
Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Kidney Cancers

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
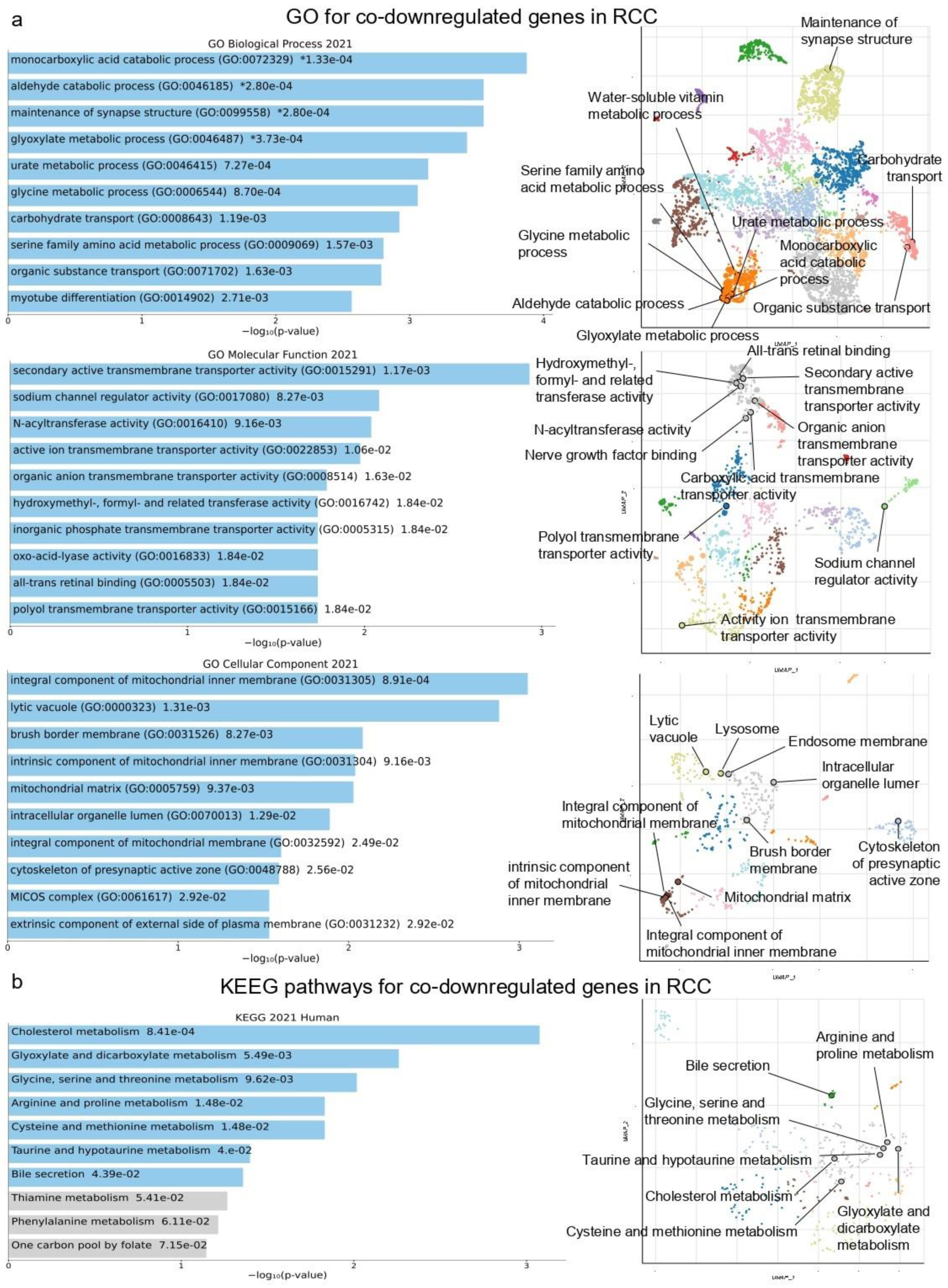
2.1. Co-Deregulated Genes in Kidney Cancers vs. the Normal Tissue
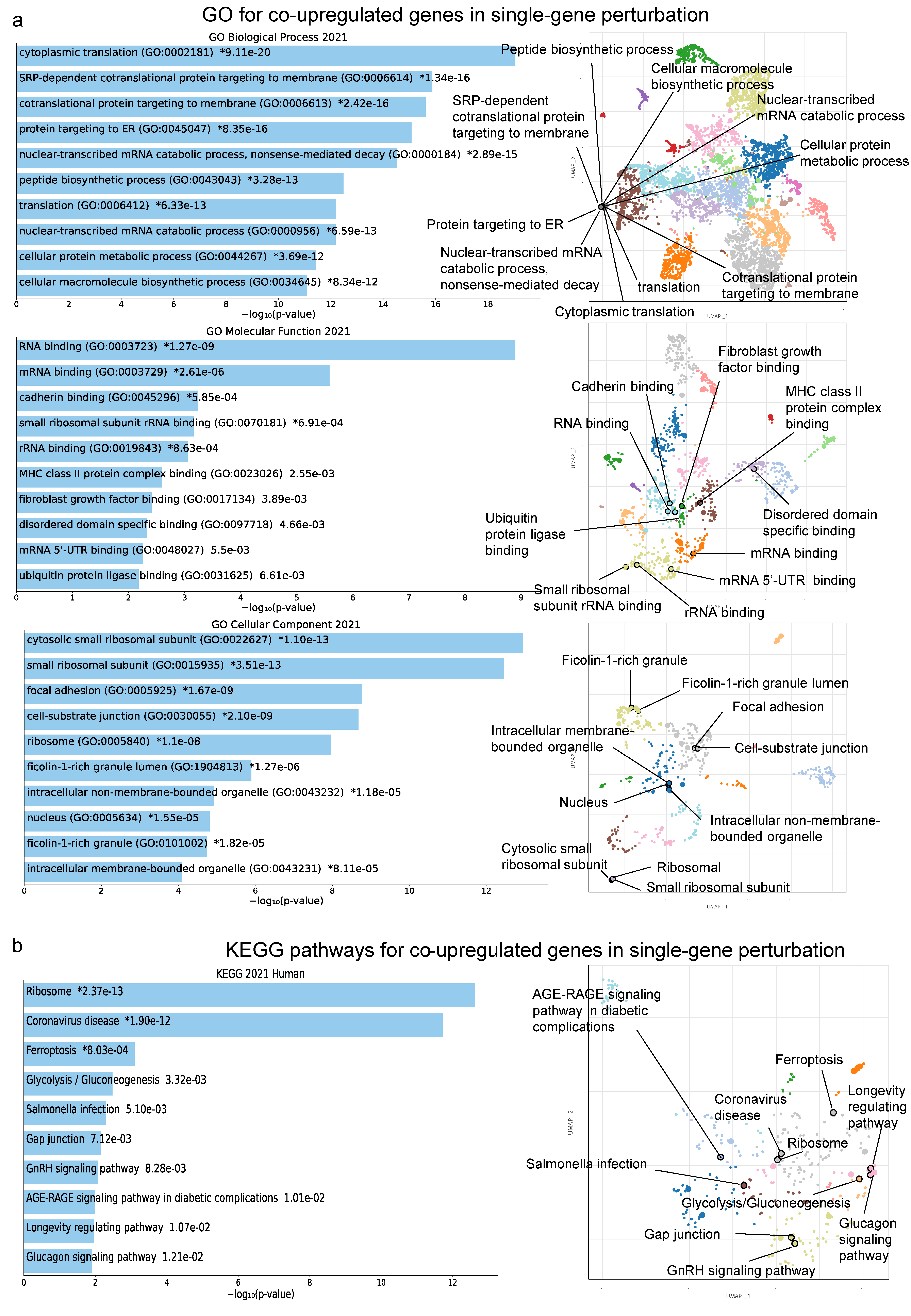
2.2. Co-Deregulated Genes within Single-Gene Perturbation Studies in Kidney Cancers
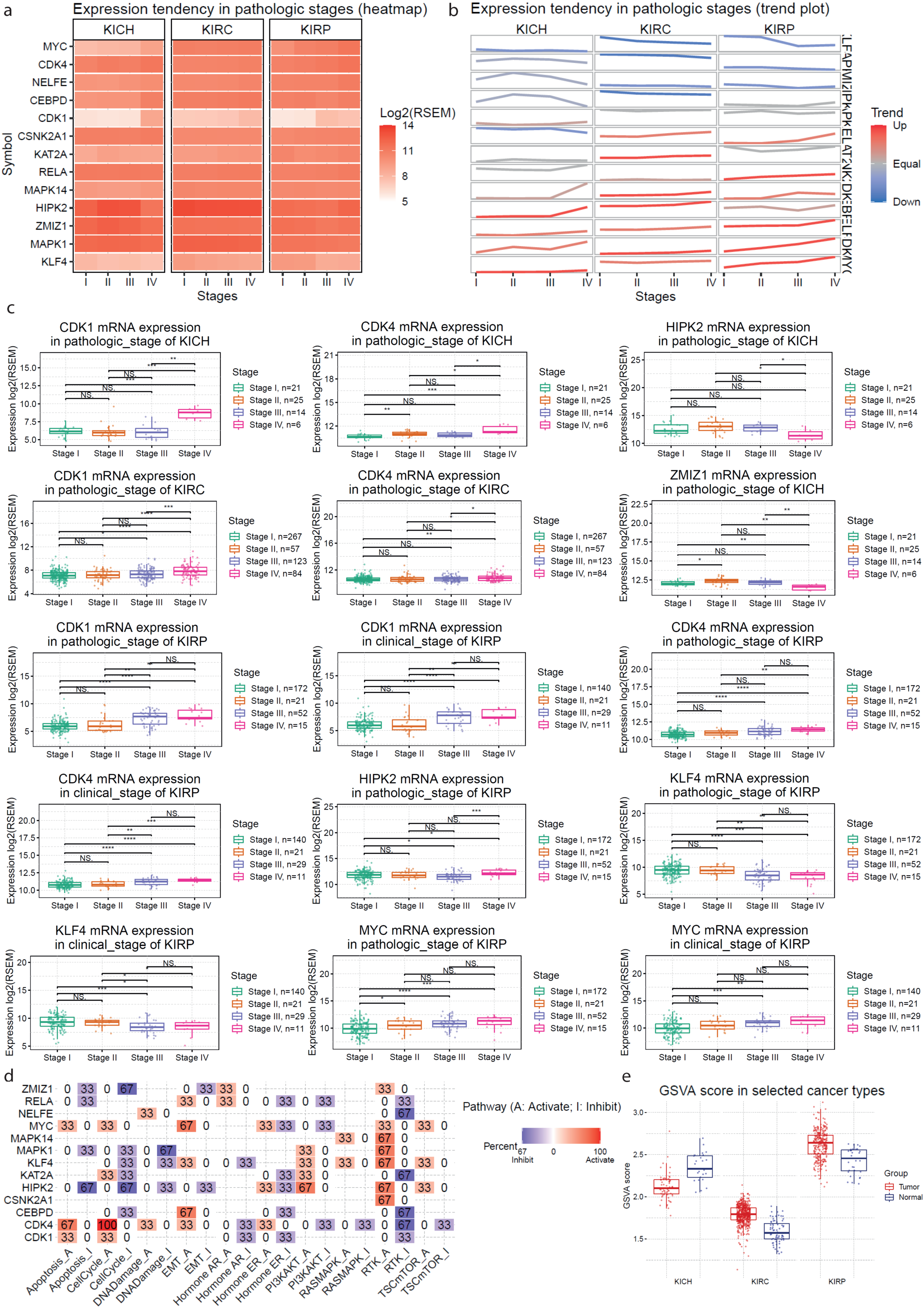
2.3. Validation of the Co-Deregulated Gene Signatures in the TCGA Database
2.4. Gene Set Cancer Analysis
2.5. Repurposed Drugs in Kidney Cancers
2.6. Repurposed Drugs in Single-Gene Perturbation Experiments in Kidney Cancers
2.7. In Vitro Validation of Repurposed Drugs
3. Discussion
4. Materials and Methods
4.1. Extraction and Filtering of Gene Expression Signatures from the GEO Database
4.2. Differential Gene Expression and the Co-Deregulated Genes across Different Studies
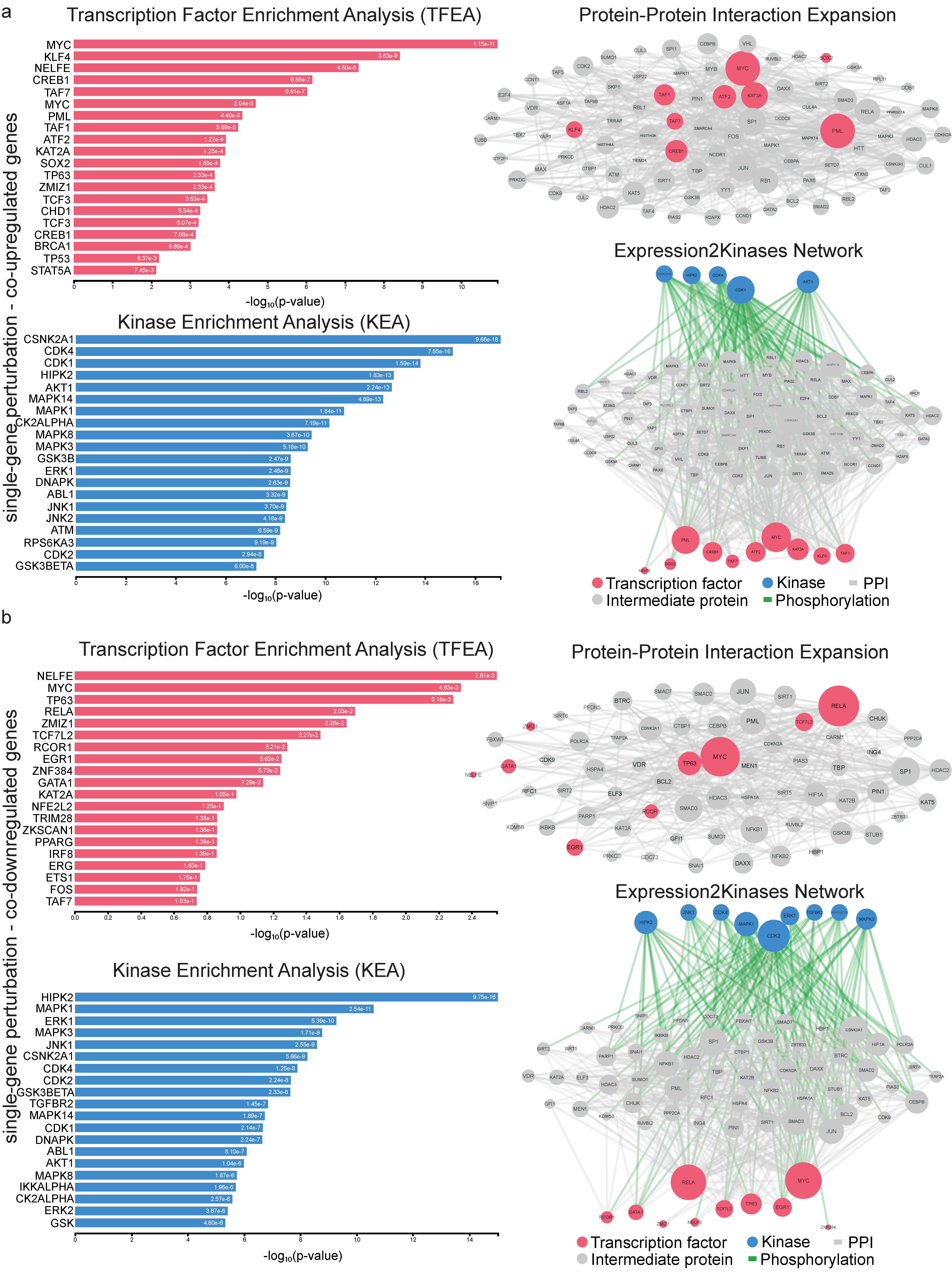
4.3. Upstream Regulators of the Co-Deregulated Genes and Protein–Protein (PPI) Interactions
4.4. Gene Ontology (GO) Enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis
4.5. Validation of the Co-Deregulated Gene Signatures in the TCGA and across Different Molecular and Immune Subtypes in Kidney Cancer
4.6. Gene Set Analysis in Kidney Cancers
4.7. Detection of Repurposing Drugs in Kidney Cancer
4.8. Cell Culture
4.9. MTT Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Safiri, S.; Kolahi, A.-A.; Mansournia, M.A.; Almasi-Hashiani, A.; Ashrafi-Asgarabad, A.; Sullman, M.J.M.; Bettampadi, D.; Qorbani, M.; Moradi-Lakeh, M.; Ardalan, M.; et al. The Burden of Kidney Cancer and Its Attributable Risk Factors in 195 Countries and Territories, 1990–2017. Sci. Rep. 2020, 10, 13862. [Google Scholar] [CrossRef]
- Moch, H. An Overview of Renal Cell Cancer: Pathology and Genetics. Semin. Cancer Biol. 2013, 23, 3–9. [Google Scholar] [CrossRef]
- Luu, V.-D.; Boysen, G.; Struckmann, K.; Casagrande, S.; von Teichman, A.; Wild, P.J.; Sulser, T.; Schraml, P.; Moch, H. Loss of VHL and Hypoxia Provokes PAX2 Up-Regulation in Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2009, 15, 3297–3304. [Google Scholar] [CrossRef]
- Gatto, F.; Nookaew, I.; Nielsen, J. Chromosome 3p Loss of Heterozygosity Is Associated with a Unique Metabolic Network in Clear Cell Renal Carcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, E866–E875. [Google Scholar] [CrossRef]
- Scelo, G.; Hofmann, J.N.; Banks, R.E.; Bigot, P.; Bhatt, R.S.; Cancel-Tassin, G.; Chew, S.K.; Creighton, C.J.; Cussenot, O.; Davis, I.J.; et al. International Cancer Seminars: A Focus on Kidney Cancer. Ann. Oncol. 2016, 27, 1382–1385. [Google Scholar] [CrossRef]
- Ho, T.H.; Kapur, P.; Joseph, R.W.; Serie, D.J.; Eckel-Passow, J.E.; Parasramka, M.; Cheville, J.C.; Wu, K.J.; Frenkel, E.; Rakheja, D.; et al. Loss of PBRM1 and BAP1 Expression Is Less Common in Non-Clear Cell Renal Cell Carcinoma than in Clear Cell Renal Cell Carcinoma. Urol. Oncol. 2015, 33, 23.e9–23.e14. [Google Scholar] [CrossRef]
