
Abundance of Transgene Transcript Variants Associated with Somatically Active Transgenic Helitrons from Multiple T-DNA Integration Sites in Maize

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Phenotypic Variation Correlated with Transgene Expression in Primary Transformants and Their Hybrids with c1 Tester Lines
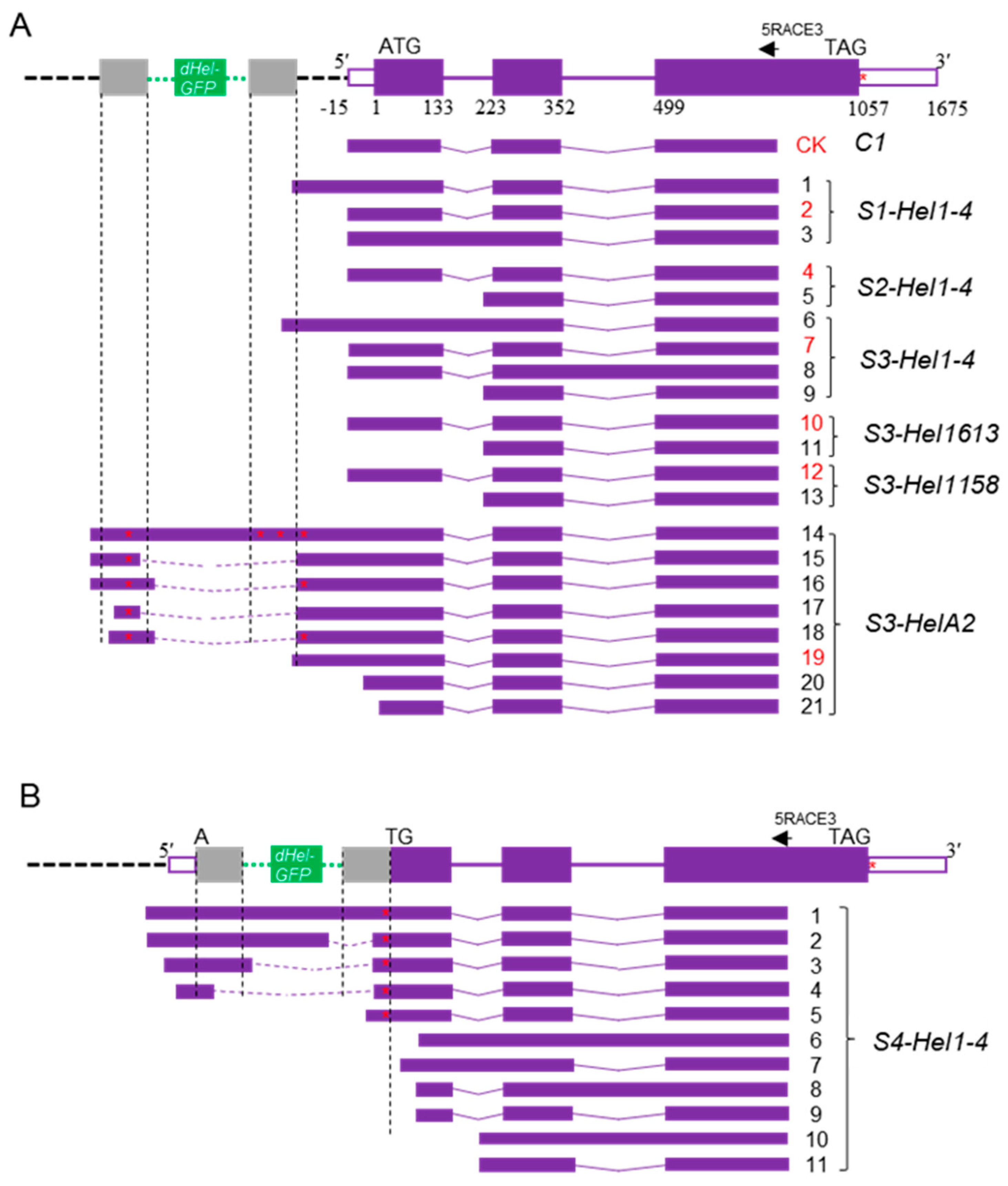
2.2. Transcript Spectrum of c1 Transgenes from Variable dHel-GFP Insertion Sites
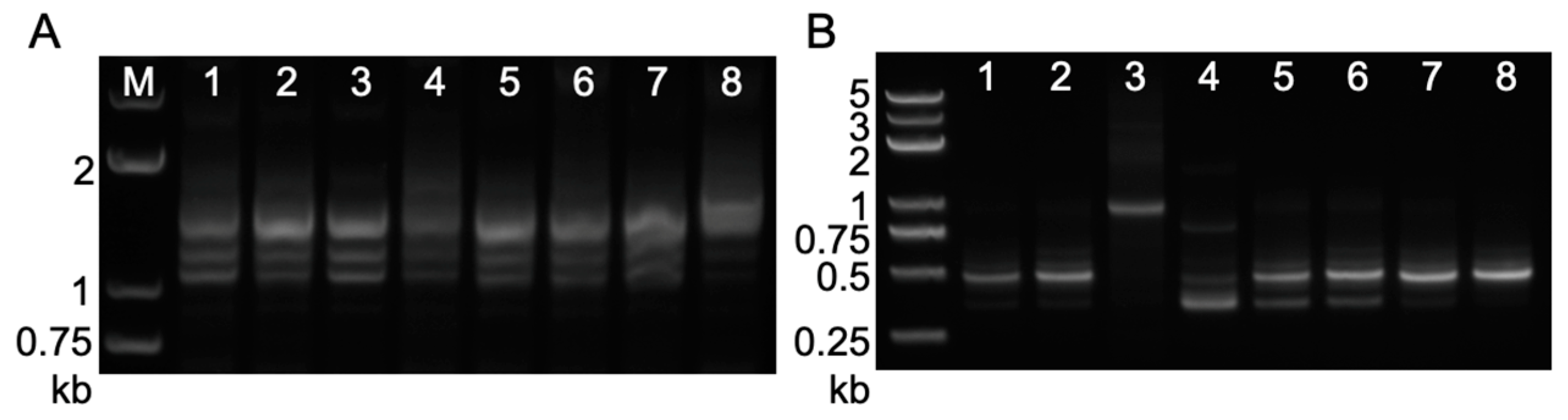
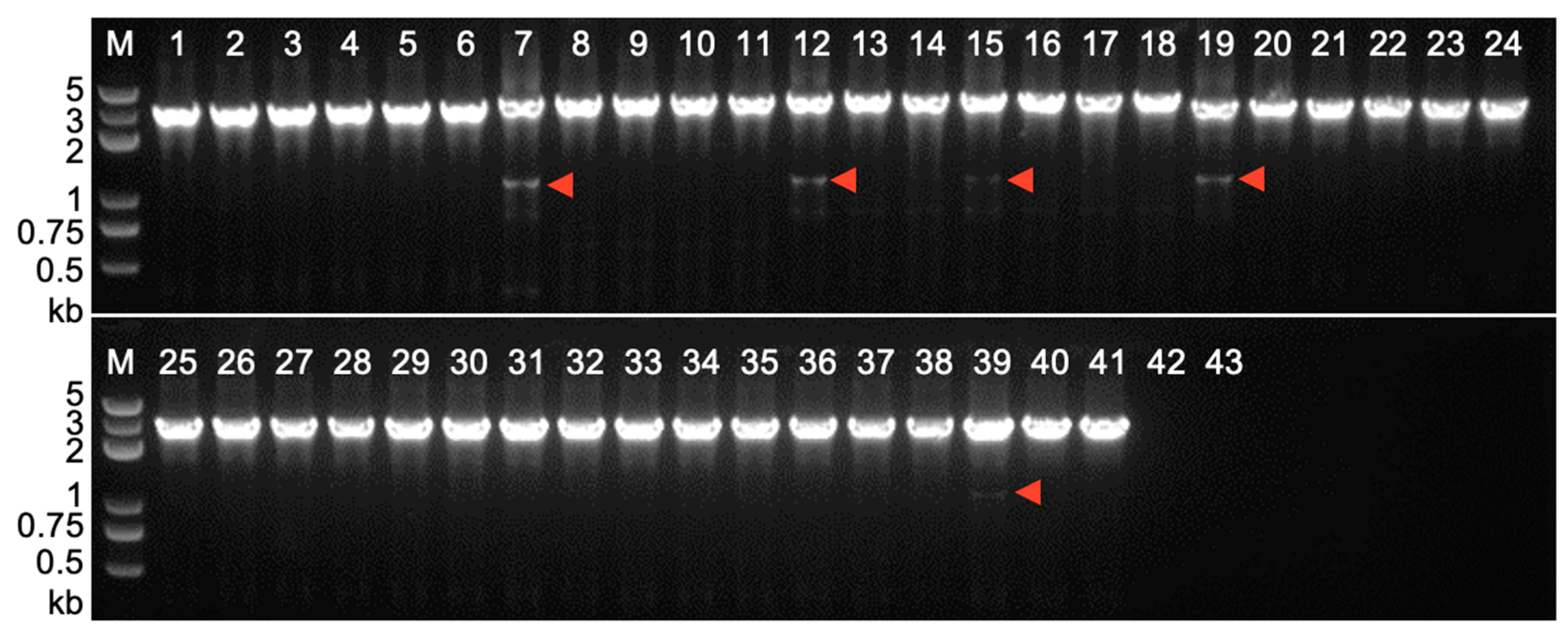
2.3. Footprint Analysis on Somatic Excision of Helitrons via the Cut-And-Paste Mechanism in Transgenic Lines and Hybrids with Maize Inbred Lines
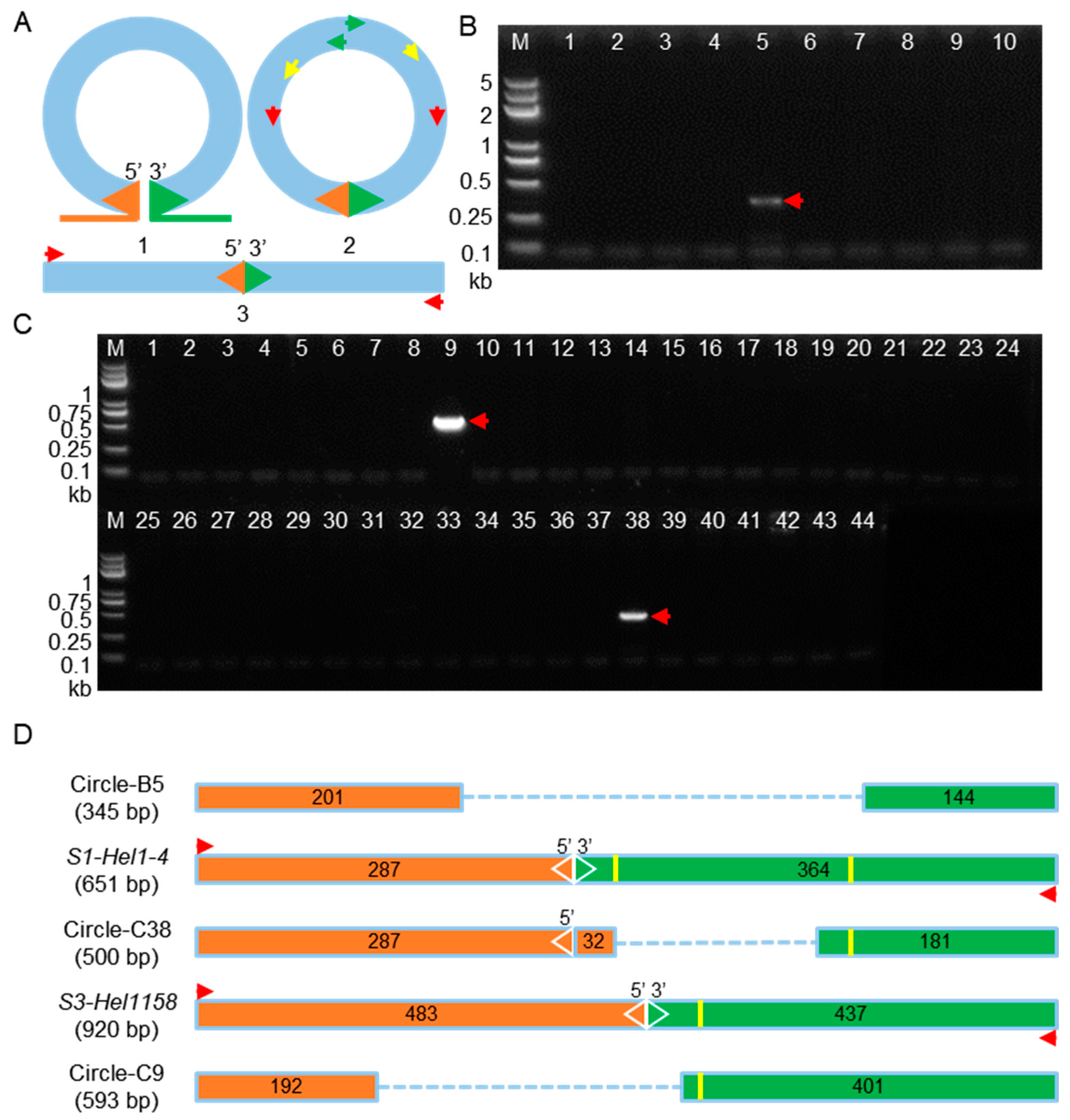
2.4. Circular Intermediates Detection in Rolling-Circle Transposition of Helitrons from Transgenic Lines and Their Hybrid Populations
3. Discussion
3.1. Helitron Transposon Families and Their Insertion Sites in the Promoter Region Alter Gene Expression through Linked Processes of Transcription or Splicing
3.2. Coexistence of RCR/Cut-And-Paste Somatic Transposition Mechanism Catalyzed by Autonomous Helitrons in Maize
4. Materials and Methods
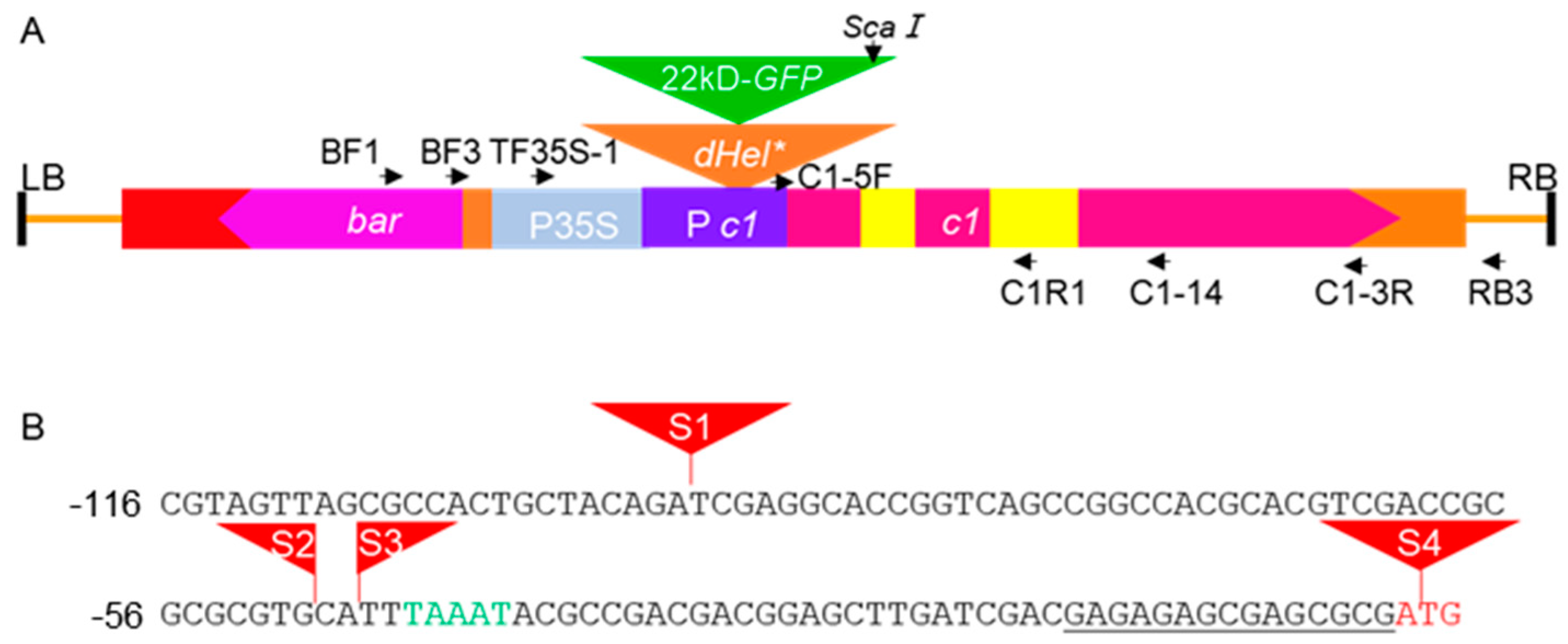
4.1. Development of the Reporter System
4.2. Genetic Activity Detection of dHel in BC1F1 Population from Test Crosses with Inbred Lines
4.3. PCR Amplification of Somatic Excision Products and Circular dHel Intermediates
4.4. Molecular Characterization of Variant Transcripts from Transgenes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kapitonov, V.V.; Jurka, J. Rolling-circle transposons in eukaryotes. Proc. Natl. Acad. Sci. USA 2001, 98, 8714–8719. [Google Scholar] [CrossRef] [PubMed]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.J.; Wortman, J.R.; Batzoglou, S.; Lee, S.I.; Basturkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Pritham, E.J.; Feschotte, C. Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc. Natl. Acad. Sci. USA 2007, 104, 1895–1900. [Google Scholar] [CrossRef] [PubMed]
