
Integrated Analysis of lncRNA–mRNA Regulatory Networks Related to Lipid Metabolism in High-Oleic-Acid Rapeseed
Abstract
1. Introduction
2. Results
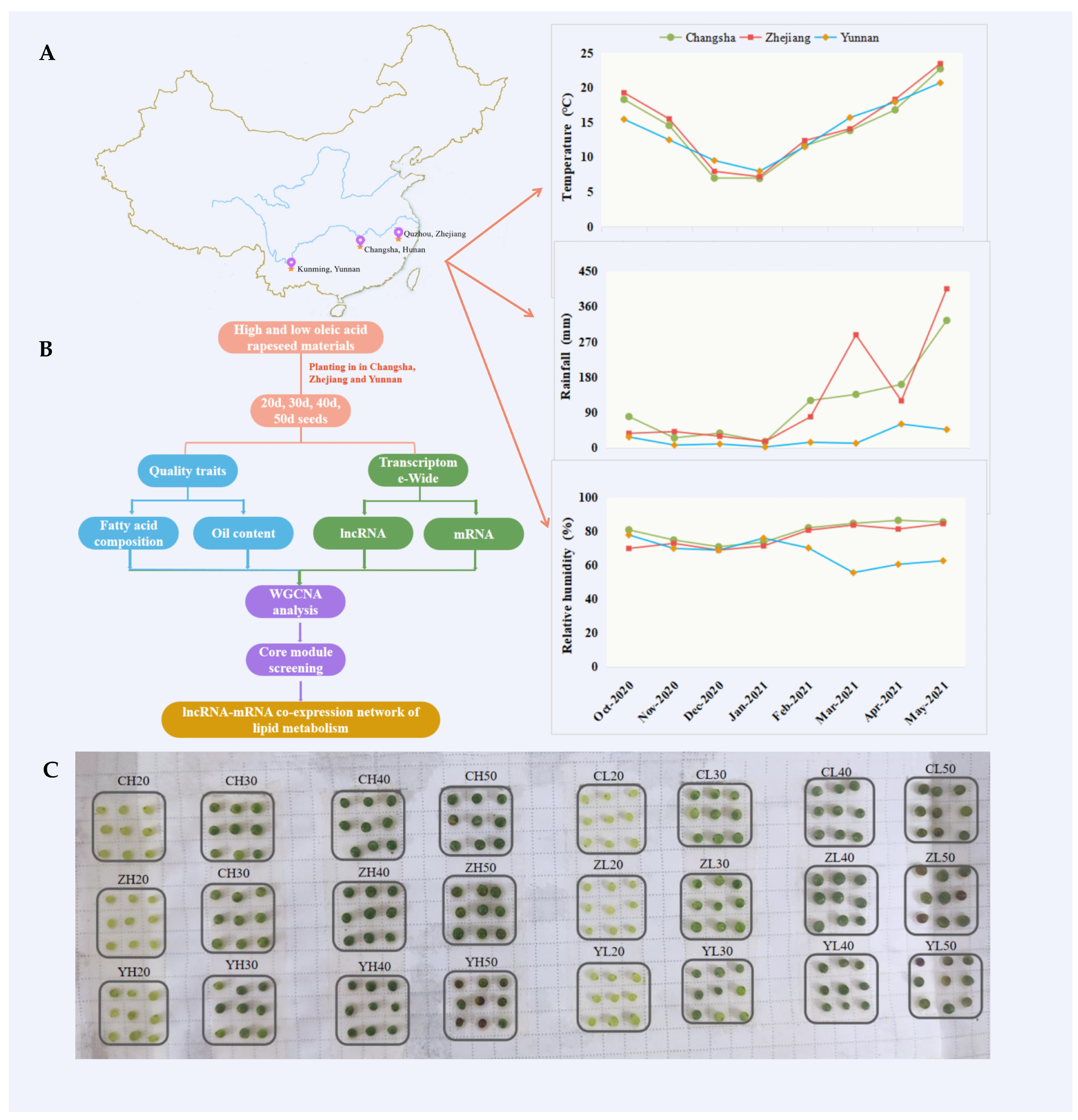
2.1. Dynamic Changes in the Oil Content and Fatty Acid Components during Seed Development in High- and Low-Oleic-Acid Rapeseed
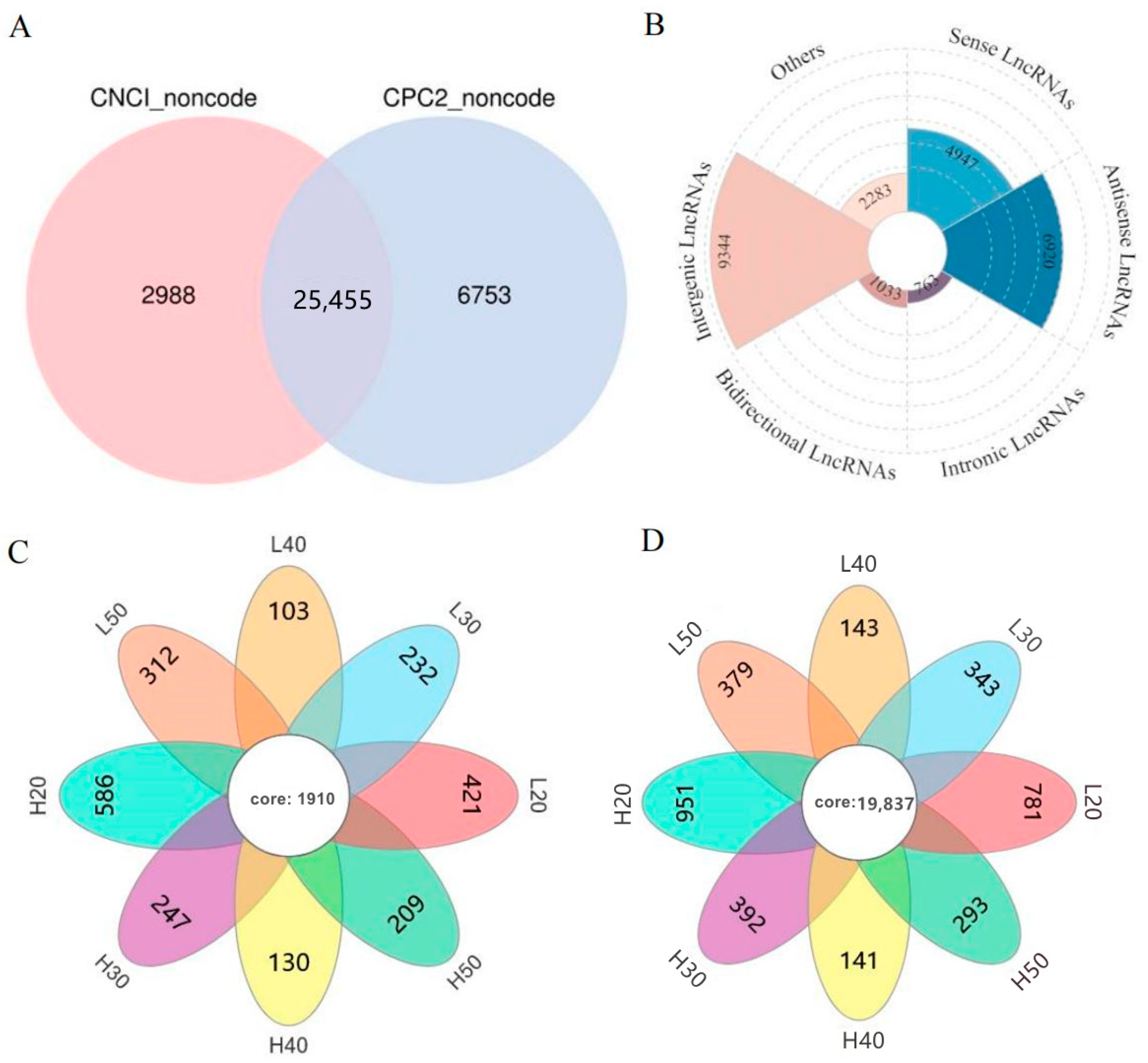
2.2. Whole-Transcriptome Identification of lncRNAs in High- and Low-Oleic-Acid Rapeseed
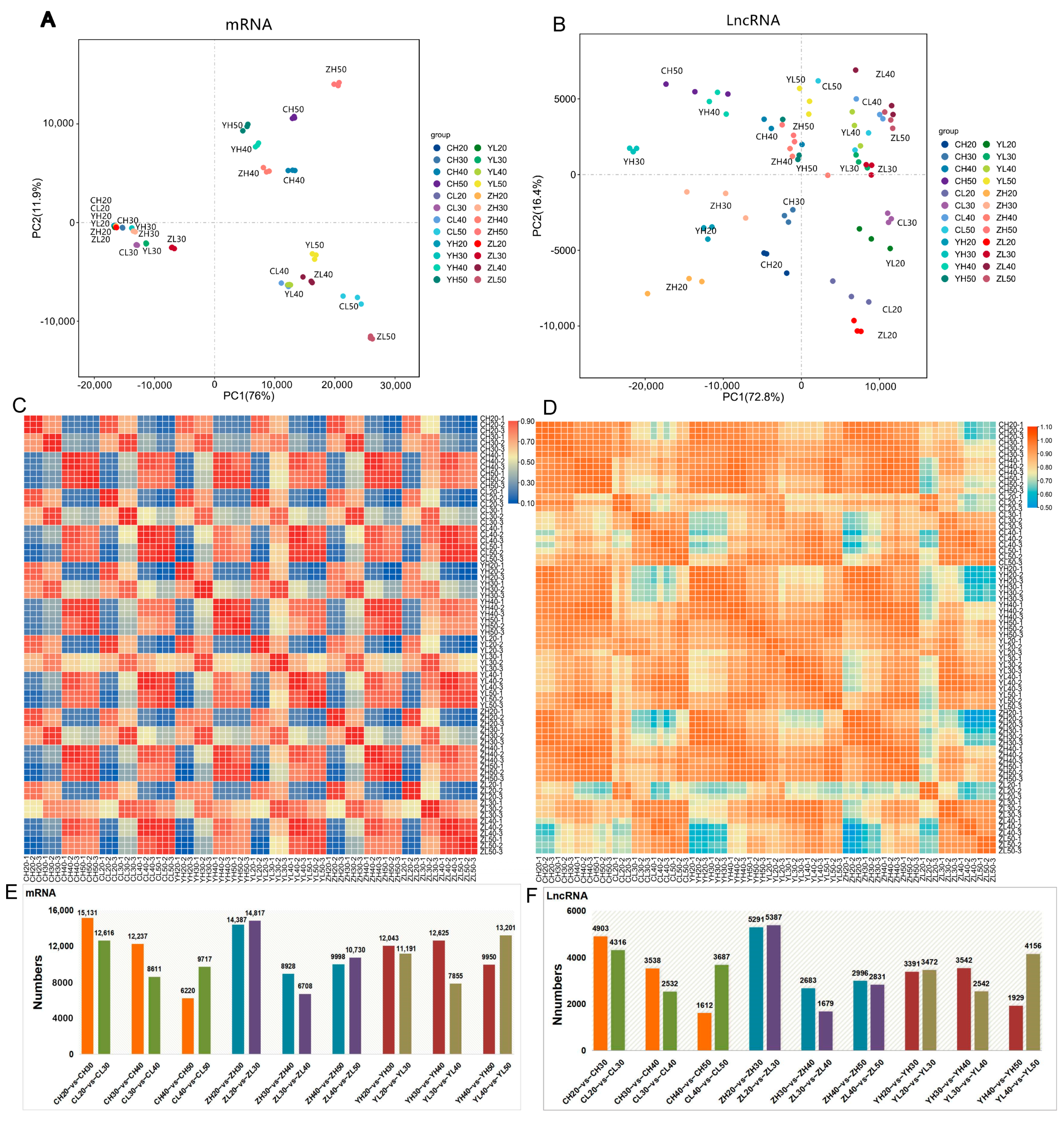
2.3. Identification of lncRNA and mRNA Expression Patterns
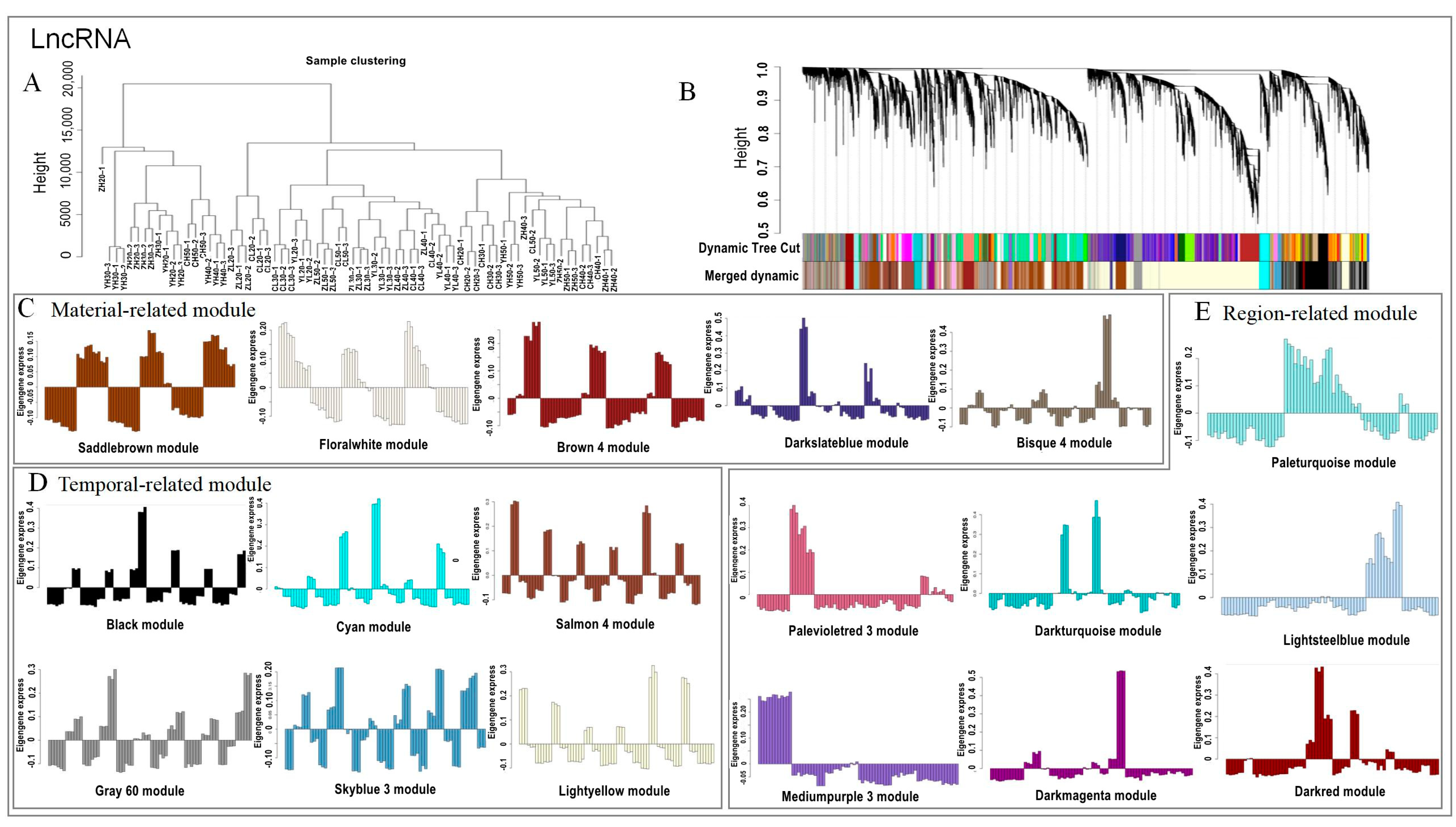
2.4. Discrete Expression Modules of lncRNA and mRNA Expression Using WGCNA Analysis
2.5. Identification of Material- and Temporal-Related lncRNAs and mRNAs
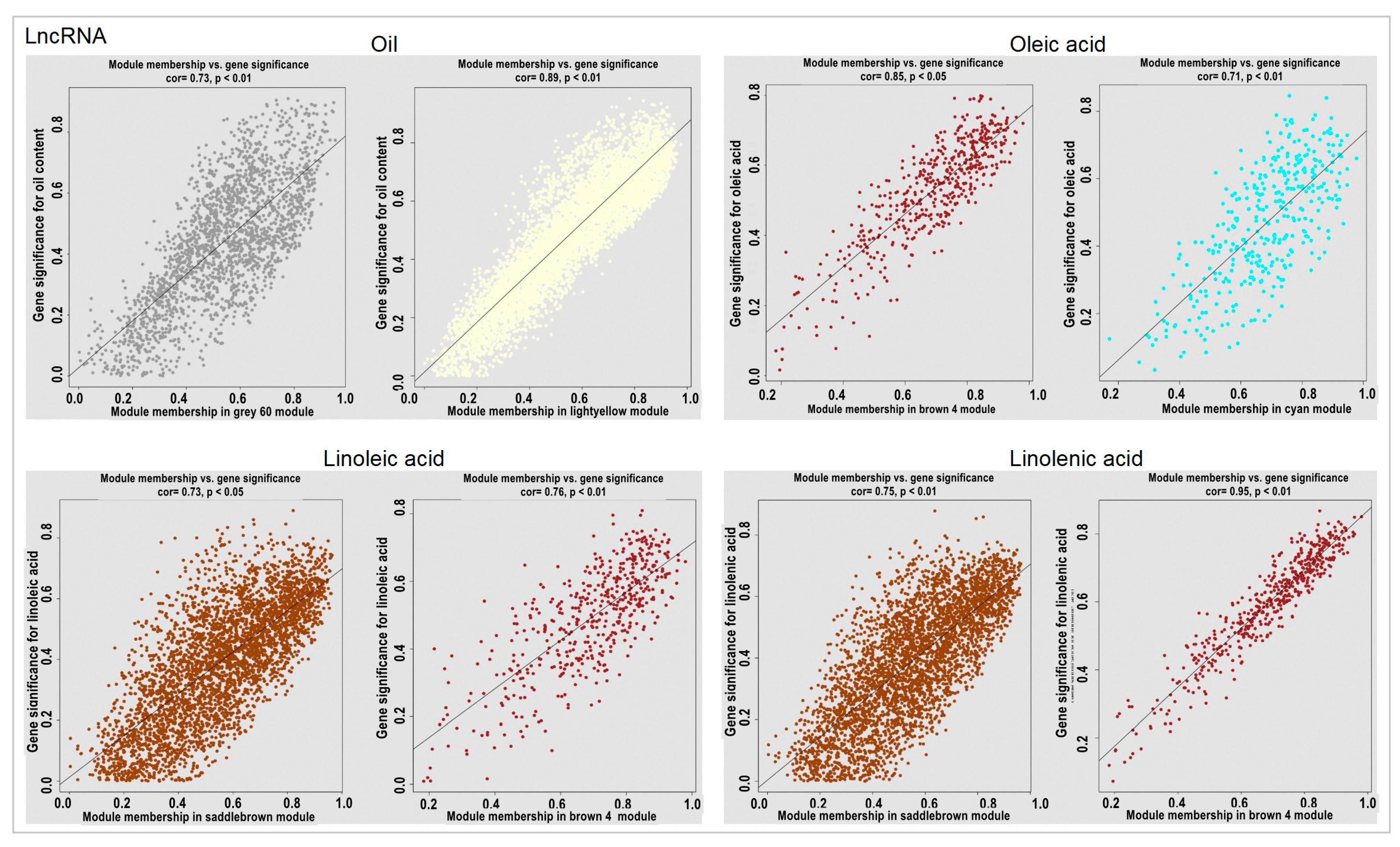
2.6. Module Analysis Related to the Oil Content and Fatty Acid Combined with Physiological Data
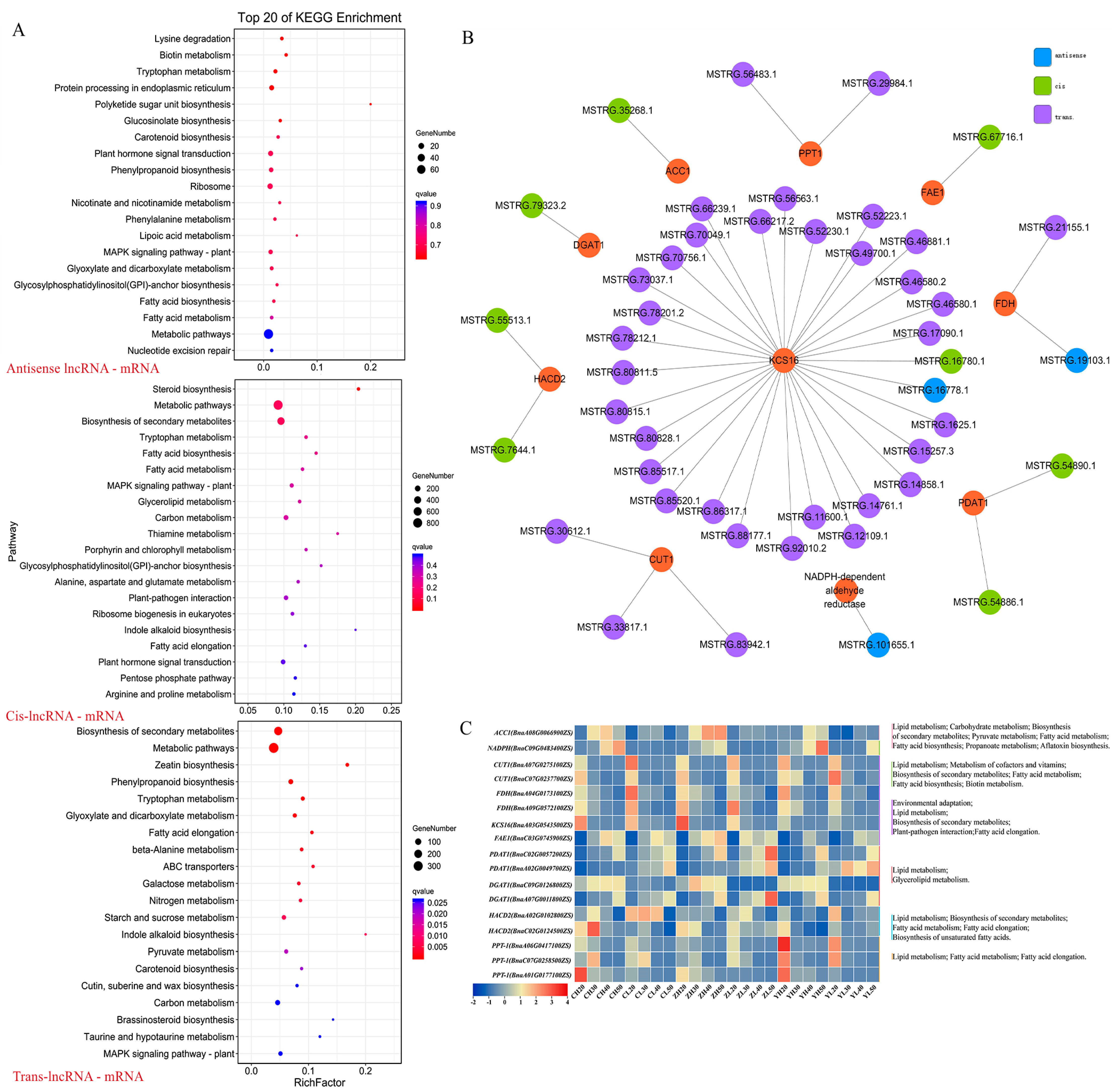
2.7. Construction of lncRNA–mRNA co-Expression Network Related to Lipid Metabolism
3. Discussion
3.1. Characteristics of lncRNAs: Low Expression Level and High Expression Specificity (in Different Materials, Regions, and Development Stages)
3.2. The lncRNA–mRNA Relationships in the Core Network Play an Important Role in Regulating Lipid Metabolism
4. Material and Methods
4.1. Plant Materials, Growth Conditions, and Sample Collection
4.2. Seed Oil Extraction and Fatty Acid Composition Analysis
4.3. RNA Extraction, Strand-Specific Library Construction, and Illumina Sequencing
4.4. Sequence Data Analysis
4.5. WGCNA Analysis
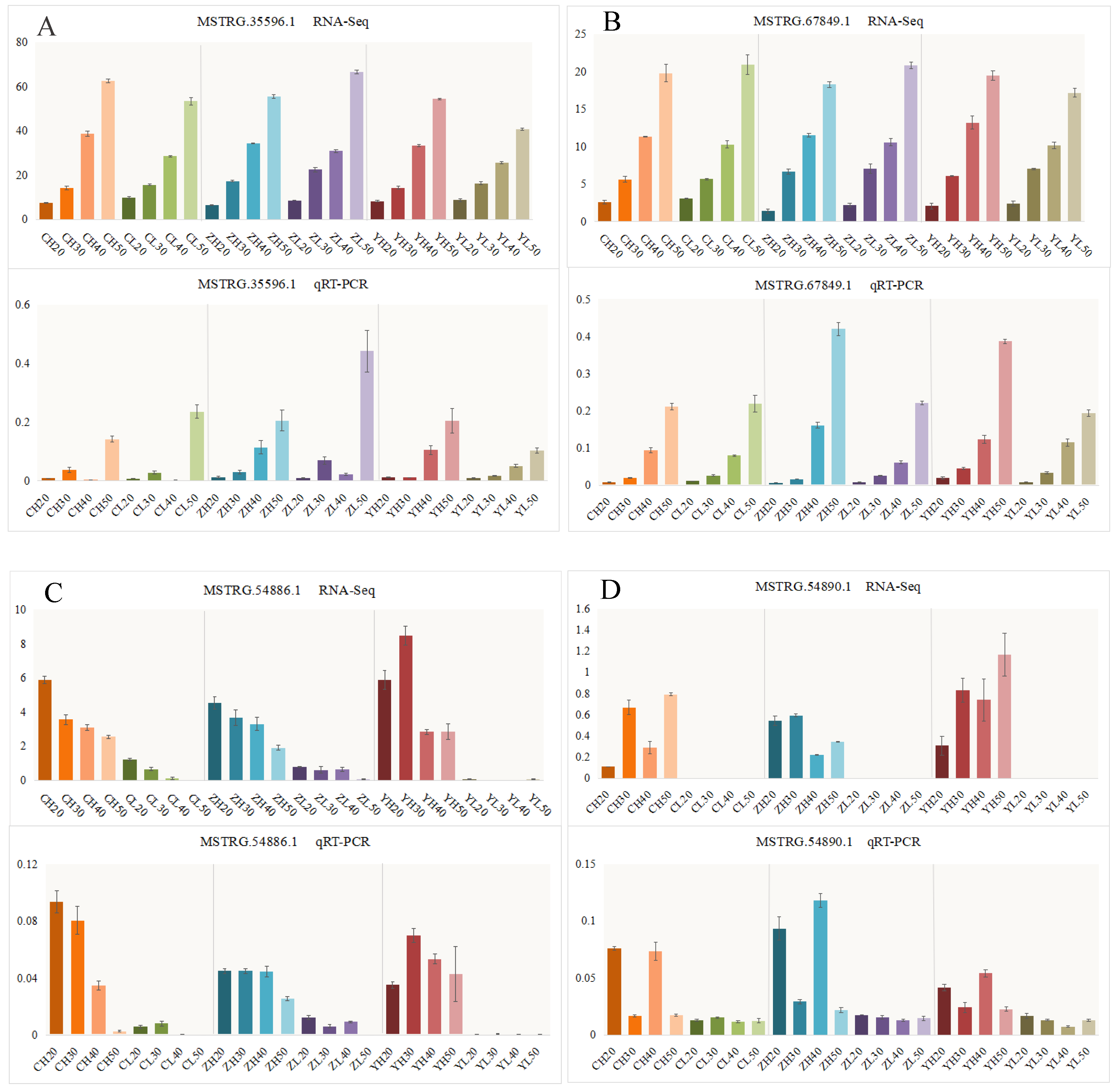
4.6. qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Zuo, Q.; Chang, H.; Bai, G.; Zhou, G. Higher density planting benefits mechanical harvesting of rapeseed in the yangtze river basin of china. Field Crops Res. 2018, 218, 97–105. [Google Scholar] [CrossRef]
- Matthaus, B.; Özcan, M.M.; Al, J.F. Some rape/canola seed oils: Fatty acid composition and tocopherols. Z. Für Nat. C 2016, 71, 73–77. [Google Scholar] [CrossRef]
