R.; Hojjat-Farsangi, M.; Yousefi, M. Berberine: A novel therapeutic strategy for cancer. IUBMB Life 2020, 72, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Du, X.; Ma, H.; Yao, J. The Anti-Cancer Mechanisms of Berberine: A Review. Cancer Manag. Res. 2020, 12, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Abu-Izneid, T.; Khalil, A.A.; Imran, M.; Shah, Z.A.; Emran, T.B.; Mitra, S.; Khan, Z.; Alhumaydhi, F.A.; Aljohani, A.S.M.; et al. Berberine as a Potential Anticancer Agent: A Comprehensive Review. Molecules 2021, 26, 7368. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Ahmadi, Z.; Tavakol, S.; Ashrafizadeh, M. Berberine as a potential autophagy modulator. J. Cell. Physiol. 2019, 234, 14914–14926. [Google Scholar] [CrossRef]
- Hu, S.; Zhao, R.; Liu, Y.; Chen, J.; Zheng, Z.; Wang, S. Preventive and Therapeutic Roles of Berberine in Gastrointestinal Cancers. Biomed. Res. Int. 2019, 2019, 6831520. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.N.; Jin, C.L.; Jang, J.H.; Zhao, Z.H.; Kim, S.J.; Zhang, Y.H. Correction to: Reduced nNOS activity is responsible for impaired fatty acid-dependent mitochondrial oxygen consumption in atrial myocardium from hypertensive rat. Pflug. Arch. 2020, 472, 1655. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Hernández-Esquivel, L.; Rivero-Segura, N.A.; Marín-Hernández, A.; Neuzil, J.; Ralph, S.J.; Rodríguez-Enríquez, S. Reactive oxygen species are generated by the respiratory complex II—Evidence for lack of contribution of the reverse electron flow in complex I. FEBS J. 2013, 280, 927–938. [Google Scholar] [CrossRef]
- Kilbride, S.M.; Telford, J.E.; Davey, G.P. Complex I Controls Mitochondrial and Plasma Membrane Potentials in Nerve Terminals. Neurochem. Res. 2021, 46, 100–107. [Google Scholar] [CrossRef]
- Heo, G.; Sun, M.H.; Jiang, W.J.; Li, X.H.; Lee, S.H.; Guo, J.; Zhou, D.; Cui, X.S. Rotenone causes mitochondrial dysfunction and prevents maturation in porcine oocytes. PLoS ONE 2022, 17, e0277477. [Google Scholar] [CrossRef]
- Chen, Y.; Suzuki, I. Effect of uncouplers on endogenous respiration and ferrous iron oxidation in a chemolithoautotrophic bacterium Acidithiobacillus (Thiobacillus) ferrooxidans. FEMS Microbiol. Lett. 2004, 237, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ye, J. Mitochondrial inhibitor as a new class of insulin sensitizer. Acta Pharm. Sin. B 2012, 2, 341–349. [Google Scholar] [CrossRef]
- Flatmark, T.; Romslo, I. Energy-dependent accumulation of iron by isolated rat liver mitochondria. Requirement of reducing equivalents and evidence for a unidirectional flux of Fe(II) across the inner membrane. J. Biol. Chem. 1975, 250, 6433–6438. [Google Scholar] [CrossRef]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Radogna, F.; Dicato, M.; Diederich, M. Cancer-type-specific crosstalk between autophagy, necroptosis and apoptosis as a pharmacological target. Biochem. Pharmacol. 2015, 94, 1–11. [Google Scholar] [CrossRef]
- Lin, L.; Baehrecke, E.H. Autophagy, cell death, and cancer. Mol. Cell. Oncol. 2015, 2, e985913. [Google Scholar] [CrossRef] [PubMed]