- Coates, B.S.; Hellmich, R.L.; Grant, D.M.; Abel, C.A. Mobilizing the Genome of Lepidoptera through Novel Sequence Gains and End Creation by Non-autonomous Lep1 Helitrons. DNA Res. 2011, 19, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Cocca, E.; De Iorio, S.; Capriglione, T. Identification of a novel helitron transposon in the genome of Antarctic fish. Mol. Phylogenet. Evol. 2011, 58, 439–446. [Google Scholar] [CrossRef]
- Tollis, M.; Boissinot, S. The transposable element profile of the anolis genome: How a lizard can provide insights into the evolution of vertebrate genome size and structure. Mob. Genet. Elem. 2011, 1, 107–111. [Google Scholar] [CrossRef]
- Lal, S.K.; Giroux, M.J.; Brendel, V.; Vallejos, C.E.; Hannah, L.C. The maize genome contains a helitron insertion. Plant Cell 2003, 15, 381–391. [Google Scholar] [CrossRef]
- Brunner, S.; Pea, G.; Rafalski, A. Origins, genetic organization and transcription of a family of non-autonomous helitron elements in maize. Plant J. 2005, 43, 799–810. [Google Scholar] [CrossRef]
- Xu, J.H.; Messing, J. Maize haplotype with a helitron-amplified cytidine deaminase gene copy. BMC Genet. 2006, 7, 52. [Google Scholar] [CrossRef]
- Grabundzija, I.; Messing, S.A.; Thomas, J.; Cosby, R.L.; Bilic, I.; Miskey, C.; Gogol-Doring, A.; Kapitonov, V.; Diem, T.; Dalda, A.; et al. A Helitron transposon reconstructed from bats reveals a novel mechanism of genome shuffling in eukaryotes. Nat. Commun. 2016, 7, 10716. [Google Scholar] [CrossRef]
- Tempel, S.; Giraud, M.; Lavenier, D.; Lerman, I.C.; Valin, A.S.; Couee, I.; El Amrani, A.; Nicolas, J. Domain organization within repeated DNA sequences: Application to the study of a family of transposable elements. Bioinformatics 2006, 22, 1948–1954. [Google Scholar] [CrossRef]