- Mei, G.; Hong, C.; Xiong, X.; Xin, L.; Guan, C. A study on triacylglycerol composition and the structure of high-oleic rapeseed oil. Engineering 2016, 2, 258–262. [Google Scholar]
- Jahreis, G.; Schäfer, U. Chapter 114—Rapeseed (Brassica napus) Oil and its Benefits for Human Health. In Nuts and Seeds in Health and Disease Prevention; Academic Press: Cambridge, MA, USA, 2011; pp. 967–974. [Google Scholar] [CrossRef]
- Rahman, M.; Michalak, D.E.; Jiménez, M. Chapter 15—Designer Oil Crops. In Breeding Oilseed Crops for Sustainable Production; Gupta, S.K., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 361–376. [Google Scholar] [CrossRef]
- Onacik-Gür, S.; Żbikowska, A. Effect of high-oleic rapeseed oil oleogels on the quality of short-dough biscuits and fat migration. J. Food Sci. Technol. 2020, 57, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Spasibionek, S.; Mikołajczyk, K.; Ćwiek-Kupczyńska, H.; Piętka, T.; Krótka, K.; Matuszczak, M.; Nowakowska, J.; Michalski, K.; Bartkowiak-Broda, I. Marker assisted selection of new high oleic and low linolenic winter oilseed rape (Brassica napus L.) inbred lines revealing good agricultural value. PLoS ONE 2020, 15, e0233959. [Google Scholar] [CrossRef]
- Chang, T.; Wu, J.; Wu, X.; Yao, M.; Zhao, D.; Guan, C.; Guan, M. Comprehensive evaluation of high-oleic rapeseed (Brassica napus) based on quality, resistance, and yield traits: A new method for rapid identification of high-oleic acid rapeseed germplasm. PLoS ONE 2022, 18, e0272798. [Google Scholar] [CrossRef]
- Xian, Z.H.; Wei, L.H.; Tan, X.Y.; Hu, M.L.; Pu, H.M. Research Progress on the Genetics and Varieties Breeding of High-oleic-acid Rapeseed. Curr. Biotechnol. 2022, 12, 641–646. (In Chinese) [Google Scholar] [CrossRef]
- Bates, P.D.; Stymne, S.; Ohlrogge, J. Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 2013, 16, 358–364. [Google Scholar] [CrossRef]
- Patel, M.S.; Harris, R.A. Metabolic Regulation. In Encyclopedia of Cell Biology; Ralph, A., Bradshaw, P., Stahl, D., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 288–297. [Google Scholar] [CrossRef]