- Joseph, R.W.; Kapur, P.; Serie, D.J.; Eckel-Passow, J.E.; Parasramka, M.; Ho, T.; Cheville, J.C.; Frenkel, E.; Rakheja, D.; Brugarolas, J.; et al. Loss of BAP1 Protein Expression Is an Independent Marker of Poor Prognosis in Patients with Low-Risk Clear Cell Renal Cell Carcinoma. Cancer 2014, 120, 1059–1067. [Google Scholar] [CrossRef]
- Clark, N.R.; Hu, K.S.; Feldmann, A.S.; Kou, Y.; Chen, E.Y.; Duan, Q.; Ma’ayan, A. The Characteristic Direction: A Geometrical Approach to Identify Differentially Expressed Genes. BMC Bioinform. 2014, 15, 79. [Google Scholar] [CrossRef]
- Terrell, E.M.; Morrison, D.K. Ras-Mediated Activation of the Raf Family Kinases. Cold. Spring Harb. Perspect. Med. 2019, 9, a033746. [Google Scholar] [CrossRef]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC Oncogene Overexpression Drives Renal Cell Carcinoma in a Mouse Model through Glutamine Metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC Transcription Factor Network: Balancing Metabolism, Proliferation and Oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef]
- Miller, A.J.; Chang, A.; Cunningham, P.N. Chronic Microangiopathy Due to DCR-MYC, a Myc-Targeted Short Interfering RNA. Am. J. Kidney Dis. 2020, 75, 513–516. [Google Scholar] [CrossRef]
- Ko, C.-Y.; Chang, W.-C.; Wang, J.-M. Biological Roles of CCAAT/Enhancer-Binding Protein Delta during Inflammation. J. Biomed. Sci. 2015, 22, 6. [Google Scholar] [CrossRef]
- Brueckner, L.M.; Hess, E.M.; Schwab, M.; Savelyeva, L. Instability at the FRA8I Common Fragile Site Disrupts the Genomic Integrity of the KIAA0146, CEBPD and PRKDC Genes in Colorectal Cancer. Cancer Lett. 2013, 336, 85–95. [Google Scholar] [CrossRef]
- Tang, D.; DeWille, J. Detection of Base Sequence Changes in the CEBPD Gene in Human Breast Cancer Cell Lines and Primary Breast Cancer Isolates. Mol. Cell Probes 2003, 17, 11–14. [Google Scholar] [CrossRef]
- Pan, Y.-C.; Li, C.-F.; Ko, C.-Y.; Pan, M.-H.; Chen, P.-J.; Tseng, J.T.; Wu, W.-C.; Chang, W.-C.; Huang, A.-M.; Sterneck, E.; et al. CEBPD Reverses RB/E2F1-Mediated Gene Repression and Participates in HMDB-Induced Apoptosis of Cancer Cells. Clin. Cancer Res. 2010, 16, 5770–5780. [Google Scholar] [CrossRef]
- Balamurugan, K.; Sterneck, E. The Many Faces of C/EBPδ and Their Relevance for Inflammation and Cancer. Int. J. Biol. Sci. 2013, 9, 917–933. [Google Scholar] [CrossRef]
- Chuang, C.-H.; Wang, W.-J.; Li, C.-F.; Ko, C.-Y.; Chou, Y.-H.; Chuu, C.-P.; Cheng, T.-L.; Wang, J.-M. The Combination of the Prodrugs Perforin-CEBPD and Perforin-Granzyme B Efficiently Enhances the Activation of Caspase Signaling and Kills Prostate Cancer. Cell Death Dis. 2014, 5, e1220. [Google Scholar] [CrossRef]
- Yu, L.-L.; Yu, H.-G.; Yu, J.-P.; Luo, H.-S.; Xu, X.-M.; Li, J.-H. Nuclear Factor-KappaB P65 (RelA) Transcription Factor Is Constitutively Activated in Human Colorectal Carcinoma Tissue. World J. Gastroenterol. 2004, 10, 3255–3260. [Google Scholar] [CrossRef]
