

Hydrogel-Inducing Graphene-Oxide-Derived Core–Shell Fiber Composite for Antibacterial Wound Dressing

 ,
,  , , ,
, , , 
 ,
,  ,
, 

Abstract
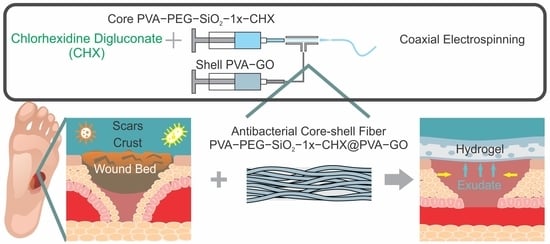
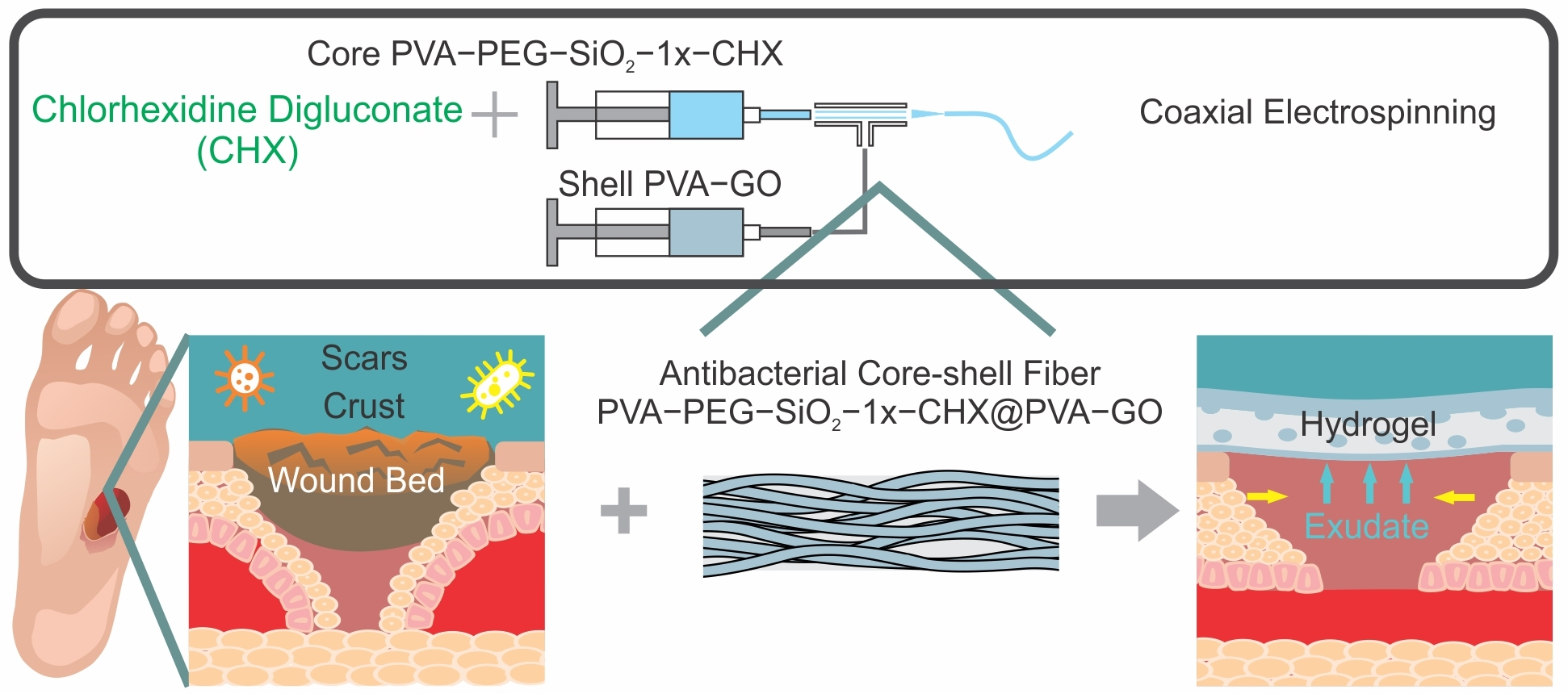
1. Introduction
2. Results and Discussion
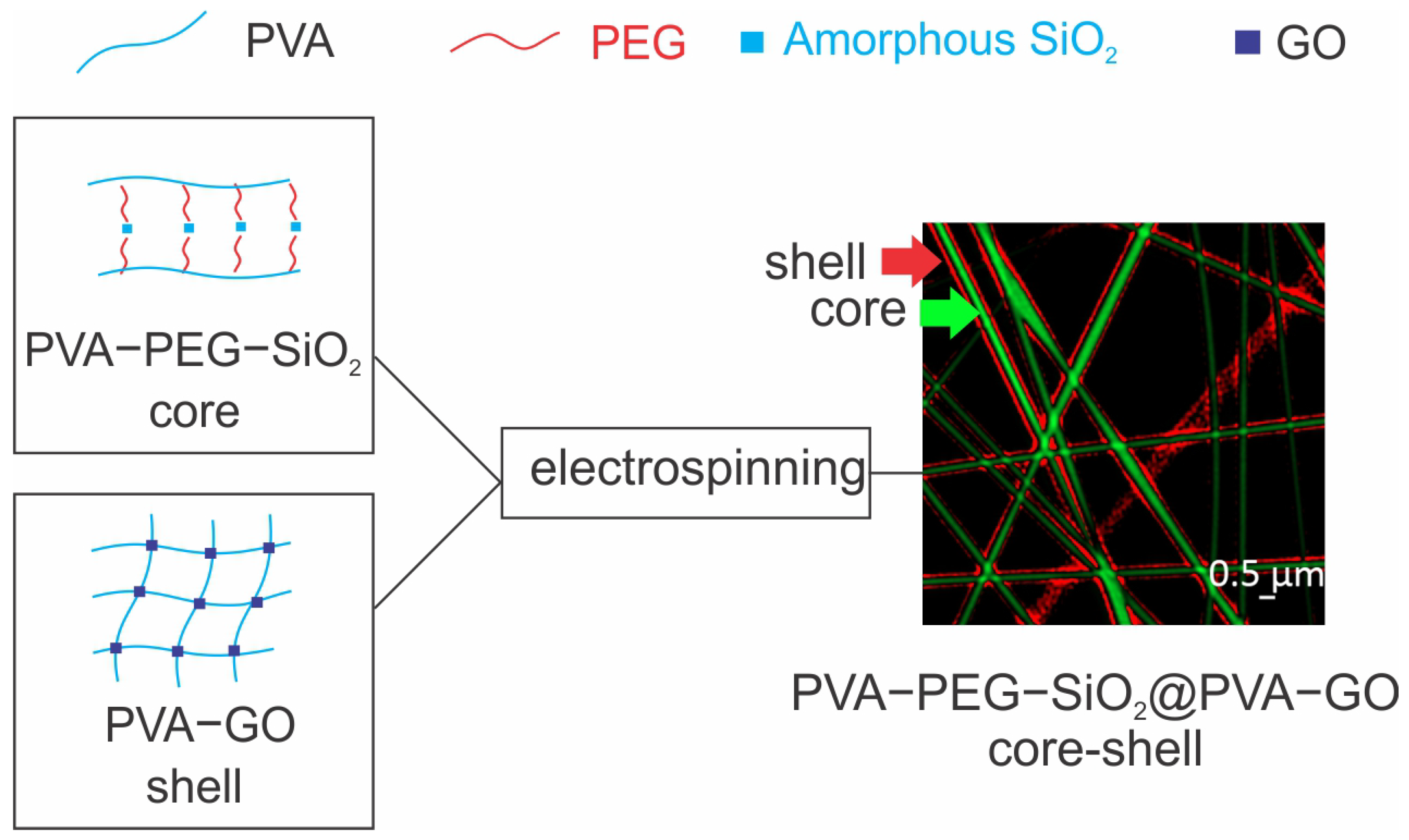
2.1. Analysis of Core–Shell Fiber PVA-PEG-SiO2@PVA-GO
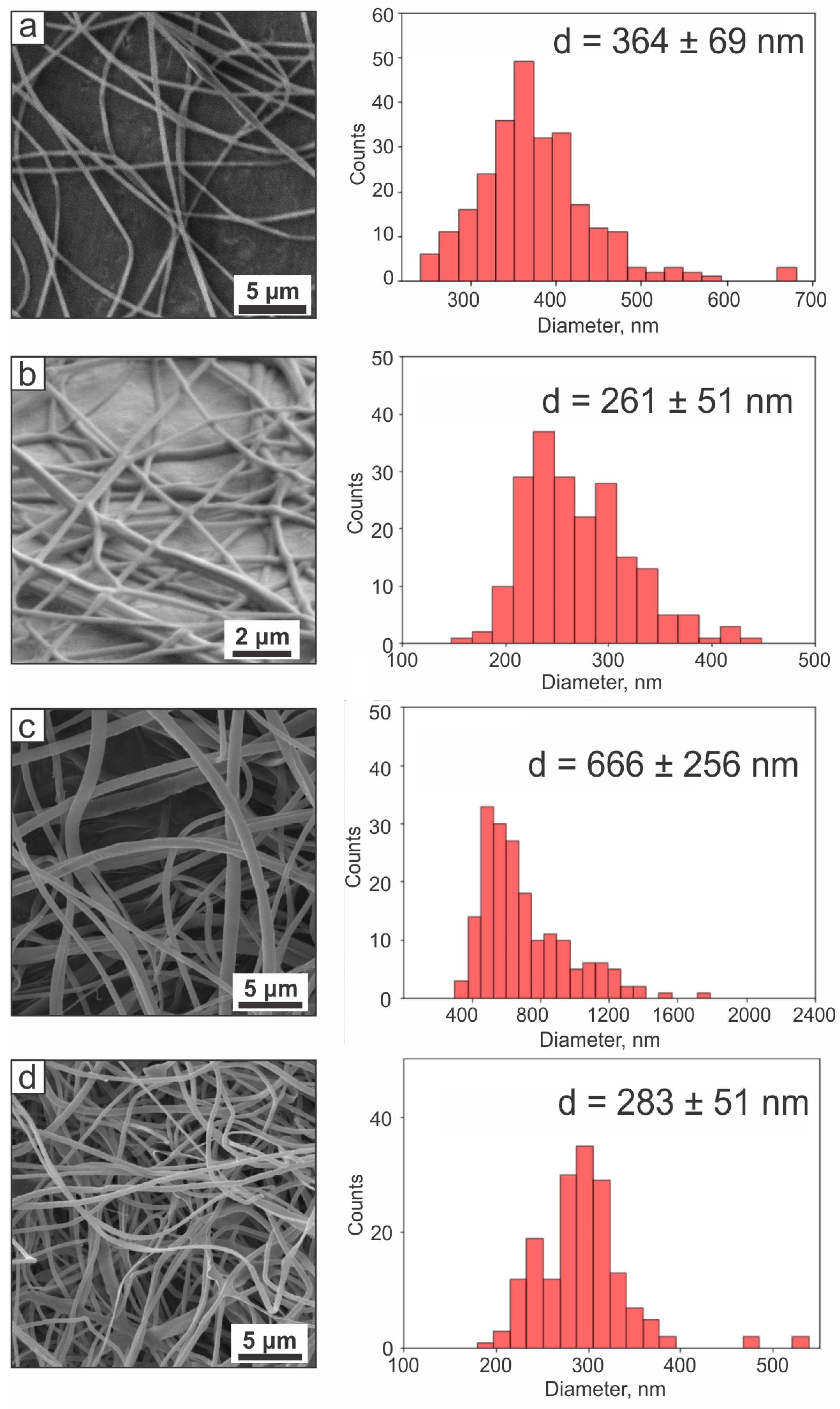
2.1.1. Fiber Morphology
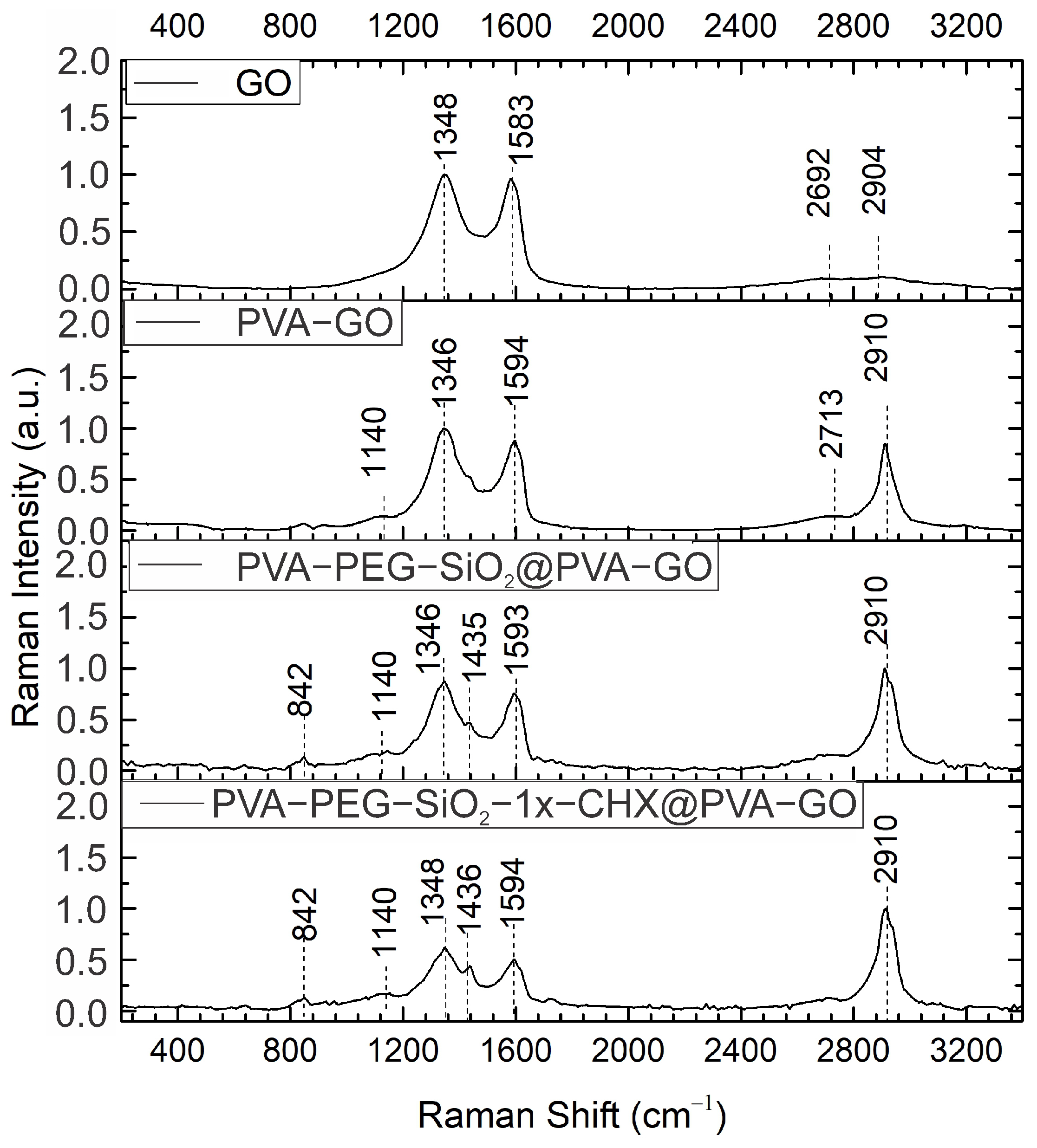
2.1.2. Raman Spectroscopy
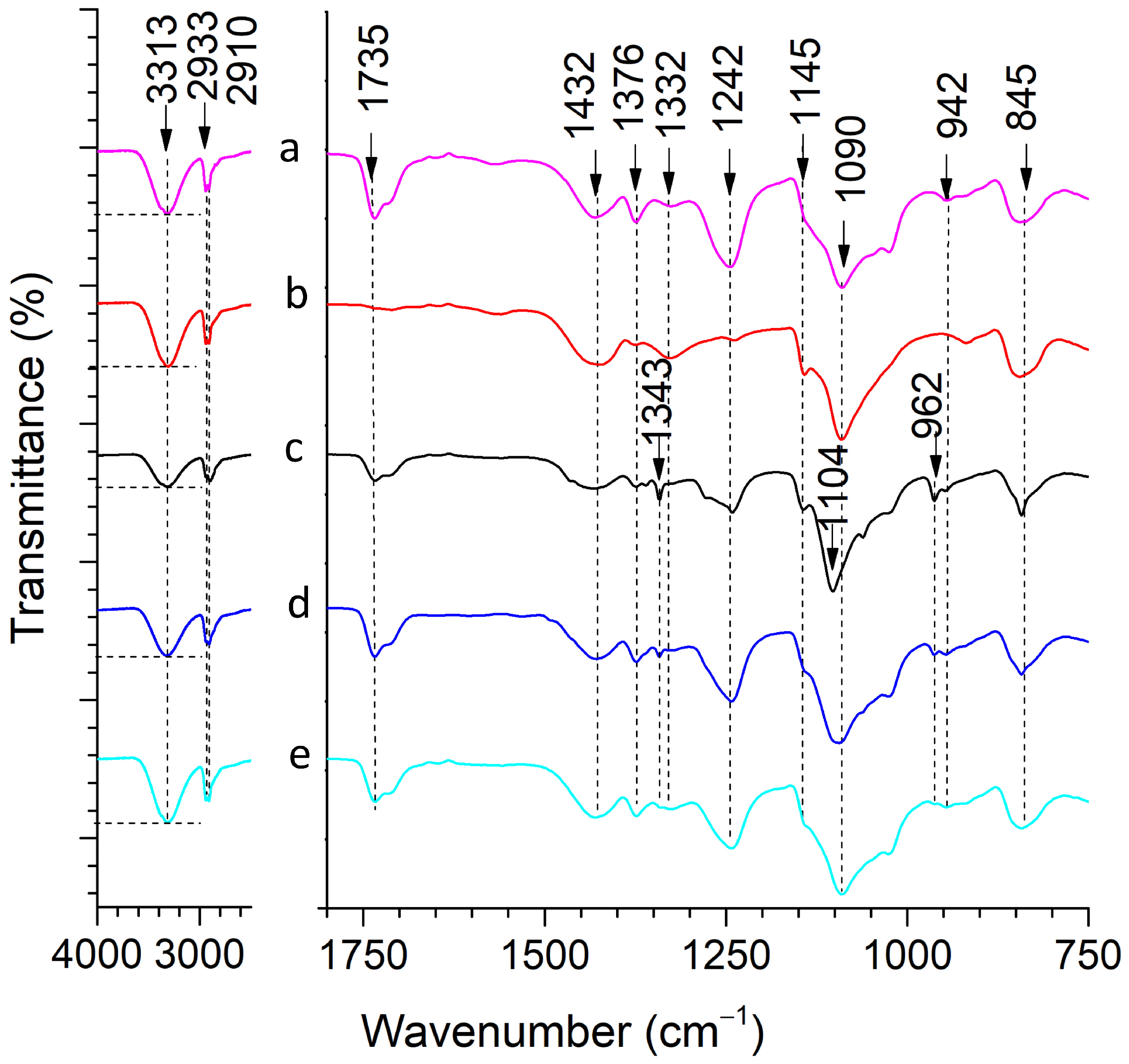
2.1.3. Fourier Transform Infrared (FTIR) Spectroscopy
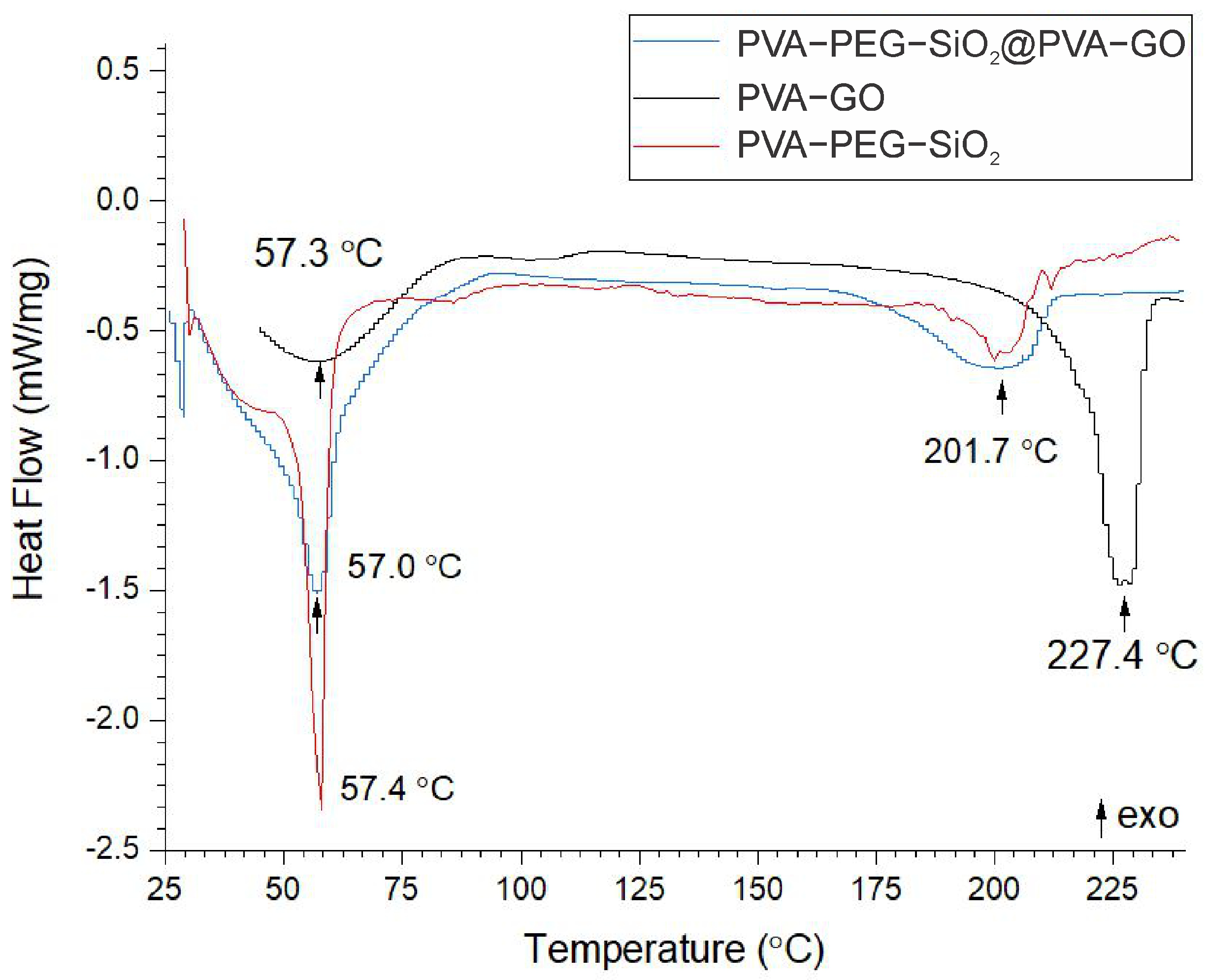
2.1.4. Thermal Analysis
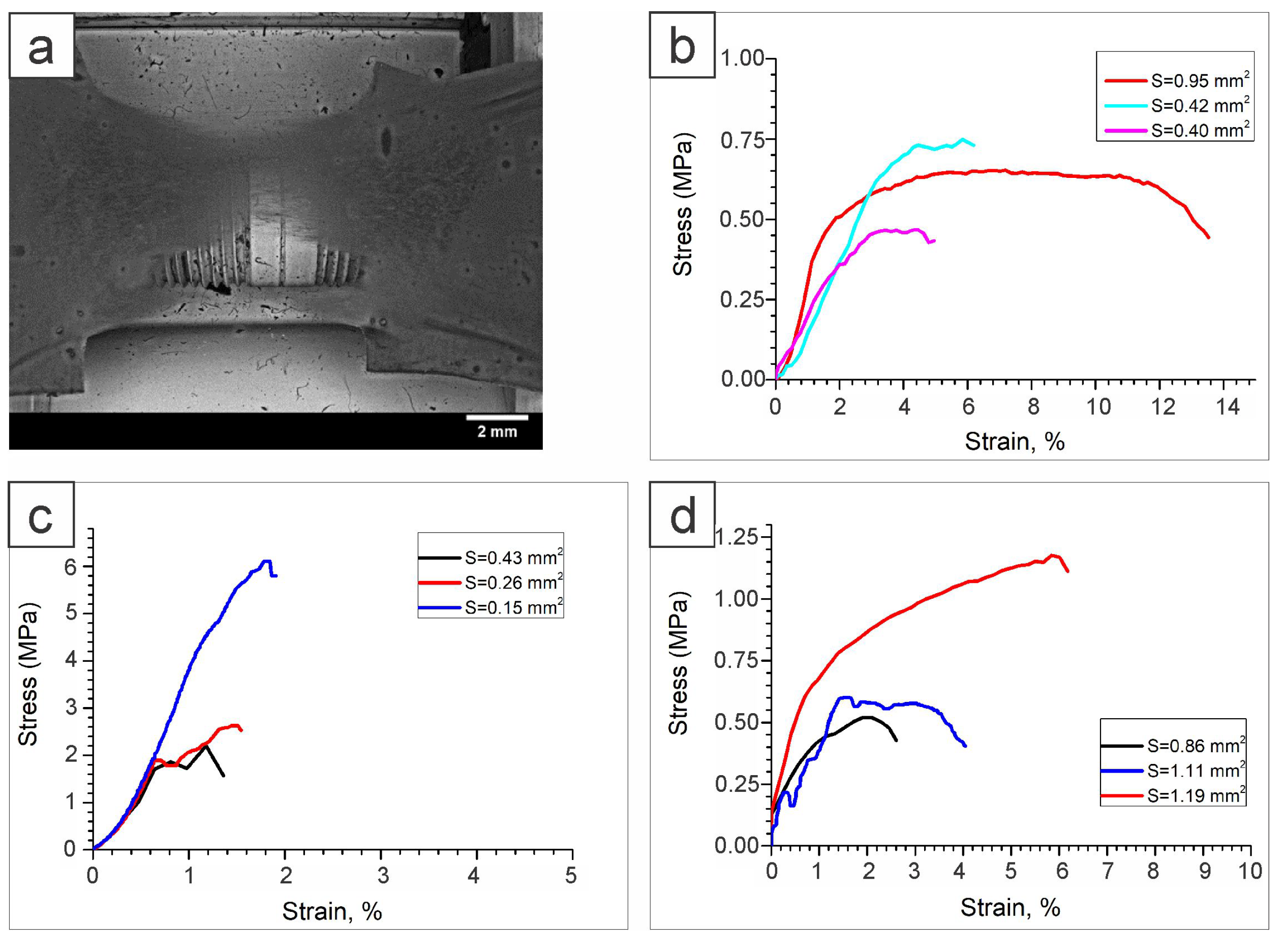
2.1.5. Mechanical Analysis
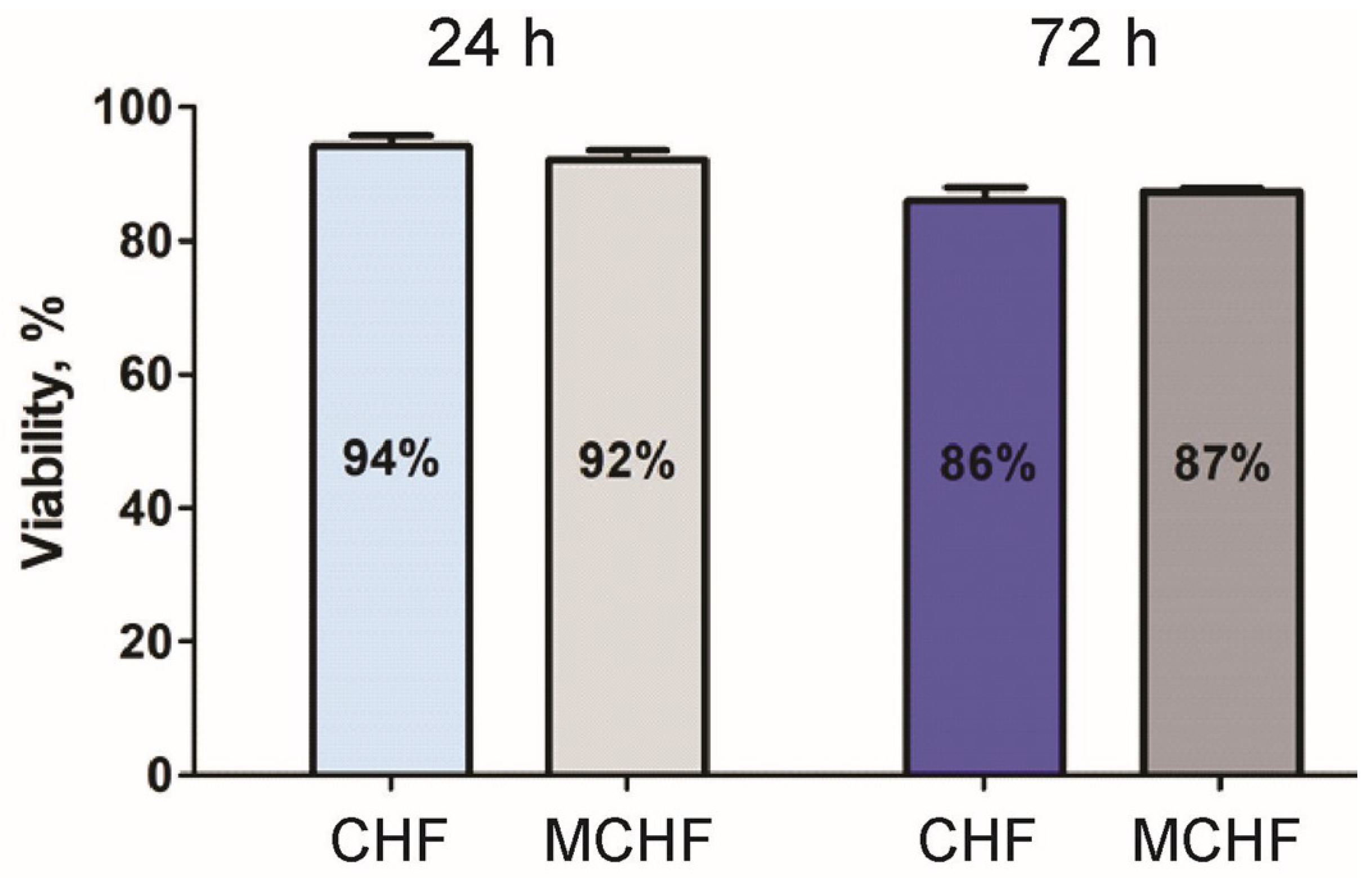
2.1.6. Biocompatibility Analysis
2.2. Analysis of Fibers Encapsulated with CHX PVA-PEG-SiO2-1x-CHX@PVA-GO
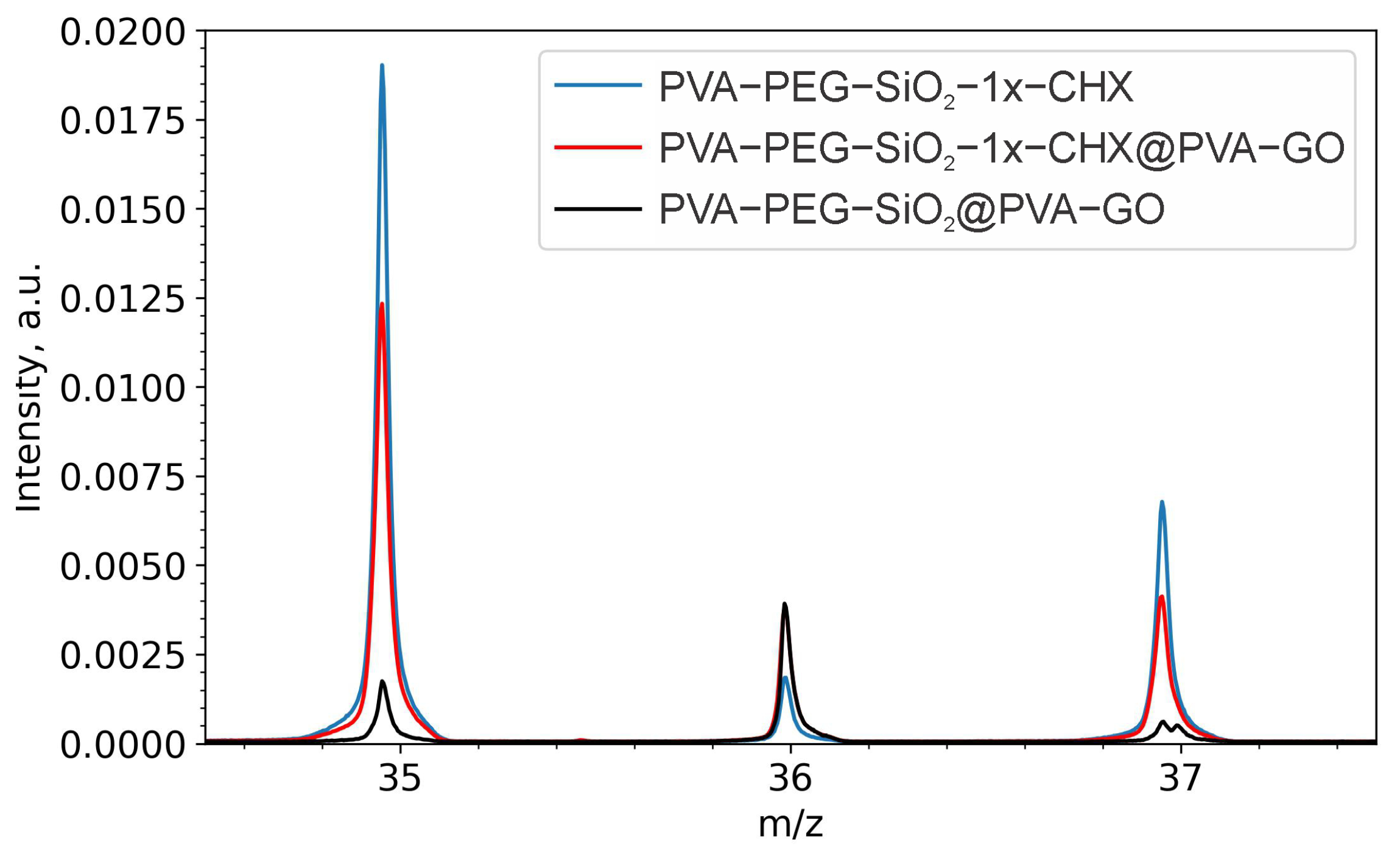
2.2.1. Drug Confirmation
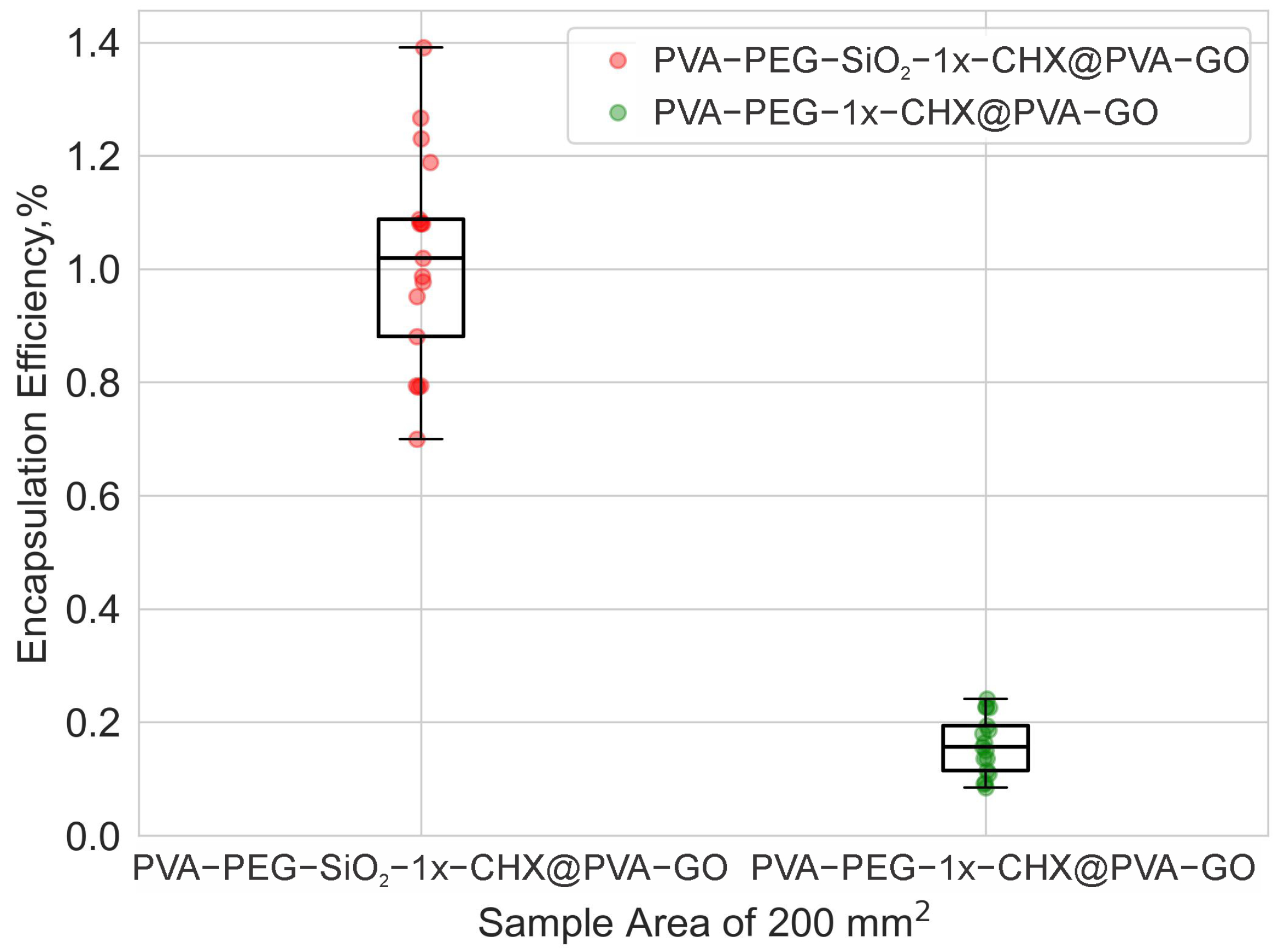
2.2.2. Drug Encapsulation Efficiency
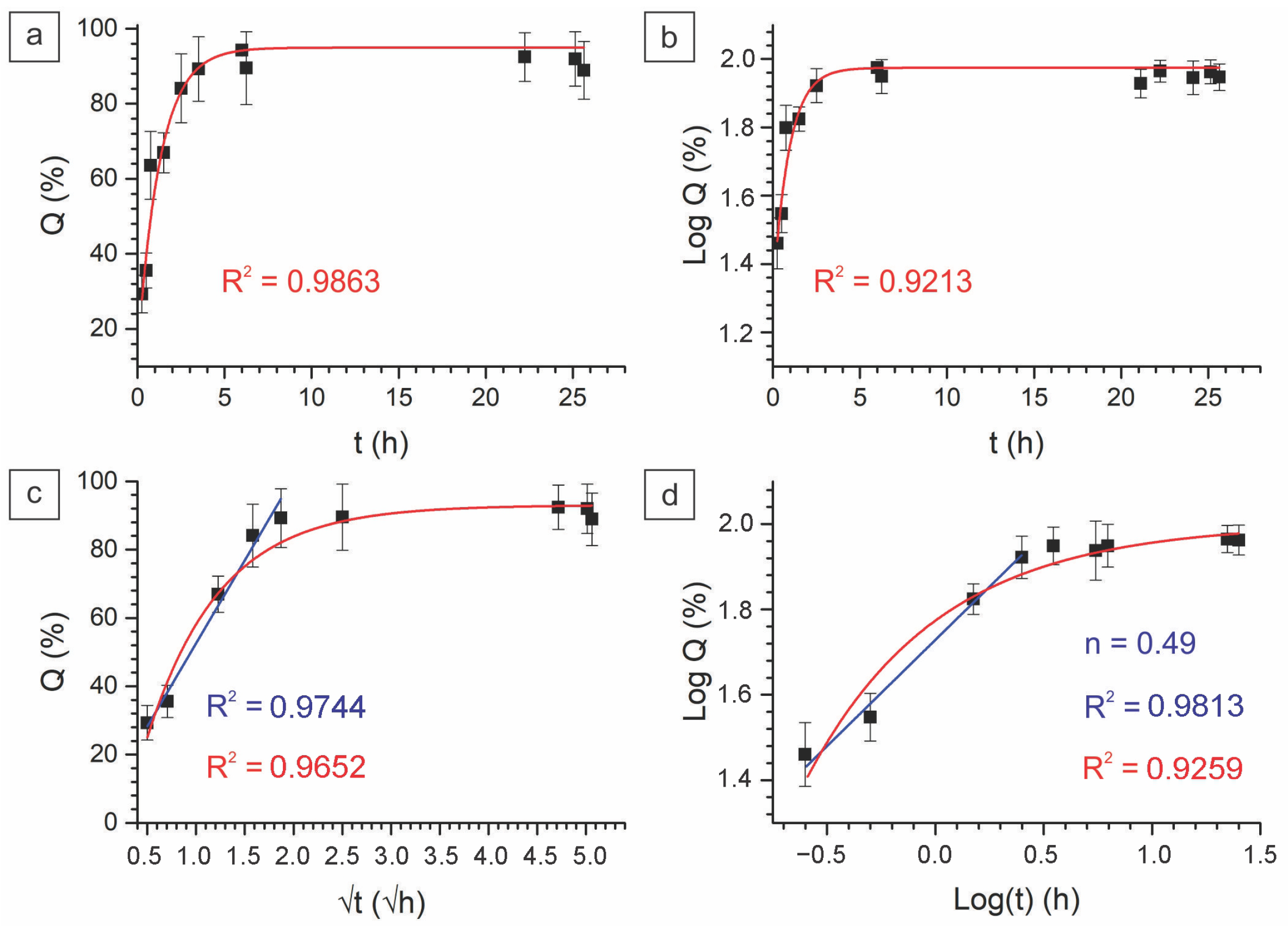
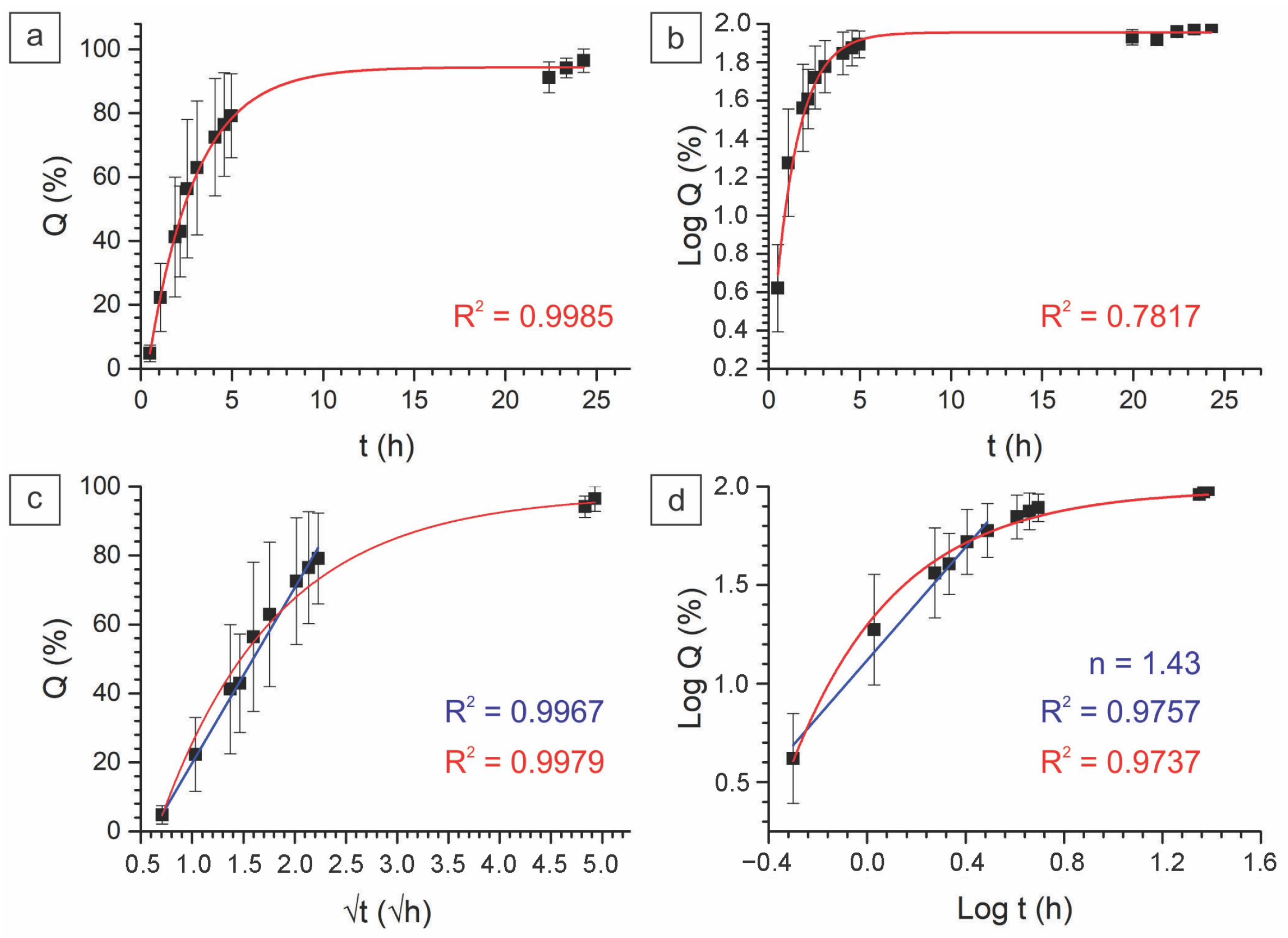
2.2.3. In Vitro Drug Kinetics
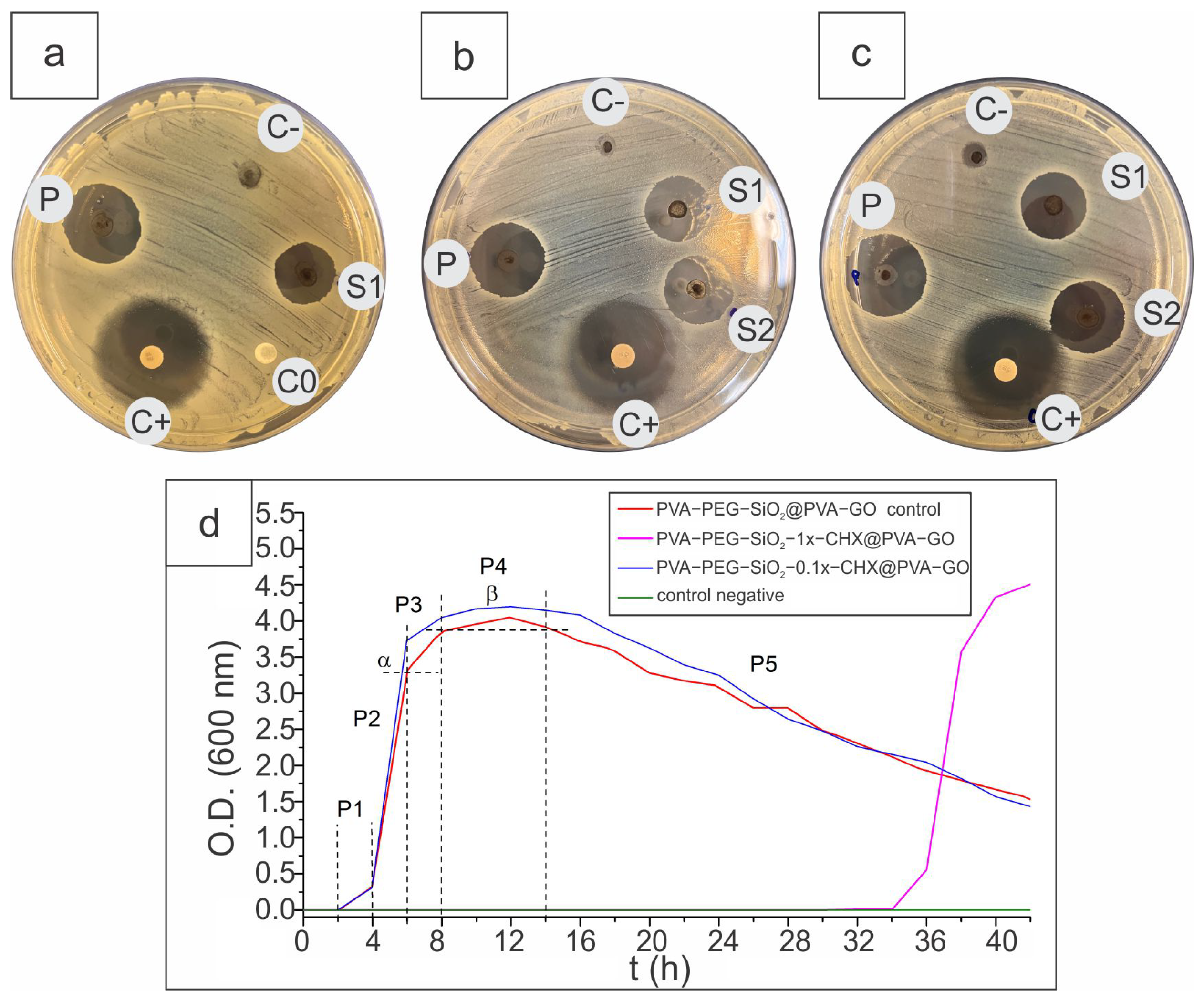
2.2.4. Antibacterial Activity
3. Materials and Methods
3.1. Materials
3.2. Fiber Fabrication
3.3. Raman and FTIR Spectroscopy
3.4. DSC
3.5. Tensile Tests
3.6. ToF-SIMS
3.7. UV Spectroscopy
3.8. Cell Viability Assay
3.9. Disk Diffusion Assay
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Thomas, R.E.; Thomas, B.C. Reducing Biofilm Infections in Burn Patients’ Wounds and Biofilms on Surfaces in Hospitals, Medical Facilities and Medical Equipment to Improve Burn Care: A Systematic Review. Int. J. Environ. Res. Public Health 2021, 18, 13195. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cheng, R.; Zhao, X.; Zhang, Y.; Tam, A.; Yan, Y.; Shen, H.; Zhang, Y.S.; Qi, J.; Feng, Y.; et al. An Injectable self-Healing Coordinative Hydrogel with Antibacterial and Angiogenic Properties for Diabetic Skin Wound Repair. NPG Asia Mater. 2019, 11, 3. [Google Scholar] [CrossRef]
- Lipsky, B.A.; Senneville, É.; Abbas, Z.G.; Aragón-Sánchez, J.; Diggle, M.; Embil, J.M.; Kono, S.; Lavery, L.A.; Malone, M.; van Asten, S.A.; et al. Guidelines on the Diagnosis and Treatment of Foot Infection in Persons with Diabetes (IWGDF 2019 Update). Diabetes/Metab. Res. Rev. 2020, 36, e3280. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.H.; Cho, Y.B.; Jang, Y.S.; Kim, M.S.; Kim, G.H. Antibacterial Effect of Electrospun Polycaprolactone/Polyethylene Oxide/Vancomycin Nanofiber Mat for Prevention of Periprosthetic Infection and Biofilm Formation. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1299–1305. [Google Scholar] [CrossRef]