- Chi, G.; Cao, X.; Li, Q.; Yao, C.; Lu, F.; Liu, Y.; Cao, M.; He, N. Computationally Guided Enzymatic Studies on Schizochytrium-Sourced Malonyl-CoA:ACP Transacylase. J. Agric. Food Chem. 2022, 70, 13922–13934. [Google Scholar] [CrossRef]
- Turgeson, A.; Morley, L.; Giles, D.; Harris, B. Simulated Docking Predicts Putative Channels for the Transport of Long-Chain Fatty Acids in Vibrio cholerae. Biomolecules 2022, 12, 1269. [Google Scholar] [CrossRef]
- Bates, P.D.; Browse, J. The pathway of triacylglycerol synthesis through phosphatidylcholine in Arabidopsis produces a bottleneck for the accumulation of unusual fatty acids in transgenic seeds. Plant J. Cell Mol. Biol. 2011, 68, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, J.; Wei, Y.; Khavari, P. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell. Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xia, X.; Jiang, B.; Ma, K.; Zhu, L.; Wang, L.; Jin, B. Identification and characterization of novel lncRNAs in Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 2017, 488, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Rigo, R.; Bazin, J.; Romero-Barrios, N.; Moison, M.; Lucero, L.; Christ, A.; Benhamed, M.; Blein, T.; Huguet, S.; Charon, C.; et al. The Arabidopsis lncRNA ASCO modulates the transcriptome through interaction with splicing factors. EMBO Rep. 2020, 21, e48977. [Google Scholar] [CrossRef] [PubMed]
- Jampala, P.; Garhewal, A.; Lodha, M. Functions of long non-coding RNA in Arabidopsis thaliana. Plant Signal Behav. 2021, 16, 1925440. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.X.; Zheng, X.W.; Li, H.B.; Mlekwa, U.A.; Gao, Y.; Xiong, J. Roles of lncRNAs in Rice: Advances and Challenges. Rice Sci. 2020, 27, 384–395. [Google Scholar] [CrossRef]