- Yu, H.-G.; Zhong, X.; Yang, Y.-N.; Luo, H.-S.; Yu, J.-P.; Meier, J.J.; Schrader, H.; Bastian, A.; Schmidt, W.E.; Schmitz, F. Increased Expression of Nuclear Factor-KappaB/RelA Is Correlated with Tumor Angiogenesis in Human Colorectal Cancer. Int. J. Color. Dis. 2004, 19, 18–22. [Google Scholar] [CrossRef]
- Weichert, W.; Boehm, M.; Gekeler, V.; Bahra, M.; Langrehr, J.; Neuhaus, P.; Denkert, C.; Imre, G.; Weller, C.; Hofmann, H.-P.; et al. High Expression of RelA/P65 Is Associated with Activation of Nuclear Factor-KappaB-Dependent Signaling in Pancreatic Cancer and Marks a Patient Population with Poor Prognosis. Br. J. Cancer 2007, 97, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.; Denkert, C.; Budczies, J.; Buckendahl, A.-C.; Darb-Esfahani, S.; Noske, A.; Müller, B.M.; Bahra, M.; Neuhaus, P.; Dietel, M.; et al. High Class I HDAC Activity and Expression Are Associated with RelA/P65 Activation in Pancreatic Cancer in Vitro and in Vivo. BMC Cancer 2009, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, N.; Zheng, H.; Lee, L.-H.; Ser, H.-L.; Jiang, L.; Chen, J.; Wang, J. ZMIZ1 Affects Invasion and Metastasis of Wilms Tumor by Regulating Notch Signaling Pathway. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Li, X.; Zhu, C.; Tu, W.H.; Yang, N.; Qin, H.; Sun, Z. ZMIZ1 Preferably Enhances the Transcriptional Activity of Androgen Receptor with Short Polyglutamine Tract. PLoS ONE 2011, 6, e25040. [Google Scholar] [CrossRef]
- Mathios, D.; Hwang, T.; Xia, Y.; Phallen, J.; Rui, Y.; See, A.P.; Maxwell, R.; Belcaid, Z.; Casaos, J.; Burger, P.C.; et al. Genome-Wide Investigation of Intragenic DNA Methylation Identifies ZMIZ1 Gene as a Prognostic Marker in Glioblastoma and Multiple Cancer Types. Int. J. Cancer 2019, 145, 3425–3435. [Google Scholar] [CrossRef]
- Zhao, A.; Wang, Y.; Lin, F.; Bai, K.; Gu, C. Long Noncoding RNA LBX2-AS1 Promotes Colorectal Cancer Progression via Binding with PTBP1 and Stabilizing KAT2A Expression. J. Biochem. Mol. Toxicol. 2022, 36, e23020. [Google Scholar] [CrossRef]
- Tang, Y.; Li, X.; Wei, Y.; Sun, Y.; Yang, Y.; Zhang, X.; Gao, Z.; Liu, J.; Zhuang, Q. A Preliminary Study of KAT2A on CGAS-Related Immunity in Inflammation Amplification of Systemic Lupus Erythematosus. Cell Death Dis. 2021, 12, 1036. [Google Scholar] [CrossRef]
- Tong, Y.; Guo, D.; Yan, D.; Ma, C.; Shao, F.; Wang, Y.; Luo, S.; Lin, L.; Tao, J.; Jiang, Y.; et al. KAT2A Succinyltransferase Activity-Mediated 14-3-3ζ Upregulation Promotes β-Catenin Stabilization-Dependent Glycolysis and Proliferation of Pancreatic Carcinoma Cells. Cancer Lett. 2020, 469, 1–10. [Google Scholar] [CrossRef]
- Han, L.; Zan, Y.; Huang, C.; Zhang, S. NELFE Promoted Pancreatic Cancer Metastasis and the Epithelial-to-mesenchymal Transition by Decreasing the Stabilization of NDRG2 MRNA. Int. J. Oncol. 2019, 55, 1313–1323. [Google Scholar] [CrossRef]