- Tsapras, P.; Nezis, I.P. Caspase involvement in autophagy. Cell Death Differ. 2017, 24, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, D.; Grinevich, V.; Arima, H. A novel mechanism of autophagy-associated cell death of vasopressin neurons in familial neurohypophysial diabetes insipidus. Cell Tissue Res. 2019, 375, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Basit, F.; van Oppen, L.M.; Schöckel, L.; Bossenbroek, H.M.; van Emst-de Vries, S.E.; Hermeling, J.C.; Grefte, S.; Kopitz, C.; Heroult, M.; Hgm Willems, P.; et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Ma, Y.; Wang, Q.; Zhang, J.; Zhu, C.; Zhang, L.; Xu, X. Atg3 Overexpression Enhances Bortezomib-Induced Cell Death in SKM-1 Cell. PLoS ONE 2016, 11, e0158761. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef]
- Qian, W.; Liu, J.; Jin, J.; Ni, W.; Xu, W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk. Res. 2007, 31, 329–339. [Google Scholar] [CrossRef]
- Basciani, S.; Vona, R.; Matarrese, P.; Ascione, B.; Mariani, S.; Cauda, R.; Gnessi, L.; Malorni, W.; Straface, E.; Lucia, M.B. Imatinib interferes with survival of multi drug resistant Kaposi’s sarcoma cells. FEBS Lett. 2007, 581, 5897–5903. [Google Scholar] [CrossRef]
- Yokoyama, T.; Miyazawa, K.; Naito, M.; Toyotake, J.; Tauchi, T.; Itoh, M.; Yuo, A.; Hayashi, Y.; Georgescu, M.M.; Kondo, Y.; et al. Vitamin K2 induces autophagy and apoptosis simultaneously in leukemia cells. Autophagy 2008, 4, 629–640. [Google Scholar] [CrossRef]
- Qu, X.; Zou, Z.; Sun, Q.; Luby-Phelps, K.; Cheng, P.; Hogan, R.N.; Gilpin, C.; Levine, B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007, 128, 931–946. [Google Scholar] [CrossRef]
- Bovellan, M.; Fritzsche, M.; Stevens, C.; Charras, G. Death-associated protein kinase (DAPK) and signal transduction: Blebbing in programmed cell death. FEBS J. 2010, 277, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, X.; Cao, S.; Sun, Y.; He, X.; Jiang, B.; Yu, Y.; Duan, J.; Qiu, F.; Kang, N. Berberine represses human gastric cancer cell growth in vitro and in vivo by inducing cytostatic autophagy via inhibition of MAPK/mTOR/p70S6K and Akt signaling pathways. Biomed. Pharmacother. 2020, 128, 110245. [Google Scholar] [CrossRef]
- Fei, Q.; Ma, H.; Zou, J.; Wang, W.; Zhu, L.; Deng, H.; Meng, M.; Tan, S.; Zhang, H.; Xiao, X.; et al. Metformin protects against ischaemic myocardial injury by alleviating autophagy-ROS-NLRP3-mediated inflammatory response in macrophages. J. Mol. Cell. Cardiol. 2020, 145, 1–13. [Google Scholar] [CrossRef]
- Gao, L.; Loveless, J.; Shay, C.; Teng, Y. Targeting ROS-Mediated Crosstalk Between Autophagy and Apoptosis in Cancer. Adv. Exp. Med. Biol. 2020, 1260, 1–12. [Google Scholar]
- Li, J.; Liu, J.; Xu, Y.; Wu, R.; Chen, X.; Song, X.; Zeh, H.; Kang, R.; Klionsky, D.J.; Wang, X.; et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy 2021, 17, 3361–3374. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, J.; Hou, W.; Kang, R.; Tang, D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front. Cell Dev. Biol. 2021, 9, 698679. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fan, J.; Ai, G.; Liu, J.; Luo, N.; Li, C.; Cheng, Z. Berberine in combination with cisplatin induces necroptosis and apoptosis in ovarian cancer cells. Biol. Res. 2019, 52, 37. [Google Scholar] [CrossRef]
- Mai, W.; Xu, Y.; Xu, J.; Zhao, D.; Ye, L.; Yu, G.; Wang, Z.; Lu, Q.; Lin, J.; Yang, T.; et al. Berberine Inhibits Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome Activation and Pyroptosis in Nonalcoholic Steatohepatitis via the ROS/TXNIP Axis. Front. Pharmacol. 2020, 11, 185. [Google Scholar] [CrossRef]