- Zhang, T.; Liang, Q.; Li, C.; Fu, S.; Kundu, J.K.; Zhou, X.; Wu, J. Transcriptome Analysis of Rice Reveals the lncRNA-mRNA Regulatory Network in Response to Rice Black-Streaked Dwarf Virus Infection. Viruses 2020, 12, 951. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Li, L.; Li, D.; Zhang, Q.; Guo, Y.; Wang, S.; Zhong, C.; Huang, H. Whole transcriptome sequencing of Pseudomonas syringae pv. actinidiae-infected kiwifruit plants reveals species-specific interaction between long non-coding RNA and coding genes. Sci. Rep. 2017, 7, 4910. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, C.; Feng, X.; Lai, R.; Gao, M.; Chen, W.; Wu, R. Integrated analysis of lncRNA and mRNA transcriptomes reveals the potential regulatory role of lncRNA in kiwifruit ripening and softening. Sci. Rep. 2021, 11, 1671. [Google Scholar] [CrossRef]
- Song, X.; Hu, J.; Wu, T.; Yang, Q.; Feng, X.; Lin, H.; Feng, S.; Cui, C.; Yu, Y.; Zhou, R.; et al. Comparative analysis of long noncoding RNAs in angiosperms and characterization of long noncoding RNAs in response to heat stress in Chinese cabbage. Hortic. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Joshi, R.K.; Megha, S.; Basu, U.; Rahman, M.H.; Kav, N. Genome Wide Identification and Functional Prediction of Long Non-Coding RNAs Responsive to Sclerotinia sclerotiorum Infection in Brassica napus. PLoS ONE 2016, 11, e0158784. [Google Scholar] [CrossRef] [PubMed]
- Summanwar, A.; Basu, U.; Kav, N.; Rahman, H. Identification of lncRNAs in response to infection by Plasmodiophora brassicae in Brassica napus and development of lncRNA-based SSR markers. Genome 2021, 64, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Li, S.; Hu, L.; Zhang, C. Genome-wide analysis of long non-coding RNAs (lncRNAs) in two contrasting rapeseed (Brassica napus L.) genotypes subjected to drought stress and re-watering. BMC Plant Biol. 2020, 20, 81. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.J.; Zhang, X.D.; Liu, X.S.; Tan, S.K.; Chu, S.S.; Meng, J.G.; Zhao, K.X.; Zheng, J.F.; Yang, Z.M. Characterization of long non-coding RNAs involved in cadmium toxic response in Brassica napus. RSC Adv. 2016, 6, 82157–82173. [Google Scholar] [CrossRef]