- Yu, S.; Li, L.; Cai, H.; He, B.; Gao, Y.; Li, Y. Overexpression of NELFE Contributes to Gastric Cancer Progression via Wnt/β-Catenin Signaling-Mediated Activation of CSNK2B Expression. J. Exp. Clin. Cancer Res. 2021, 40, 54. [Google Scholar] [CrossRef]
- Ghaleb, A.M.; Yang, V.W. Krüppel-like Factor 4 (KLF4): What We Currently Know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef]
- Sellak, H.; Wu, S.; Lincoln, T.M. KLF4 and SOX9 Transcription Factors Antagonize β-Catenin and Inhibit TCF-Activity in Cancer Cells. Biochim. Biophys. Acta 2012, 1823, 1666–1675. [Google Scholar] [CrossRef]
- Liang, S.; Nayak, B.K.; Vogel, K.S.; Habib, S.L. TP63 Is Significantly Upregulated in Diabetic Kidney. Int. J. Mol. Sci. 2021, 22, 4070. [Google Scholar] [CrossRef]
- Hu, Q.-Y.; Jin, T.-B.; Wang, L.; Zhang, L.; Geng, T.; Liang, G.; Kang, L.-L. Genetic Variation in the TP63 Gene Is Associated with Lung Cancer Risk in the Han Population. Tumour Biol. 2014, 35, 1863–1866. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, Y.-Y.; Xie, J.-J.; Mayakonda, A.; Hazawa, M.; Chen, L.; Xiao, J.-F.; Li, C.-Q.; Huang, M.-L.; Ding, L.-W.; et al. Co-Activation of Super-Enhancer-Driven CCAT1 by TP63 and SOX2 Promotes Squamous Cancer Progression. Nat. Commun. 2018, 9, 3619. [Google Scholar] [CrossRef]
- Palmbos, P.L.; Wang, Y.; Bankhead Iii, A.; Kelleher, A.J.; Wang, L.; Yang, H.; Ahmet, M.L.; Gumkowski, E.R.; Welling, S.D.; Magnuson, B.; et al. ATDC Mediates a TP63-Regulated Basal Cancer Invasive Program. Oncogene 2019, 38, 3340–3354. [Google Scholar] [CrossRef]
- Shi, Z.; Wu, D.; Xu, H.; Yang, J.; Sun, X. CSNK2A1-Mediated Phosphorylation of HMGA2 Modulates Cisplatin Resistance in Cervical Cancer. FEBS Open Bio 2021, 11, 2245–2255. [Google Scholar] [CrossRef]
- Bae, J.S.; Park, S.-H.; Jamiyandorj, U.; Kim, K.M.; Noh, S.J.; Kim, J.R.; Park, H.J.; Kwon, K.S.; Jung, S.H.; Park, H.S.; et al. CK2α/CSNK2A1 Phosphorylates SIRT6 and Is Involved in the Progression of Breast Carcinoma and Predicts Shorter Survival of Diagnosed Patients. Am. J. Pathol. 2016, 186, 3297–3315. [Google Scholar] [CrossRef]
- Wang, P.; Sun, G.-B.; Dou, G.-X.; Wang, B.-Q. Long Non-Coding RNA B3GALT5-AS1 Contributes to the Progression of Gastric Cancer via Interacting with CSNK2A1. Exp. Ther. Med. 2021, 22, 927. [Google Scholar] [CrossRef]
- Liu, J.; Yu, X.; Yu, H.; Liu, B.; Zhang, Z.; Kong, C.; Li, Z. Knockdown of MAPK14 Inhibits the Proliferation and Migration of Clear Cell Renal Cell Carcinoma by Downregulating the Expression of CDC25B. Cancer Med. 2020, 9, 1183–1195. [Google Scholar] [CrossRef]
- El-Hameed, R.H.A.; El-Shanbaky, H.M.; Mohamed, M.S. Utility of Certain 2-Furanone Derivatives for Synthesis of Different Heterocyclic Compounds and Testing Their Anti-Cancer Activity. Med. Chem. 2022, 18, 323–336. [Google Scholar] [CrossRef]
- Furukawa, T.; Kanai, N.; Shiwaku, H.O.; Soga, N.; Uehara, A.; Horii, A. AURKA Is One of the Downstream Targets of MAPK1/ERK2 in Pancreatic Cancer. Oncogene 2006, 25, 4831–4839. [Google Scholar] [CrossRef]