- Teixeira, M.O.; Antunes, J.C.; Felgueiras, H.P. Recent Advances in Fiber–Hydrogel Composites for Wound Healing and Drug Delivery Systems. Antibiotics 2021, 10, 248. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Wang, Y.; Cui, W. Advanced Electrospun Hydrogel Fibers for Wound Healing. Compos. Part B Eng. 2021, 223, 109101. [Google Scholar] [CrossRef]
- Razali, N.A.M.; Lin, W.C. Accelerating the Excisional Wound Closure by Using the Patterned Microstructural Nanofibrous Mats/Gentamicin-loaded Hydrogel Composite Scaffold. Mater. Today Bio 2022, 16, 100347. [Google Scholar] [CrossRef]
- Ahmadian, Z.; Correia, A.; Hasany, M.; Figueiredo, P.; Dobakhti, F.; Eskandari, M.R.; Hosseini, S.H.; Abiri, R.; Khorshid, S.; Hirvonen, J.; et al. A Hydrogen-bonded Extracellular Matrix-mimicking Bactericidal Hydrogel with Radical Scavenging and Hemostatic Function for pH-Responsive Wound Healing Acceleration. Adv. Healthc. Mater. 2021, 10, 2001122. [Google Scholar] [CrossRef]
- Liang, Y.; He, J.; Guo, B. Functional Hydrogels as Wound Dressing to Enhance Wound Healing. ACS Nano 2021, 15, 12687–12722. [Google Scholar] [CrossRef]
- Mirzaeei, S.; Taghe, S.; Asare-Addo, K.; Nokhodchi, A. Polyvinyl Alcohol/Chitosan Single-Layered and Polyvinyl Alcohol/Chitosan/Eudragit RL100 Multi-layered Electrospun Nanofibers as an Ocular Matrix for the Controlled Release of Ofloxacin: An In Vitro and In Vivo Evaluation. AAPS PharmSciTech 2021, 22, 170. [Google Scholar] [CrossRef]
- Kimmins, S.D.; Hanay, S.B.; Murphy, R.; O’Dwyer, J.; Ramalho, J.; Ryan, E.J.; Kearney, C.J.; O’Brien, F.J.; Cryan, S.A.; Fitzgerald-Hughes, D.; et al. Antimicrobial and Degradable Triazolinedione (TAD) Crosslinked Polypeptide Hydrogels. J. Mater. Chem. B 2021, 9, 5456–5464. [Google Scholar] [CrossRef]
- Ahmed, A.S.; Mandal, U.K.; Taher, M.; Susanti, D.; Jaffri, J.M. PVA-PEG Physically Cross-linked Hydrogel Film as a Wound Dressing: Experimental Design and Optimization. Pharm. Dev. Technol. 2018, 23, 751–760. [Google Scholar] [CrossRef]
- Uchino, Y.; Shimmura, S.; Miyashita, H.; Taguchi, T.; Kobayashi, H.; Shimazaki, J.; Tanaka, J.; Tsubota, K. Amniotic Membrane Immobilized Poly (vinyl alcohol) Hybrid Polymer as an Artificial Cornea Scaffold that Supports a Stratified and Differentiated Corneal Epithelium. J. Biomed. Mater. Res. Part B Appl. Biomater. Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2007, 81, 201–206. [Google Scholar] [CrossRef]
- Hu, Y.; Yan, P.; Fu, L.; Zheng, Y.; Kong, W.; Wu, H.; Yu, X. Polyvinyl Alcohol/Polyvinyl Pyrrolidone Crosslinked Hydrogels Induce Wound Healing through Cell Proliferation and Migration. J. Biomater. Tissue Eng. 2017, 7, 310–318. [Google Scholar] [CrossRef]
- Horiguchi, S.; Adachi, T.; Rondinella, A.; Boschetto, F.; Marin, E.; Zhu, W.; Tahara, Y.; Yamamoto, T.; Kanamura, N.; Akiyoshi, K.; et al. Osteogenic Response of Mesenchymal Progenitor Cells to Natural Polysaccharide Nanogel and Atelocollagen Scaffolds: A Spectroscopic Study. Mater. Sci. Eng. C 2019, 99, 1325–1340. [Google Scholar] [CrossRef]