- Shen, E.; Zhu, X.; Hua, S.; Chen, H.; Ye, C.; Zhou, L.; Liu, Q.; Zhu, Q.H.; Fan, L.; Chen, X. Genome-wide identification of oil biosynthesis-related long non-coding RNAs in allopolyploid Brassica napus. BMC Genom. 2018, 19, 745. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35 (Suppl. S2), W345–W349. [Google Scholar] [CrossRef]
- Johnsson, P.; Ziegenhain, C.; Hartmanis, L.; Hendriks, G.J.; Hagemann-Jensen, M.; Reinius, B.; Sandberg, R. Transcriptional kinetics and molecular functions of long noncoding RNAs. Nat. Genet. 2022, 54, 306–317. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Q.H.; Kaufmann, K. Long non-coding RNAs in plants: Emerging modulators of gene activity in development and stress responses. Planta 2020, 252, 92. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Song, Z.; Zhu, C.; Tao, C.; Kang, L.; Liu, W.; He, F.; Yan, J.; Sang, T. Systematic comparison of lncRNAs with protein coding mRNAs in population expression and their response to environmental change. BMC Plant Biol. 2017, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Schilbert, H.M.; Pucker, B.; Ries, D.; Viehöver, P.; Micic, Z.; Dreyer, F.; Beckmann, K.; Wittkop, B.; Weisshaar, B.; Holtgräwe, D. Mapping-by-Sequencing Reveals Genomic Regions Associated with Seed Quality Parameters in Brassica napus. Genes 2022, 13, 1131. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Zhang, Y.Y.; Huang, L.T.; Yu, X.L.; Luo, Y.; Jiang, L.; Sun, Y.F.; Liu, S.; Huang, H. Differences in Fatty Acids and Lipids of Massive and Branching Reef-Building Corals and Response to Environmental Changes. Front. Mar. Sci. 2022, 9, 882663. [Google Scholar] [CrossRef]
- Rui, C.; Chen, X.G.; Xu, N.; Wang, J.; Zhang, H.; Li, S.M.; Huang, H.; Fan, Y.P.; Zhang, Y.X.; Lu, X.L.; et al. Identification and Structure Analysis of KCS Family Genes Suggest Their Reponding to Regulate Fiber Development in Long-Staple Cotton Under Salt-Alkaline Stress. Front. Genet. 2022, 13, 812449. [Google Scholar] [CrossRef]