- Fei, B.; Wu, H. MiR-378 Inhibits Progression of Human Gastric Cancer MGC-803 Cells by Targeting MAPK1 in Vitro. Oncol. Res. 2012, 20, 557–564. [Google Scholar] [CrossRef]
- Li, X.-W.; Tuergan, M.; Abulizi, G. Expression of MAPK1 in Cervical Cancer and Effect of MAPK1 Gene Silencing on Epithelial-Mesenchymal Transition, Invasion and Metastasis. Asian Pac. J. Trop. Med. 2015, 8, 937–943. [Google Scholar] [CrossRef]
- Sun, F.; Li, N.; Tong, X.; Zeng, J.; He, S.; Gai, T.; Bai, Y.; Liu, L.; Lu, K.; Shen, J.; et al. Ara-c Induces Cell Cycle G1/S Arrest by Inducing Upregulation of the INK4 Family Gene or Directly Inhibiting the Formation of the Cell Cycle-Dependent Complex CDK4/Cyclin D1. Cell Cycle 2019, 18, 2293–2306. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Wettersten, H.I.; Weiss, R.H. Inhibition of PPARα Induces Cell Cycle Arrest and Apoptosis, and Synergizes with Glycolysis Inhibition in Kidney Cancer Cells. PLoS ONE 2013, 8, e71115. [Google Scholar] [CrossRef]
- Sager, R.A.; Backe, S.J.; Ahanin, E.; Smith, G.; Nsouli, I.; Woodford, M.R.; Bratslavsky, G.; Bourboulia, D.; Mollapour, M. Therapeutic Potential of CDK4/6 Inhibitors in Renal Cell Carcinoma. Nat. Rev. Urol. 2022, 19, 305–320. [Google Scholar] [CrossRef]
- Yang, C.-A.; Huang, H.-Y.; Yen, J.-C.; Chang, J.-G. Prognostic Value of RNASEH2A-, CDK1-, and CD151-Related Pathway Gene Profiling for Kidney Cancers. Int. J. Mol. Sci. 2018, 19, 1586. [Google Scholar] [CrossRef]
- Pan, X.-W.; Chen, L.; Hong, Y.; Xu, D.-F.; Liu, X.; Li, L.; Huang, Y.; Cui, L.-M.; Gan, S.-S.; Yang, Q.-W.; et al. EIF3D Silencing Suppresses Renal Cell Carcinoma Tumorigenesis via Inducing G2/M Arrest through Downregulation of Cyclin B1/CDK1 Signaling. Int. J. Oncol. 2016, 48, 2580–2590. [Google Scholar] [CrossRef]
- Li, Y.; Quan, J.; Chen, F.; Pan, X.; Zhuang, C.; Xiong, T.; Zhuang, C.; Li, J.; Huang, X.; Ye, J.; et al. MiR-31-5p Acts as a Tumor Suppressor in Renal Cell Carcinoma by Targeting Cyclin-Dependent Kinase 1 (CDK1). Biomed. Pharmacother. 2019, 111, 517–526. [Google Scholar] [CrossRef]
- Al-Beiti, M.A.M.; Lu, X. Expression of HIPK2 in Cervical Cancer: Correlation with Clinicopathology and Prognosis. Aust. N. Z. J. Obstet. Gynaecol. 2008, 48, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Givol, D.; D’Orazi, G. Regulation of P53 Activity by HIPK2: Molecular Mechanisms and Therapeutical Implications in Human Cancer Cells. Oncogene 2010, 29, 4378–4387. [Google Scholar] [CrossRef]
- Roskoski, R. ERK1/2 MAP Kinases: Structure, Function, and Regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Pathania, S.; Rawal, R.K. An Update on Chemical Classes Targeting ERK1/2 for the Management of Cancer. Future Med. Chem. 2020, 12, 593–611. [Google Scholar] [CrossRef]