- Qi, X.; Huang, Y.; You, S.; Xiang, Y.; Cai, E.; Mao, R.; Pan, W.; Tong, X.; Dong, W.; Ye, F.; et al. Engineering Robust Ag-Decorated Polydopamine Nano-Photothermal Platforms to Combat Bacterial Infection and Prompt Wound Healing. Adv. Sci. 2022, 9, 2106015. [Google Scholar] [CrossRef]
- Wang, M.; Hou, J.; Yu, D.G.; Li, S.; Zhu, J.; Chen, Z. Electrospun Tri-layer Nanodepots for Sustained Release of Acyclovir. J. Alloys Compd. 2020, 846, 156471. [Google Scholar] [CrossRef]
- Elyaderani, A.K.; Lama-Odría, D.; del Carmen, M.; Valle, L.J.d.; Puiggalí, J. Multifunctional Scaffolds Based on Emulsion and Coaxial Electrospinning Incorporation of Hydroxyapatite for Bone Tissue Regeneration. Int. J. Mol. Sci. 2022, 23, 15016. [Google Scholar] [CrossRef]
- Darbasizadeh, B.; Fatahi, Y.; Feyzi-Barnaji, B.; Arabi, M.; Motasadizadeh, H.; Farhadnejad, H.; Moraffah, F.; Rabiee, N. Crosslinked-Polyvinyl Alcohol-Carboxymethyl Cellulose/ZnO Nanocomposite Fibrous Mats Containing Erythromycin (PVA-CMC/ZnO-EM): Fabrication, Characterization and In-Vitro Release and Anti-Bacterial Properties. Int. J. Biol. Macromol. 2019, 141, 1137–1146. [Google Scholar] [CrossRef]
- Fan, Z.; Guan, J. Antifibrotic Therapies to Control Cardiac Fibrosis. Biomater. Res. 2016, 20, 13. [Google Scholar] [CrossRef]
- Morimune, S.; Kotera, M.; Nishino, T.; Goto, K.; Hata, K. Poly (vinyl Alcohol) Nanocomposites with Nanodiamond. Macromolecules 2011, 44, 4415–4421. [Google Scholar] [CrossRef]
- Jia, R.J.; Teng, K.Y.; Huang, J.Y.; Wei, X.; Qin, Z.Y. Hydrogen Bonding Crosslinking of Starch-Polyvinyl Alcohol Films Reinforced by Ultrasound-Assisted and Cellulose Nanofibers Dispersed Cellulose Nanocrystals. Starch-Stärke 2022, 74, 2100227. [Google Scholar] [CrossRef]
- Zhang, P.; Guo, W.; Guo, Z.H.; Ma, Y.; Gao, L.; Cong, Z.; Zhao, X.J.; Qiao, L.; Pu, X.; Wang, Z.L. Dynamically Crosslinked Dry Ion-Conducting Elastomers for Soft Iontronics. Adv. Mater. 2021, 33, 2101396. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Cui, K.; Xu, F.; Jiang, T.; Ma, Z. Synthesis of New Ionic Crosslinked Polymer Hydrogel Combining Polystyrene and Poly (4-vinyl Pyridine) and Its Self-Healing through a Reshuffling Reaction of the Trithiocarbonate Moiety under Irradiation of Ultraviolet Light. Polym. Int. 2018, 67, 868–873. [Google Scholar] [CrossRef]
- Cheng, S.; Wang, H.; Pan, X.; Zhang, C.; Zhang, K.; Chen, Z.; Dong, W.; Xie, A.; Qi, X. Dendritic Hydrogels with Robust Inherent Antibacterial Properties for Promoting Bacteria-infected Wound Healing. ACS Appl. Mater. Interfaces 2022, 14, 11144–11155. [Google Scholar] [CrossRef]