- Joubès, J.; Raffaele, S.; Bourdenx, B.; Garcia, C.; Laroche-Traineau, J.; Moreau, P.; Domergue, F.; Lessire, R. The VLCFA elongase gene family in Arabidopsis thaliana: Phylogenetic analysis, 3D modelling and expression pro-filing. Plant Mol. Biol. 2008, 67, 547–566. [Google Scholar] [CrossRef]
- Blacklock, B.J.; Jaworski, J. Substrate specificity of Arabidopsis 3-ketoacyl-CoA synthases. Biochem. Bioph. Res. Co. 2006, 346, 583–590. [Google Scholar] [CrossRef]
- Hegebarth, D.; Buschhaus, C.; Joubès, J.; Thoraval, D.; Bird, D.; Jetter, R. Arabidopsis ketoacyl-CoA synthase 16 (KCS16) forms C36/C38 acyl precursors for leaf trichome and pavement surface wax. Plant Cell Environ. 2017, 40, 1761–1776. [Google Scholar] [CrossRef]
- Rizwan, H.M.; Shaozhong, F.; Li, X.; Arshad, M.B.; Yousef, A.F.; Chenglong, Y.; Shi, M.; Jaber, M.Y.M.; Anwar, M.; Hu, S.Y.; et al. Genome-Wide Identification and Expression Profiling of KCS Gene Family in Passion Fruit (Passiflora edulis) Under Fusarium kyushuense and Drought Stress Conditions. Front. Plant Sci. 2022, 13, 872263. [Google Scholar] [CrossRef]
- Sun, X.; Pang, H.; Li, M.; Peng, B.; Guo, H.; Yan, Q.; Yueyu, H. Evolutionary Pattern of the FAE1 Gene in Brassicaceae and Its Correlation with the Erucic Acid Trait. PLoS ONE 2013, 8, e83535. [Google Scholar] [CrossRef]
- Zhan, Z.; Jiang, Y.; Shah, N.; Hou, Z.; Zhou, Y.; Dun, B.; Li, S.; Zhu, L.; Li, Z.; Piao, Z.; et al. Association of Clubroot Resistance Locus PbBa8.1 With a Linkage Drag of High Erucic Acid Content in the Seed of the European Turnip. Front. Plant Sci. 2020, 11, 810. [Google Scholar] [CrossRef] [PubMed]
- James, D.W., Jr.; Lim, E.; Keller, J.; Plooy, I.; Ralston, E.; Dooner, H.K. Directed tagging of the Arabidopsis FATTY ACID ELONGATION1 (FAE1) gene with the maize transposon activator. Plant Cell. 1995, 7, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Ozseyhan, M.E.; Kang, J.; Mu, X.; Lu, C. Mutagenesis of the fae1 genes significantly changes fatty acid composition in seeds of Camelina sativa. Plant Physiol. Biochem. 2018, 123, 1–7. [Google Scholar] [CrossRef]