- Du, C.G.; Caronna, J.; He, L.M.; Dooner, H.K. Computational prediction and molecular confirmation of Helitron transposons in the maize genome. BMC Genom. 2008, 9, 51. [Google Scholar] [CrossRef]
- Xiong, W.; He, L.; Lai, J.; Dooner, H.K.; Du, C. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc. Natl. Acad. Sci. USA 2014, 111, 10263–10268. [Google Scholar] [CrossRef]
- Hu, K.; Xu, K.; Wen, J.; Yi, B.; Shen, J.; Ma, C.; Fu, T.; Ouyang, Y.; Tu, J. Helitron distribution in Brassicaceae and whole Genome Helitron density as a character for distinguishing plant species. BMC Bioinform. 2019, 20, 354. [Google Scholar] [CrossRef]
- Gupta, S.; Gallavotti, A.; Stryker, G.A.; Schmidt, R.J.; Lal, S.K. A novel class of Helitron-related transposable elements in maize contain portions of multiple pseudogenes. Plant Mol. Biol. 2005, 57, 115–127. [Google Scholar] [CrossRef]
- Chuck, G.; Meeley, R.; Irish, E.; Sakai, H.; Hake, S. The maize tasselseed4 microRNA controls sex determination and meristem cell fate by targeting Tasselseed6/indeterminate spikelet1. Nat. Genet. 2007, 39, 1517–1521. [Google Scholar] [CrossRef]
- Gao, C.; Zhou, G.; Ma, C.; Zhai, W.; Zhang, T.; Liu, Z.; Yang, Y.; Wu, M.; Yue, Y.; Duan, Z.; et al. Helitron-like transposons contributed to the mating system transition from out-crossing to self-fertilizing in polyploid Brassica napus L. Sci. Rep. 2016, 6, 33785. [Google Scholar] [CrossRef]
- Choi, J.D.; Hoshino, A.; Park, K.I.; Park, I.S.; Iida, S. Spontaneous mutations caused by a Helitron transposon, Hel-It1, in morning glory, Ipomoea tricolor. Plant J. 2007, 49, 924–934. [Google Scholar] [CrossRef]
- Tsukamoto, T.; Hauck, N.R.; Tao, R.; Jiang, N.; Iezzoni, A.F. Molecular and genetic analyses of four nonfunctional S haplotype variants derived from a common ancestral S haplotype identified in sour cherry (Prunus cerasus L.). Genetics 2010, 184, 411–427. [Google Scholar] [CrossRef]