- Bardonová, L.; Mamulová Kutláková, K.; Kotzianová, A.; Kulhánek, J.; Zidek, O.; Velebný, V.; Tokarský, J. Electrospinning of Fibrous Layers Containing an Antibacterial Chlorhexidine/Kaolinite Composite. ACS Appl. Bio Mater. 2020, 3, 3028–3038. [Google Scholar] [CrossRef]
- Massarelli, E.; Silva, D.; Pimenta, A.; Fernandes, A.; Mata, J.; Armês, H.; Salema-Oom, M.; Saramago, B.; Serro, A. Polyvinyl Alcohol/Chitosan Wound Dressings Loaded with Antiseptics. Int. J. Pharm. 2021, 593, 120110. [Google Scholar] [CrossRef]
- Pilloni, A.; Ceccarelli, S.; Bosco, D.; Gerini, G.; Marchese, C.; Marini, L.; Rojas, M.A. Effect of Chlorhexidine Digluconate in Early Wound Healing of Human Gingival Tissues. A Histological, Immunohistochemical and Biomolecular Analysis. Antibiotics 2021, 10, 1192. [Google Scholar] [CrossRef]
- Abdel-Sayed, P.; Tornay, D.; Hirt-Burri, N.; de Buys Roessingh, A.; Raffoul, W.; Applegate, L.A. Implications of Chlorhexidine Use in Burn Units for Wound Healing. Burns 2020, 46, 1150–1156. [Google Scholar] [CrossRef]
- Kan, Y.; Bondareva, J.V.; Statnik, E.S.; Cvjetinovic, J.; Lipovskikh, S.; Abdurashitov, A.S.; Kirsanova, M.A.; Sukhorukhov, G.B.; Evlashin, S.A.; Salimon, A.I.; et al. Effect of Graphene Oxide and Nanosilica Modifications on Electrospun Core-Shell PVA–PEG–SiO2@ PVA–GO Fiber Mats. Nanomaterials 2022, 12, 998. [Google Scholar] [CrossRef]
- James, P.; Worthington, H.V.; Parnell, C.; Harding, M.; Lamont, T.; Cheung, A.; Whelton, H.; Riley, P. Chlorhexidine Mouthrinse as an Adjunctive Treatment for Gingival Health. Cochrane Database Syst. Rev. 2017. [Google Scholar] [CrossRef]
- Repanas, A.; Wolkers, W.; Gryshkov, O.; Müller, M.; Glasmacher, B. PCL/PEG Electrospun Fibers as Drug Carriers for the Controlled Delivery of Dipyridamole. Silico In Vitro Pharm. 2015, 1, 1–10. [Google Scholar]
- Zubair, N.A.; Rahman, N.A.; Lim, H.N.; Zawawi, R.M.; Sulaiman, Y. Electrochemical Properties of PVA–GO/PEDOT Nanofibers Prepared Using Electrospinning and Electropolymerization Techniques. RSC Adv. 2016, 6, 17720–17727. [Google Scholar] [CrossRef]
- Sagitova, E.; Prokhorov, K.; Nikolaeva, G.Y.; Baimova, A.; Pashinin, P.; Yarysheva, A.Y.; Mendeleev, D. Raman Analysis of Polyethylene Glycols and Polyethylene Oxides. Proc. J. Phys. Conf. Ser. 2018, 999, 012002. [Google Scholar] [CrossRef]
- Pawar, P.B.; Shukla, S.; Saxena, S. Graphene Oxide–Polyvinyl Alcohol Nanocomposite Based Electrode Material for Dupercapacitors. J. Power Sour. 2016, 321, 102–105. [Google Scholar] [CrossRef]
- Somesh, T.; Demappa, T. Tailoring of Ternary Nanocomposite Films of Poly (vinyl Alcohol)/AgAlO2@ Reduced Graphene Oxide: An Active Material for Flexible Supercapacitors. J. Solid State Chem. 2022, 309, 122824. [Google Scholar] [CrossRef]
- Wadhwa, H.; Kandhol, G.; Deshpande, U.P.; Mahendia, S.; Kumar, S. Thermal Stability and Dielectric Relaxation Behavior of In Situ Prepared Poly (vinyl Alcohol)(PVA)-reduced Graphene Oxide (RGO) Composites. Colloid Polym. Sci. 2020, 298, 1319–1333. [Google Scholar] [CrossRef]