- Judd, N.P.; Winkler, A.E.; Murillo-Sauca, O.; Brotman, J.J.; Law, J.H.; Lewis, J.S.; Dunn, G.P.; Bui, J.D.; Sunwoo, J.B.; Uppaluri, R. ERK1/2 Regulation of CD44 Modulates Oral Cancer Aggressiveness. Cancer Res. 2012, 72, 365–374. [Google Scholar] [CrossRef]
- Mendes, O.; Kim, H.-T.; Lungu, G.; Stoica, G. MMP2 Role in Breast Cancer Brain Metastasis Development and Its Regulation by TIMP2 and ERK1/2. Clin. Exp. Metastasis 2007, 24, 341–351. [Google Scholar] [CrossRef]
- Yang, J.; Bogni, A.; Schuetz, E.G.; Ratain, M.; Dolan, M.E.; McLeod, H.; Gong, L.; Thorn, C.; Relling, M.V.; Klein, T.E.; et al. Etoposide Pathway. Pharmacogenet. Genom. 2009, 19, 552–553. [Google Scholar] [CrossRef]
- Li, X.; Yun, J.-K.; Choi, J.-S. Effects of Morin on the Pharmacokinetics of Etoposide in Rats. Biopharm. Drug Dispos. 2007, 28, 151–156. [Google Scholar] [CrossRef]
- Seibel, N.L.; Chi, Y.-Y.; Perlman, E.J.; Tian, J.; Sun, J.; Anderson, J.R.; Ritchey, M.L.; Thomas, P.R.; Miser, J.; Kalapurakal, J.A.; et al. Impact of Cyclophosphamide and Etoposide on Outcome of Clear Cell Sarcoma of the Kidney Treated on the National Wilms Tumor Study-5 (NWTS-5). Pediatr. Blood Cancer 2019, 66, e27450. [Google Scholar] [CrossRef]
- Hardy, J.R.; O’Shea, A.; White, C.; Gilshenan, K.; Welch, L.; Douglas, C. The Efficacy of Haloperidol in the Management of Nausea and Vomiting in Patients with Cancer. J. Pain Symptom Manag. 2010, 40, 111–116. [Google Scholar] [CrossRef]
- Radha Krishna, L.K.; Poulose, V.J.; Goh, C. The Use of Midazolam and Haloperidol in Cancer Patients at the End of Life. Singap. Med. J. 2012, 53, 62–66. [Google Scholar]
- Friedman, G.D.; Habel, L.A.; Achacoso, N.; Sanders, C.M.; Oyer, H.M.; Fireman, B.; Van Den Eeden, S.K.; Kim, F.J. Haloperidol and Prostate Cancer Prevention: More Epidemiologic Research Needed. Perm. J. 2020, 24, 18.313. [Google Scholar] [CrossRef]
- Asong, G.M.; Amissah, F.; Voshavar, C.; Nkembo, A.T.; Ntantie, E.; Lamango, N.S.; Ablordeppey, S.Y. A Mechanistic Investigation on the Anticancer Properties of SYA013, a Homopiperazine Analogue of Haloperidol with Activity against Triple Negative Breast Cancer Cells. ACS Omega 2020, 5, 32907–32918. [Google Scholar] [CrossRef]
- Yeadon, M.; Dougan, F.L.; Petrovic, A.; Beesley, J.E.; Payne, A.N. Effect of BW B70C, a Novel Inhibitor of Arachidonic Acid 5-Lipoxygenase, on Allergen-Induced Bronchoconstriction and Late-Phase Lung Eosinophil Accumulation in Sensitised Guinea-Pigs. Agents Actions 1993, 38, 8–18. [Google Scholar] [CrossRef]
- Villegas, S.N.; Gombos, R.; García-López, L.; Gutiérrez-Pérez, I.; García-Castillo, J.; Vallejo, D.M.; Da Ros, V.G.; Ballesta-Illán, E.; Mihály, J.; Dominguez, M. PI3K/Akt Cooperates with Oncogenic Notch by Inducing Nitric Oxide-Dependent Inflammation. Cell Rep. 2018, 22, 2541–2549. [Google Scholar] [CrossRef]
- Baba, W.I.; Tudhope, G.R.; Wilson, G.M. Triamterene, a New Diuretic Drug. I. Studies in Normal Men and in Adrenalectomized Rats. Br. Med. J. 1962, 2, 756–760. [Google Scholar] [CrossRef]