- Liu, Q.; Deng, S.; Liu, B.; Tao, Y.; Xu, M. A helitron-induced RabGDIα variant causes quantitative recessive resistance to maize rough dwarf disease. Nat. Commun. 2020, 11, 495. [Google Scholar] [CrossRef]
- Morgante, M.; Brunner, S.; Pea, G.; Fengler, K.; Zuccolo, A.; Rafalski, A. Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat. Genet. 2005, 37, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bennetzen, J.L. Distribution, diversity, evolution, and survival of Helitrons in the maize genome. Proc. Natl. Acad. Sci. USA 2009, 106, 19922–19927. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bennetzen, J.L. Structure-based discovery and description of plant and animal Helitrons. Proc. Natl. Acad. Sci. USA 2009, 106, 12832–12837. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Grant, D.; Tian, Z.; Nelson, R.T.; Zhu, L.; Shoemaker, R.C.; Ma, J. SoyTEdb: A comprehensive database of transposable elements in the soybean genome. BMC Genom. 2010, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar] [CrossRef]
- Thomas, J.; Phillips, C.D.; Baker, R.J.; Pritham, E.J. Rolling-circle transposons catalyze genomic innovation in a mammalian lineage. Genome Biol. Evol. 2014, 6, 2595–2610. [Google Scholar] [CrossRef]
- Wang, Q.H.; Dooner, H.K. Remarkable variation in maize genome structure inferred from haplotype diversity at the bz locus. Proc. Natl. Acad. Sci. USA 2006, 103, 17644–17649. [Google Scholar] [CrossRef]
- Volfovsky, N.; Haas, B.J.; Salzberg, S.L. A clustering method for repeat analysis in DNA sequences. Genome Biol. 2001, 2, RESEARCH0027. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. S1), i351–i358. [Google Scholar] [CrossRef]
- Flutre, T.; Duprat, E.; Feuillet, C.; Quesneville, H. Considering transposable element diversification in de novo annotation approaches. PLoS ONE 2011, 6, e16526. [Google Scholar] [CrossRef]
- Du, C.G.; Fefelova, N.; Caronna, J.; He, L.M.; Dooner, H.K. The polychromatic Helitron landscape of the maize genome. Proc. Natl. Acad. Sci. USA 2009, 106, 19916–19921. [Google Scholar] [CrossRef]