- Kim, S.G.; Park, O.K.; Lee, J.H.; Ku, B.C. Layer-by-layer Assembled Graphene Oxide Films and Barrier Properties of Thermally Reduced Graphene Oxide Membranes. Carbon Lett. 2013, 14, 247–250. [Google Scholar] [CrossRef]
- Khabibullin, A.; Alizadehgiashi, M.; Khuu, N.; Prince, E.; Tebbe, M.; Kumacheva, E. Injectable Shear-Thinning Fluorescent Hydrogel Formed by Cellulose Nanocrystals and Graphene Quantum Dots. Langmuir 2017, 33, 12344–12350. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Q.; Liu, X.; Xia, S.; Gao, Y.; Gao, G. Flexible and Wearable Strain Sensors Based on Conductive Hydrogels. J. Polym. Sci. 2022, 60, 2663–2678. [Google Scholar] [CrossRef]
- Zidan, H.M.; Abdelrazek, E.M.; Abdelghany, A.M.; Tarabiah, A.E. Characterization and Some Physical Studies of PVA/PVP Filled with MWCNTs. J. Mater. Res. Technol. 2019, 8, 904–913. [Google Scholar] [CrossRef]
- Kharazmi, A.; Faraji, N.; Hussin, R.M.; Saion, E.; Yunus, W.M.M.; Behzad, K. Structural, Optical, Opto-thermal and Thermal Properties of ZnS–PVA Nanofluids Synthesized through a Radiolytic Approach. Beilstein J. Nanotechnol. 2015, 6, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Ramirez, D.O.; Cruz-Maya, I.; Vineis, C.; Tonetti, C.; Varesano, A.; Guarino, V. Design of Asymmetric Nanofibers-Membranes Based on Polyvinyl Alcohol and Wool-Keratin for Wound Healing Applications. J. Funct. Biomater. 2021, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Tretinnikov, O.; Zagorskaya, S. Determination of the Degree of Crystallinity of Poly (vinyl Alcohol) by FTIR Spectroscopy. J. Appl. Spectrosc. 2012, 79, 521–526. [Google Scholar] [CrossRef]
- Halima, N.B. Poly (vinyl Alcohol): Review of its Promising Applications and Insights into Biodegradation. RSC Adv. 2016, 6, 39823–39832. [Google Scholar] [CrossRef]
- Jessie Lue, S.; Chen, J.Y.; Ming Yang, J. Crystallinity and Stability of Poly (vinyl Alcohol)-Fumed Silica Mixed Matrix Membranes. J. Macromol. Sci. Part B 2007, 47, 39–51. [Google Scholar] [CrossRef]
- Abdelaziz, M.; Abdelrazek, E. Effect of Dopant Mixture on Structural, Optical and Electron Spin Resonance Properties of Polyvinyl Alcohol. Phys. B Condens. Matter 2007, 390, 1–9. [Google Scholar] [CrossRef]
- Alghunaim, N.S. Optimization and Spectroscopic Studies on Carbon Nanotubes/PVA Nanocomposites. Results Phys. 2016, 6, 456–460. [Google Scholar] [CrossRef]
- Budi, H.S.; Ansari, M.J.; Jasim, S.A.; Abdelbasset, W.K.; Bokov, D.; Mustafa, Y.F.; Najm, M.A.; Kazemnejadi, M. Preparation of Antibacterial Gel/PCL Nanofibers Reinforced by Dicalcium Phosphate-modified Graphene Oxide with Control Release of Clindamycin for Possible Application in Bone Tissue Engineering. Inorg. Chem. Commun. 2022, 139, 109336. [Google Scholar] [CrossRef]