- Moghadam, N.H.; Salehzadeh, S.; Tanzadehpanah, H.; Saidijam, M.; Karimi, J.; Khazalpour, S. In Vitro Cytotoxicity and DNA/HSA Interaction Study of Triamterene Using Molecular Modelling and Multi-Spectroscopic Methods. J. Biomol. Struct. Dyn. 2019, 37, 2242–2253. [Google Scholar] [CrossRef]
- Johnson, W.; Bergfeld, W.F.; Belsito, D.V.; Hill, R.A.; Klaassen, C.D.; Liebler, D.C.; Marks, J.G.; Shank, R.C.; Slaga, T.J.; Snyder, P.W.; et al. Safety Assessment of Chlorphenesin as Used in Cosmetics. Int. J. Toxicol. 2014, 33, 5S–15S. [Google Scholar] [CrossRef]
- Spencer, H.J.; Runser, R.H.; Berger, F.M.; Tarnowski, G.S.; Mathé, G. Attenuation of Certain Neoplasias by Chlorphenesin. Proc. Soc. Exp. Biol. Med. 1972, 140, 1156–1161. [Google Scholar] [CrossRef]
- Pan, Y.; Zhao, S.; Chen, F. The Potential Value of Dequalinium Chloride in the Treatment of Cancer: Focus on Malignant Glioma. Clin. Exp. Pharmacol. Physiol 2021, 48, 445–454. [Google Scholar] [CrossRef]
- Gamboa-Vujicic, G.; Emma, D.A.; Liao, S.Y.; Fuchtner, C.; Manetta, A. Toxicity of the Mitochondrial Poison Dequalinium Chloride in a Murine Model System. J. Pharm. Sci. 1993, 82, 231–235. [Google Scholar] [CrossRef]
- Li, M.; Sala, V.; De Santis, M.C.; Cimino, J.; Cappello, P.; Pianca, N.; Di Bona, A.; Margaria, J.P.; Martini, M.; Lazzarini, E.; et al. Phosphoinositide 3-Kinase Gamma Inhibition Protects From Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation 2018, 138, 696–711. [Google Scholar] [CrossRef]
- Kong, D.; Yamori, T. Phosphatidylinositol 3-Kinase Inhibitors: Promising Drug Candidates for Cancer Therapy. Cancer Sci. 2008, 99, 1734–1740. [Google Scholar] [CrossRef]
- Chang, N.-W.; Pei, R.-J.; Tseng, H.-C.; Yeh, K.-T.; Chan, H.-C.; Lee, M.-R.; Lin, C.; Hsieh, W.-T.; Kao, M.-C.; Tsai, M.-H.; et al. Co-Treating with Arecoline and 4-Nitroquinoline 1-Oxide to Establish a Mouse Model Mimicking Oral Tumorigenesis. Chem. Biol. Interact. 2010, 183, 231–237. [Google Scholar] [CrossRef]
- Tsai, Y.-S.; Lee, K.-W.; Huang, J.-L.; Liu, Y.-S.; Juo, S.-H.H.; Kuo, W.-R.; Chang, J.-G.; Lin, C.-S.; Jong, Y.-J. Arecoline, a Major Alkaloid of Areca Nut, Inhibits P53, Represses DNA Repair, and Triggers DNA Damage Response in Human Epithelial Cells. Toxicology 2008, 249, 230–237. [Google Scholar] [CrossRef]
- Mathema, V.B.; Koh, Y.-S.; Thakuri, B.C.; Sillanpää, M. Parthenolide, a Sesquiterpene Lactone, Expresses Multiple Anti-Cancer and Anti-Inflammatory Activities. Inflammation 2012, 35, 560–565. [Google Scholar] [CrossRef]
- Dong, L.; Shi, W.; Li, W.-H.; Li, Y.; Sun, Y.-Q. Parthenolide Induces Apoptosis and Inhibits Proliferation of Human 786-O Kidney Cancer Cells in Vitro. Int. J. Clin. Exp. Med. 2019, 12, 7056–7064. [Google Scholar]
- Gundersen, G.W.; Jones, M.R.; Rouillard, A.D.; Kou, Y.; Monteiro, C.D.; Feldmann, A.S.; Hu, K.S.; Ma’ayan, A. GEO2Enrichr: Browser Extension and Server App to Extract Gene Sets from GEO and Analyze Them for Biological Functions. Bioinformatics 2015, 31, 3060–3062. [Google Scholar] [CrossRef]