- Feschotte, C.; Pritham, E.J. A cornucopia of Helitrons shapes the maize genome. Proc. Natl. Acad. Sci. USA 2009, 106, 19747–19748. [Google Scholar] [CrossRef]
- Grabundzija, I.; Hickman, A.B.; Dyda, F. Helraiser intermediates provide insight into the mechanism of eukaryotic replicative transposition. Nat. Commun. 2018, 9, 1278. [Google Scholar] [CrossRef]
- Kosek, D.; Grabundzija, I.; Lei, H.; Bilic, I.; Wang, H.; Jin, Y.; Peaslee, G.F.; Hickman, A.B.; Dyda, F. The large bat Helitron DNA transposase forms a compact monomeric assembly that buries and protects its covalently bound 5′-transposon end. Mol. Cell 2021, 81, 4271–4286. [Google Scholar] [CrossRef]
- Li, Y.; Dooner, H.K. Excision of Helitron transposons in maize. Genetics 2009, 182, 399–402. [Google Scholar] [CrossRef]
- Xiong, W.; Dooner, H.K.; Du, C. Rolling-circle amplification of centromeric Helitrons in plant genomes. Plant J. 2016, 88, 1038–1045. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, G.; Yang, Q.; Gao, L.; Liu, C.; Ru, Z.; Wang, D.; Jia, J.; Cui, D. Helitron and CACTA DNA transposons actively reshape the common wheat—AK58 genome. Genomics 2022, 114, 110288. [Google Scholar] [CrossRef]
- Chaikam, V.; Nair, S.K.; Babu, R.; Martinez, L.; Tejomurtula, J.; Boddupalli, P.M. Analysis of effectiveness of R1-nj anthocyanin marker for in vivo haploid identification in maize and molecular markers for predicting the inhibition of R1-nj expression. Theor. Appl. Genet. 2015, 128, 159–171. [Google Scholar] [CrossRef]
- Murray, A.; Mendieta, J.P.; Vollmers, C.; Schmitz, R.J. Simple and accurate transcriptional start site identification using Smar2C2 and examination of conserved promoter features. Plant J. 2022, 112, 583–596. [Google Scholar] [CrossRef]
- Paz-Ares, J.; Wienand, U.; Peterson, P.A.; Saedler, H. Molecular cloning of the c locus of Zea mays: A locus regulating the anthocyanin pathway. EMBO J. 1986, 5, 829–833. [Google Scholar] [CrossRef]