- Luo, Q.; Shan, Y.; Zuo, X.; Liu, J. Anisotropic Tough Poly (vinyl Alcohol)/Graphene Oxide Nanocomposite Hydrogels for Potential Biomedical Applications. RSC Adv. 2018, 8, 13284–13291. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, Y.; Wang, Z.; Zhang, X.; Zhao, Y.; Sun, L. Electrospinning of Ag Nanowires/Polyvinyl Alcohol Hybrid Nanofibers for their Antibacterial Properties. Mater. Sci. Eng. C 2017, 78, 706–714. [Google Scholar] [CrossRef]
- Bondareva, J.V.; Aslyamov, T.F.; Kvashnin, A.G.; Dyakonov, P.V.; Kuzminova, Y.O.; Mankelevich, Y.A.; Voronina, E.N.; Dagesyan, S.A.; Egorov, A.V.; Khmelnitsky, R.A.; et al. Environmentally Friendly Method of Silicon Recycling: Synthesis of Silica Nanoparticles in an Aqueous Solution. ACS Sustain. Chem. Eng. 2020, 8, 14006–14012. [Google Scholar] [CrossRef]
- Feifel, S.C.; Lisdat, F. Silica Nanoparticles for the Layer-by-Layer Assembly of Fully Electro-active Cytochrome c Multilayers. J. Nanobiotechnol. 2011, 9, 1–12. [Google Scholar] [CrossRef]
- Alharbi, H.F.; Luqman, M.; Fouad, H.; Khalil, K.A.; Alharthi, N.H. Viscoelastic Behavior of Core-shell Structured Nanofibers of PLA and PVA Produced by Coaxial Electrospinning. Polym. Test. 2018, 67, 136–143. [Google Scholar] [CrossRef]
- Flores-Arriaga, J.C.; Chavarría-Bolaños, D.; Pozos-Guillén, A.d.J.; Escobar-Barrios, V.A.; Cerda-Cristerna, B.I. Synthesis of a PVA Drug Delivery System for Controlled Release of a Tramadol–Dexketoprofen Combination. J. Mater. Sci. Mater. Med. 2021, 32, 56. [Google Scholar] [CrossRef]
- Kołodziej, A.; Długoń, E.; Świętek, M.; Ziąbka, M.; Dawiec, E.; Gubernat, M.; Michalec, M.; Wesełucha-Birczyńska, A. A Raman Spectroscopic Analysis of Polymer Membranes with Graphene Oxide and Reduced Graphene Oxide. J. Compos. Sci. 2021, 5, 20. [Google Scholar] [CrossRef]
- Kovaleva, P.A.; Pariy, I.O.; Chernozem, R.V.; Zadorozhnyy, M.Y.; Permyakova, E.S.; Kolesnikov, E.A.; Surmeneva, M.A.; Surmenev, R.A.; Senatov, F.S. Shape Memory Effect in Hybrid Polylactide-based Polymer Scaffolds Functionalized with Reduced Graphene Oxide for Tissue Engineering. Eur. Polym. J. 2022, 181, 111694. [Google Scholar] [CrossRef]
- Chew, S.Y.; Hufnagel, T.C.; Lim, C.T.; Leong, K.W. Mechanical Properties of Single Electrospun Drug-Encapsulated Nanofibres. Nanotechnology 2006, 17, 3880. [Google Scholar] [CrossRef]
- Daelemans, L.; Verschatse, O.; Heirman, L.; Van Paepegem, W.; De Clerck, K. Toughening Mechanisms Responsible for Excellent Crack Resistance in Thermoplastic Nanofiber Reinforced Epoxies Through In-Situ Optical and Scanning Electron Microscopy. Compos. Sci. Technol. 2021, 201, 108504. [Google Scholar] [CrossRef]
- Fu, Z.Z.; Guo, S.J.; Li, C.X.; Wang, K.; Zhang, Q.; Fu, Q. Hydrogen-bond-dominated Mechanical Stretchability in PVA Films: From Phenomenological to Numerical Insights. Phys. Chem. Chem. Phys. 2022, 24, 1885–1895. [Google Scholar] [CrossRef]