- Wang, Z.; Monteiro, C.D.; Jagodnik, K.M.; Fernandez, N.F.; Gundersen, G.W.; Rouillard, A.D.; Jenkins, S.L.; Feldmann, A.S.; Hu, K.S.; McDermott, M.G.; et al. Extraction and Analysis of Signatures from the Gene Expression Omnibus by the Crowd. Nat. Commun. 2016, 7, 12846. [Google Scholar] [CrossRef]
- Leek, J.T.; Storey, J.D. Capturing Heterogeneity in Gene Expression Studies by Surrogate Variable Analysis. PLoS Genet. 2007, 3, 1724–1735. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing Well-Connected Communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef] [PubMed]
- Zaravinos, A.; Roufas, C.; Nagara, M.; de Lucas Moreno, B.; Oblovatskaya, M.; Efstathiades, C.; Dimopoulos, C.; Ayiomamitis, G.D. Cytolytic Activity Correlates with the Mutational Burden and Deregulated Expression of Immune Checkpoints in Colorectal Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 364. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-J.; Hu, F.-F.; Xia, M.-X.; Han, L.; Zhang, Q.; Guo, A.-Y. GSCALite: A Web Server for Gene Set Cancer Analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef] [PubMed]
- Musa, A.; Ghoraie, L.S.; Zhang, S.-D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A Review of Connectivity Map and Computational Approaches in Pharmacogenomics. Brief. Bioinform. 2018, 19, 506–523. [Google Scholar] [CrossRef]
- Duan, Q.; Reid, S.P.; Clark, N.R.; Wang, Z.; Fernandez, N.F.; Rouillard, A.D.; Readhead, B.; Tritsch, S.R.; Hodos, R.; Hafner, M.; et al. L1000CDS2: LINCS L1000 Characteristic Direction Signatures Search Engine. NPJ Syst. Biol. Appl. 2016, 2, 16015. [Google Scholar] [CrossRef]
- van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell Sensitivity Assays: The MTT Assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ioannou, I.; Chatziantoniou, A.; Drenios, C.; Christodoulou, P.; Kourti, M.; Zaravinos, A. Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Kidney Cancers. Int. J. Mol. Sci. 2023, 24, 6577. https://doi.org/10.3390/ijms24076577
Ioannou I, Chatziantoniou A, Drenios C, Christodoulou P, Kourti M, Zaravinos A. Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Kidney Cancers. International Journal of Molecular Sciences. 2023; 24(7):6577. https://doi.org/10.3390/ijms24076577
Chicago/Turabian StyleIoannou, Ioanna, Angeliki Chatziantoniou, Constantinos Drenios, Panayiota Christodoulou, Malamati Kourti, and Apostolos Zaravinos. 2023. "Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Kidney Cancers" International Journal of Molecular Sciences 24, no. 7: 6577. https://doi.org/10.3390/ijms24076577
APA StyleIoannou, I., Chatziantoniou, A., Drenios, C., Christodoulou, P., Kourti, M., & Zaravinos, A. (2023). Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Kidney Cancers. International Journal of Molecular Sciences, 24(7), 6577. https://doi.org/10.3390/ijms24076577