- Cone, K.C.; Burr, F.A.; Burr, B. Molecular analysis of the maize anthocyanin regulatory locus C1. Proc. Natl. Acad. Sci. USA 1986, 83, 9631–9635. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, J.; Ghosal, D.; Saedler, H. Molecular analysis of the C1-I allele from Zea mays: A dominant mutant of the regulatory C1 locus. EMBO J. 1990, 9, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, B.; Franken, P.; Schutt, E.; Schrell, A.; Saedler, H.; Wienand, U. Molecular analysis of C1 alleles in Zea mays defines regions involved in the expression of this regulatory gene. Mol. Gen. Genet. 1994, 242, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.R.; Peterson, P.A. Transposable elements of maize--genetic basis of pattern differentiation of some mutable c alleles of the enhancer system. Mol. Gen. Genet. MGG 1983, 192, 21–31. [Google Scholar] [CrossRef]
- Li, Y.; Dooner, H.K. Helitron Proliferation and Gene-Fragment Capture. In Plant Transposable Elements: Impact on Genome Structure and Function; Grandbastien, M.-A., Casacuberta, J.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 193–217. [Google Scholar] [CrossRef]
- Li, Y.; Harris, L.; Dooner, H.K. TED, an autonomous and rare maize transposon of the mutator superfamily with a high gametophytic excision frequency. Plant Cell 2013, 25, 3251–3265. [Google Scholar] [CrossRef]
- Sundaresan, V.; Freeling, M. An extrachromosomal form of the Mu transposons of maize. Proc. Natl. Acad. Sci. USA 1987, 84, 4924–4928. [Google Scholar] [CrossRef]
- Gorbunova, V.; Levy, A.A. Circularized Ac/Ds transposons: Formation, structure and fate. Genetics 1997, 145, 1161–1169. [Google Scholar] [CrossRef]
- Lai, J.S.; Li, Y.B.; Messing, J.; Dooner, H.K. Gene movement by Helitron transposons contributes to the haplotype variability of maize. Proc. Natl. Acad. Sci. USA 2005, 102, 9068–9073. [Google Scholar] [CrossRef]