- Millar, A.A.; Clemens, S.; Zachgo, S.; Giblin, E.M.; Taylor, D.C.; Kunst, L. CUT1, an Arabidopsis gene required for cuticular wax biosynthesis and pollen fertility, encodes a very-long-chain fatty acid condensing enzyme. Plant Cell. 1999, 11, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Li, Y.; Liu, Y.; He, L.; Liu, A.; Wen, J.; Mysore, K.S.; Tadege, M.; Chen, J. The 3-ketoacyl-CoA synthase WFL is involved in lateral organ development and cuticular wax synthesis in Medicago truncatula. Plant Mol. Biol. 2021, 105, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Lolle, S.J.; Berlyn, G.P.; Engstrom, E.M.; Krolikowski, K.A.; Reiter, W.D.; Pruitt, R.E. Developmental regulation of cell interactions in the Arabidopsis fiddlehead-1 mutant: A role for the epidermal cell wall and cuticle. Dev. Biol. 1997, 189, 311–321. [Google Scholar] [CrossRef]
- Pruitt, R.E.; Vielle-Calzada, J.P.; Ploense, S.E.; Grossniklaus, U.; Lolle, S.J. Fiddlehead, a gene required to suppress epidermal cell interactions in arabidopsis, encodes a putative lipid biosynthetic enzyme. Proc. Natl. Acad. Sci. USA 2000, 97, 1311–1316. [Google Scholar] [CrossRef]
- Lu, J.; Xu, Y.; Wang, J.; Singer, S.D.; Chen, G. The Role of Triacylglycerol in Plant Stress Response. Plants 2020, 9, 472. [Google Scholar] [CrossRef]
- Lee, H.G.; Seo, P.J. Interaction of DGAT1 and PDAT1 to enhance TAG assembly in Arabidopsis. Plant Signal Behav. 2019, 14, 1554467. [Google Scholar] [CrossRef]
- Wang, L.; Li, Q.; Xia, Q.; Shen, W.; Selvaraj, G.; Zou, J. On the Role of DGAT1 in Seed Glycerolipid Metabolic Network and Critical Stages of Plant Development in Arabidopsis. Lipids 2020, 55, 457–467. [Google Scholar] [CrossRef]
- Torabi, S.; Sukumaran, A.; Dhaubhadel, S.; Johnson, S.E.; La Fayette, P.; Parrott, W.A.; Rajcan, I.; Eskandari, M. Effects of type I Diacylglycerol O-acyltransferase (DGAT1) genes on soybean (Glycine max L.) seed composition. Sci. Rep. 2021, 11, 2556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Fan, J.; Taylor, D.C.; Ohlrogge, J.B. DGAT1 and PDAT1 acyltransferases have overlapping functions in Arabidopsis triacylglycerol biosynthesis and are essential for normal pollen and seed development. Plant Cell. 2009, 21, 3885–3901. [Google Scholar] [CrossRef] [PubMed]