- Duan, G.; Jin, M.; Wang, F.; Greiner, A.; Agarwal, S.; Jiang, S. Core Effect on Mechanical Properties of One Dimensional Electrospun Core-Sheath Composite Fibers. Compos. Commun. 2021, 25, 100773. [Google Scholar] [CrossRef]
- Miraftab, M.; Saifullah, A.N.; Çay, A. Physical Stabilisation of Electrospun Poly (vinyl Alcohol) Nanofibres: Comparative Study on Methanol and Heat-based Crosslinking. J. Mater. Sci. 2015, 50, 1943–1957. [Google Scholar] [CrossRef]
- Judd, A.M.; Scurr, D.J.; Heylings, J.R.; Wan, K.W.; Moss, G.P. Distribution and Visualisation of Chlorhexidine within the Skin Using ToF-SIMS: A Potential Platform for the Design of More Efficacious Skin Antiseptic Formulations. Pharm. Res. 2013, 30, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Quagliarini, E.; Di Santo, R.; Pozzi, D.; Tentori, P.; Cardarelli, F.; Caracciolo, G. Mechanistic Insights into the Release of Doxorubicin from Graphene Oxide in Cancer Cells. Nanomaterials 2020, 10, 1482. [Google Scholar] [CrossRef]
- Cui, Z.; Zheng, Z.; Lin, L.; Si, J.; Wang, Q.; Peng, X.; Chen, W. Electrospinning and Crosslinking of Polyvinyl Alcohol/Chitosan Composite Nanofiber for Transdermal Drug Delivery. Adv. Polym. Technol. 2018, 37, 1917–1928. [Google Scholar] [CrossRef]
- Barani, H.; Khorashadizadeh, M.; Haseloer, A.; Klein, A. Characterization and Release Behavior of a Thiosemicarbazone from Electrospun Polyvinyl Alcohol Core–shell Nanofibers. Polymers 2020, 12, 1488. [Google Scholar] [CrossRef]
- Mirzaie, Z.; Reisi-Vanani, A.; Barati, M.; Atyabi, S.M. The Drug Release Kinetics and Anticancer Activity of the GO/PVA-curcumin Nanostructures: The Effects of the Preparation Method and the GO Amount. J. Pharm. Sci. 2021, 110, 3715–3725. [Google Scholar] [CrossRef]
- Evlashin, S.A.; Svyakhovskiy, S.E.; Fedorov, F.S.; Mankelevich, Y.A.; Dyakonov, P.V.; Minaev, N.V.; Dagesyan, S.A.; Maslakov, K.I.; Khmelnitsky, R.A.; Suetin, N.V.; et al. Ambient Condition Production of High Quality Reduced Graphene Oxide. Adv. Mater. Interfaces 2018, 5, 1800737. [Google Scholar] [CrossRef]
- Guan, Y.; Li, W.; Zhang, Y.; Shi, Z.; Tan, J.; Wang, F.; Wang, Y. Aramid Nanofibers and Poly (vinyl Alcohol) Nanocomposites for Ideal Combination of Strength and Toughness via Hydrogen Bonding Interactions. Compos. Sci. Technol. 2017, 144, 193–201. [Google Scholar] [CrossRef]
- Blaber, J.; Adair, B.; Antoniou, A. Ncorr: Open-source 2D Digital Image Correlation Matlab Software. Exp. Mech. 2015, 55, 1105–1122. [Google Scholar] [CrossRef]
- Pan, W.; Qi, X.; Xiang, Y.; You, S.; Cai, E.; Gao, T.; Tong, X.; Hu, R.; Shen, J.; Deng, H. Facile Formation of Injectable Quaternized Chitosan/Tannic Acid Hydrogels with Antibacterial and ROS Scavenging Capabilities for Diabetic Wound Healing. Int. J. Biol. Macromol. 2022, 195, 190–197. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavenumber, cm−1 | Band | Reference |