- Ludwig, S.R.; Habera, L.F.; Dellaporta, S.L.; Wessler, S.R. Lc, a member of the maize R gene family responsible for tissue-specific anthocyanin production, encodes a protein similar to transcriptional activators and contains the myc-homology region. Proc. Natl. Acad. Sci. USA 1989, 86, 7092–7096. [Google Scholar] [CrossRef]
- Coe, E.H. Spontaneous Mutation of the Aleurone Color Inhibitor in Maize. Genetics 1962, 47, 779. [Google Scholar] [CrossRef]
- Paz-Ares, J.; Ghosal, D.; Wienand, U.; Peterson, P.A.; Saedler, H. The regulatory c1 locus of Zea mays encodes a protein with homology to myb proto-oncogene products and with structural similarities to transcriptional activators. EMBO J. 1987, 6, 3553–3558. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct | Site (nt) | dHel-GFP (bp) | Donor Line | T1 Events | T1 Single Copy | T2 Coleoptiles | T2 Anthers | F1 Kernel |
|---|---|---|---|---|---|---|---|---|
| S1-Hel1-4 | −93 | 1723 | B104 | 3 | 1 | Purple | P | Mottled |
| S2-Hel1-4 | −49 | 1723 | B104 | 5 | 4 | Purple | LP * | Mottled |
| S3-Hel1-4 | −47 | 1723 | B104 | 5 | 4 | Purple | LP | Mottled |
| S4-Hel1-4 | 1 | 1723 | B104 | 6 | 2 | Colorless | Colorless | Colorless |
| S3-Hel1613 | −47 | 1781 | B104 | 5 | 4 | Purple | LP | Colorless |
| S3-Hel1158 | −47 | 1996 | B104 | 5 | 4 | Purple | LP | Mottled |
| S3-HelA2 | −47 | 2038 | HiⅡ | 5 | 4 | Purple | LP | Mottled |
| Constructs | Examined Segregants | Segregants with Detected SE | T-DNA Copy Numbers in Transformants |
|---|---|---|---|
| S1-Hel1-4 | 14 | 6 | 2 (Arrayed in tandem) |
| S2-Hel1-4 | 36 | 0 | 1 |
| S3-Hel1-4 | 34 | 0 | / |
| S4-Hel1-4 | 15 | 3 | 1 |
| S3-Hel1613 | 34 | 0 | / |
| S3-Hel1158 | 15 | 1 | 1 |
| S3-HelA2 | 17 | 2 | 1 |
| dHel-GFP Construct | dHel 5′-End Junction dHel 3′-End Junction |
|---|---|
| S3-Hel1158 | ACGTCGACCGCGCGCGTGCATCTATACTAT…AACCGACTAGTTTAAATACGCCGACGACGG |