- GB/T2906-1982; Method for Determination of Crude Fat of Seed in Oil Crops and Cereals. Bureau of Quality and Technical Supervision, the People’s Republic of China: Beijing, China, 1982. (In Chinese)
- Hartman, L.; Lago, R. Rapid preparation of fatty acid methyl esters. Lab. Pract. 1973, 22, 475–476. [Google Scholar] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2008, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg S, L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Yang, H.; Liu, J.; Huang, S.; Guo, T.; Deng, L.; Hua, W. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene 2014, 538, 113–122. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, 45. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhao, D.; Li, X.; Zhou, B.; Chang, T.; Hong, B.; Guan, C.; Guan, M. Integrated Analysis of lncRNA–mRNA Regulatory Networks Related to Lipid Metabolism in High-Oleic-Acid Rapeseed. Int. J. Mol. Sci. 2023, 24, 6277. https://doi.org/10.3390/ijms24076277
Wang X, Zhao D, Li X, Zhou B, Chang T, Hong B, Guan C, Guan M. Integrated Analysis of lncRNA–mRNA Regulatory Networks Related to Lipid Metabolism in High-Oleic-Acid Rapeseed. International Journal of Molecular Sciences. 2023; 24(7):6277. https://doi.org/10.3390/ijms24076277
Chicago/Turabian StyleWang, Xiaodan, Dongfang Zhao, Xi Li, Bingqian Zhou, Tao Chang, Bo Hong, Chunyun Guan, and Mei Guan. 2023. "Integrated Analysis of lncRNA–mRNA Regulatory Networks Related to Lipid Metabolism in High-Oleic-Acid Rapeseed" International Journal of Molecular Sciences 24, no. 7: 6277. https://doi.org/10.3390/ijms24076277
APA StyleWang, X., Zhao, D., Li, X., Zhou, B., Chang, T., Hong, B., Guan, C., & Guan, M. (2023). Integrated Analysis of lncRNA–mRNA Regulatory Networks Related to Lipid Metabolism in High-Oleic-Acid Rapeseed. International Journal of Molecular Sciences, 24(7), 6277. https://doi.org/10.3390/ijms24076277