|---|---|---|
| 3313 | –OH stretching (PVA) | [41] |
| 2933, 2910 | CH2 stretching (PVA) | [43] |
| 1735 | C=O stretching of acetate group (PVA) | [47] |
| 1432 | CH2 bending (PVA) | [43] |
| 1376 | (CH–OH) bending (PVA) | [41] |
| 1242 | CH wagging (PVA) | [48] |
| 1145 | C–O and C–C stretching bonds (PVA) | [46] |
| 1090 | C–O stretching of an acetyl group (PVA) | [44] |
| 942 | CH2 rocking (PVA) | [48] |
| 845 | CC stretching (PVA) | [43] |
| 1343 | CH3 bending (PEG) | [32] |
| Name | Tm, °C | Tg, °C | Heat of Melting (), J/g | Degree of Crystallinity, % |
|---|---|---|---|---|
| Shell | 227.4 | 57.3 | 83.02 | 59.90 |
| Core | 201.7 | 57.0 | 24.49 | 17.67 |
| Core–shell | 201.7 | 57.4 | 48.18 | 33.32 |
| Name | Elstic Modulus, MPa | Strength, MPa | Elongation at Break, % | Mean Diameter, nm |
|---|---|---|---|---|
| Core | 263.17 ± 96.24 | 3.65 ± 1.75 | 1.49 ± 0.25 | 381 ± 131 |
| Shell | 19.91 ± 1.67 | 0.62 ± 0.10 | 7.07 ± 2.29 | 174 ± 31 |
| Core–shell | 35.89 ± 9.70 | 0.77 ± 0.29 | 3.97 ± 1.37 | 261 ± 51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kan, Y.; Bondareva, J.V.; Statnik, E.S.; Koudan, E.V.; Ippolitov, E.V.; Podporin, M.S.; Kovaleva, P.A.; Kapaev, R.R.; Gordeeva, A.M.; Cvjetinovic, J.; et al. Hydrogel-Inducing Graphene-Oxide-Derived Core–Shell Fiber Composite for Antibacterial Wound Dressing. Int. J. Mol. Sci. 2023, 24, 6255. https://doi.org/10.3390/ijms24076255
Kan Y, Bondareva JV, Statnik ES, Koudan EV, Ippolitov EV, Podporin MS, Kovaleva PA, Kapaev RR, Gordeeva AM, Cvjetinovic J, et al. Hydrogel-Inducing Graphene-Oxide-Derived Core–Shell Fiber Composite for Antibacterial Wound Dressing. International Journal of Molecular Sciences. 2023; 24(7):6255. https://doi.org/10.3390/ijms24076255
Chicago/Turabian StyleKan, Yuliya, Julia V. Bondareva, Eugene S. Statnik, Elizaveta V. Koudan, Evgeniy V. Ippolitov, Mikhail S. Podporin, Polina A. Kovaleva, Roman R. Kapaev, Alexandra M. Gordeeva, Julijana Cvjetinovic, and et al. 2023. "Hydrogel-Inducing Graphene-Oxide-Derived Core–Shell Fiber Composite for Antibacterial Wound Dressing" International Journal of Molecular Sciences 24, no. 7: 6255. https://doi.org/10.3390/ijms24076255
APA StyleKan, Y., Bondareva, J. V., Statnik, E. S., Koudan, E. V., Ippolitov, E. V., Podporin, M. S., Kovaleva, P. A., Kapaev, R. R., Gordeeva, A. M., Cvjetinovic, J., Gorin, D. A., Evlashin, S. A., Salimon, A. I., Senatov, F. S., & Korsunsky, A. M. (2023). Hydrogel-Inducing Graphene-Oxide-Derived Core–Shell Fiber Composite for Antibacterial Wound Dressing. International Journal of Molecular Sciences, 24(7), 6255. https://doi.org/10.3390/ijms24076255