| S3-Hel1158 (e) | ACGTCGACCGCGCGCGTGCA---------------------------------------------TTTAAATACGCCGACGACGG |
| S3-HelA2 | ACGTCGACCGCGCGCGTGCATCTCTACTAC…ACTCACCTAGTTTAAATACGCCGACGACGG |
| S3-HelA2 (e) | ACGTCGACCGCGCGCGTGCA---------------------------------------------TTTAAATACGCCGACGACGG |
| S1-Hel1-4 | GTTAGCGCCACTGCTACAGATCTATACTAC…ACCTAACTAGTCGAGGCACCGGTCAGCCGG |
| S1-Hel1-4 (e) | GTTAGCGCCACTGCTACAGA---------------------------------------------TCGAGGCACCGGTCAGCCGG |
| S4-Hel1-4 | CGACGAGAGAGCGAGCGCGATCTATACTAC…ACCTAACTAGTGGGGAGGAGGGCGTGTTGC |
| S4-Hel1-4 (e) | CGACGAGAGAGCGAGCGCGA---------------------------------------------TGGGGAGGAGGGCGTGTTGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Cong, C.; Liu, F.; Yu, Q.; Zhan, Y.; Zhu, L.; Li, Y. Abundance of Transgene Transcript Variants Associated with Somatically Active Transgenic Helitrons from Multiple T-DNA Integration Sites in Maize. Int. J. Mol. Sci. 2023, 24, 6574. https://doi.org/10.3390/ijms24076574
Li C, Cong C, Liu F, Yu Q, Zhan Y, Zhu L, Li Y. Abundance of Transgene Transcript Variants Associated with Somatically Active Transgenic Helitrons from Multiple T-DNA Integration Sites in Maize. International Journal of Molecular Sciences. 2023; 24(7):6574. https://doi.org/10.3390/ijms24076574
Chicago/Turabian StyleLi, Chuxi, Chunsheng Cong, Fangyuan Liu, Qian Yu, Yuan Zhan, Li Zhu, and Yubin Li. 2023. "Abundance of Transgene Transcript Variants Associated with Somatically Active Transgenic Helitrons from Multiple T-DNA Integration Sites in Maize" International Journal of Molecular Sciences 24, no. 7: 6574. https://doi.org/10.3390/ijms24076574
APA StyleLi, C., Cong, C., Liu, F., Yu, Q., Zhan, Y., Zhu, L., & Li, Y. (2023). Abundance of Transgene Transcript Variants Associated with Somatically Active Transgenic Helitrons from Multiple T-DNA Integration Sites in Maize. International Journal of Molecular Sciences, 24(7), 6574. https://doi.org/10.3390/ijms24076574