- Jin, Y.; Qiu, J.; Lu, X.; Li, G. C-MYC Inhibited Ferroptosis and Promoted Immune Evasion in Ovarian Cancer Cells through NCOA4 Mediated Ferritin Autophagy. Cells 2022, 11, 4127. [Google Scholar] [CrossRef] [PubMed]
- Ferber, E.C.; Peck, B.; Delpuech, O.; Bell, G.P.; East, P.; Schulze, A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968–979. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Wiese, E.K.; Hitosugi, S.; Loa, S.T.; Sreedhar, A.; Andres-Beck, L.G.; Kurmi, K.; Pang, Y.P.; Karnitz, L.M.; Gonsalves, W.I.; Hitosugi, T. Enzymatic activation of pyruvate kinase increases cytosolic oxaloacetate to inhibit the Warburg effect. Nat. Metab. 2021, 3, 954–968. [Google Scholar] [CrossRef]
- Qaisiya, M.; Coda Zabetta, C.D.; Bellarosa, C.; Tiribelli, C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell. Signal. 2014, 26, 512–520. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Crespo, F.L.; Sobrado, V.R.; Gomez, L.; Cervera, A.M.; McCreath, K.J. Mitochondrial reactive oxygen species mediate cardiomyocyte formation from embryonic stem cells in high glucose. Stem Cells 2010, 28, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Cancer stem cell in breast cancer therapeutic resistance. Cancer Treat. Rev. 2018, 69, 152–163. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Oue, N.; Wakikawa, A.; Shigeishi, H.; Matsutani, N.; Kuraoka, K.; Ito, R.; Yokozaki, H.; Yasui, W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J. Pathol. 2002, 196, 163–170. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene ID | Species | Forward | Reverse |
|---|---|---|---|---|
| PKM1 | NM_001411081.1 | Human | GGAGAAACAGCCAAAGGGGA (EX9) | ACCCGGAGGTCCACGTCCTC (EX11) [50] |
| pkm1 | NM_001253883.2 | Mouse | TGTTCCACCGTCTGCTGTTT (EX9) | ACACGAAGGTCGACATCCTC (EX11) |
| ME1 | NM_002395.6 | Human | CCCTAGGGATTGCACACCTG | AGGAGGATAAAGCCGACCCT |
| me1 | NM_001198933.1 | Mouse | GGAGTTGCTGCAATTGGTGG | TGCAGGCCACGGATAACAAT |
| ACACA | NM_198836.3 | Human | GGAGGAGGAGGGAAGGGAAT | CGAGCAGCAATAACATGGCC |
| acaca | NM_133360.3 | Mouse | TTGCCATGGGGATCCCTCTA | GCTGTTCCTCAGGCTCACAT |
| G6PD | NM_001042351.3 | Human | CTACCGCATCGACCACTACC | ACTGCTGGTGGAAGATGTCG |
| g6pd | X53617.1 | Mouse | ATGGCAGAGCAGGTGGCC | AATATGTGTGTATCAGCTTGGTGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, S.; Fujiwara-Tani, R.; Gyoten, M.; Nukaga, S.; Sasaki, R.; Ikemoto, A.; Ogata, R.; Kishi, S.; Fujii, K.; Kuniyasu, H. Berberine Induces Combined Cell Death in Gastrointestinal Cell Lines. Int. J. Mol. Sci. 2023, 24, 6588. https://doi.org/10.3390/ijms24076588
Mori S, Fujiwara-Tani R, Gyoten M, Nukaga S, Sasaki R, Ikemoto A, Ogata R, Kishi S, Fujii K, Kuniyasu H. Berberine Induces Combined Cell Death in Gastrointestinal Cell Lines. International Journal of Molecular Sciences. 2023; 24(7):6588. https://doi.org/10.3390/ijms24076588
Chicago/Turabian StyleMori, Shiori, Rina Fujiwara-Tani, Momoko Gyoten, Shota Nukaga, Rika Sasaki, Ayaka Ikemoto, Ruiko Ogata, Shingo Kishi, Kiyomu Fujii, and Hiroki Kuniyasu. 2023. "Berberine Induces Combined Cell Death in Gastrointestinal Cell Lines" International Journal of Molecular Sciences 24, no. 7: 6588. https://doi.org/10.3390/ijms24076588
APA StyleMori, S., Fujiwara-Tani, R., Gyoten, M., Nukaga, S., Sasaki, R., Ikemoto, A., Ogata, R., Kishi, S., Fujii, K., & Kuniyasu, H. (2023). Berberine Induces Combined Cell Death in Gastrointestinal Cell Lines. International Journal of Molecular Sciences, 24(7), 6588. https://doi.org/10.3390/ijms24076588