

Mitochondrial Methionyl-tRNA Formyltransferase Deficiency Alleviates Metaflammation by Modulating Mitochondrial Activity in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
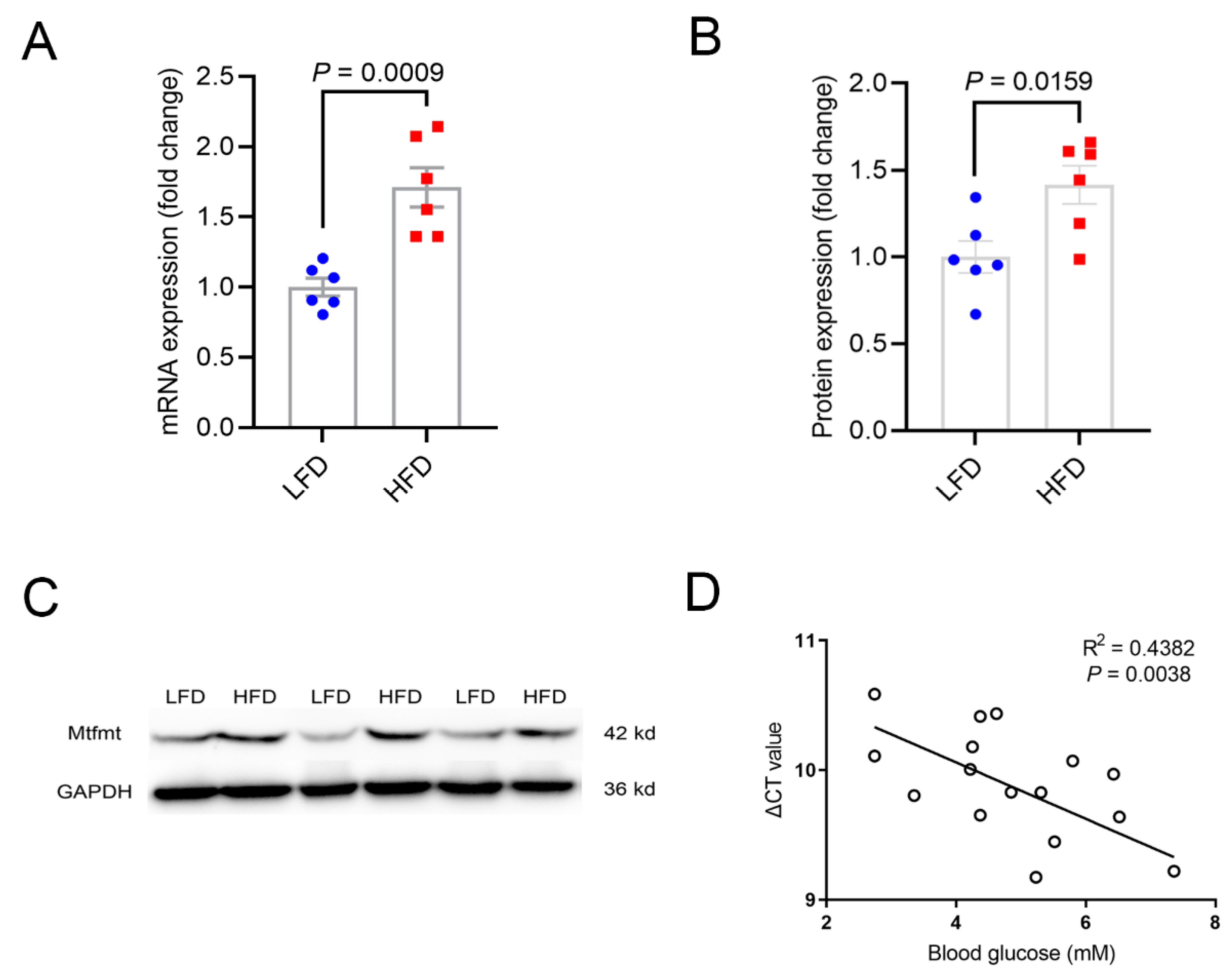
2.1. Expression Levels of Mtfmt Were Correlated with Diet and Glucose Levels
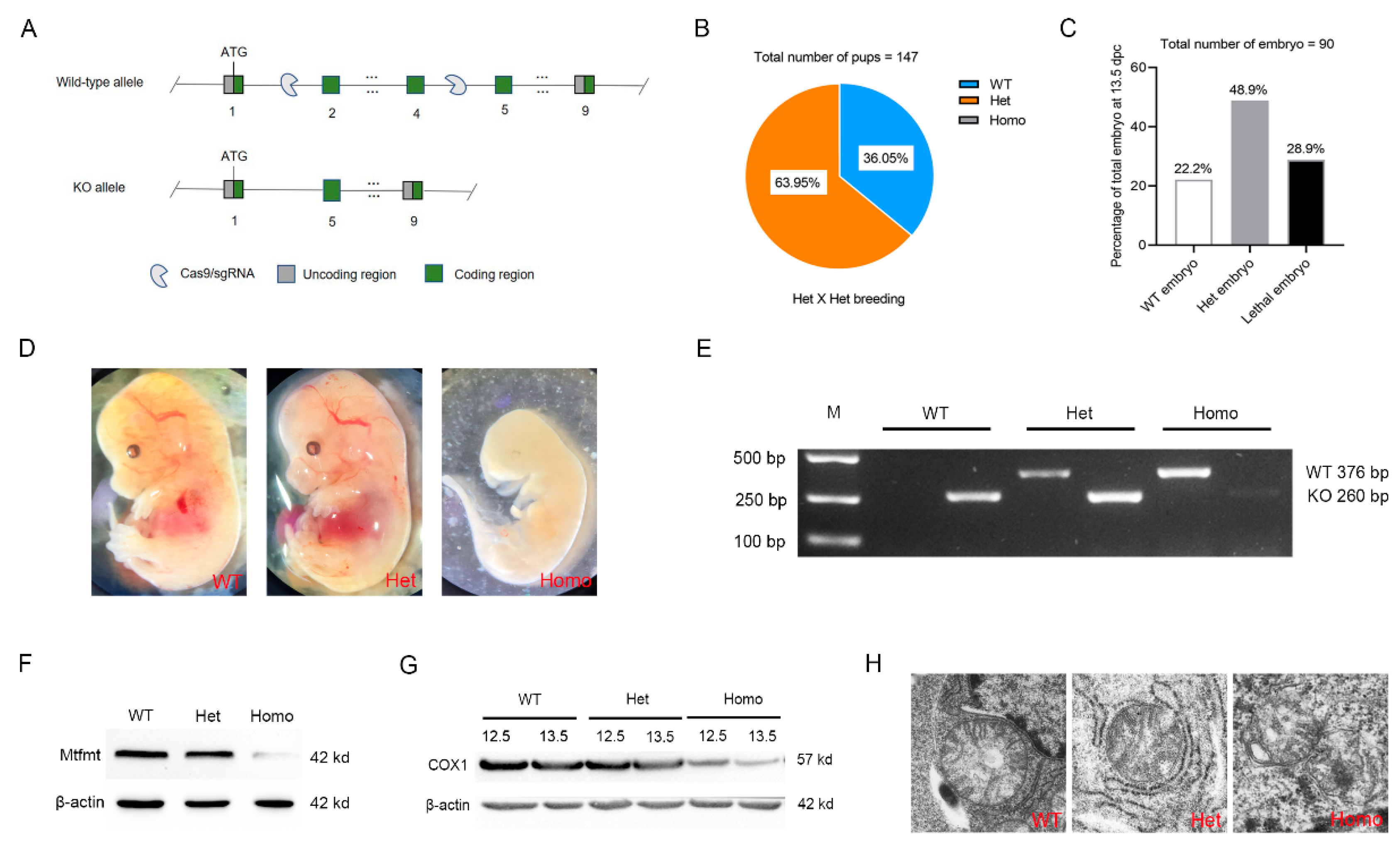
2.2. Mtfmt Deletion Caused Embryonic Lethality in Mice
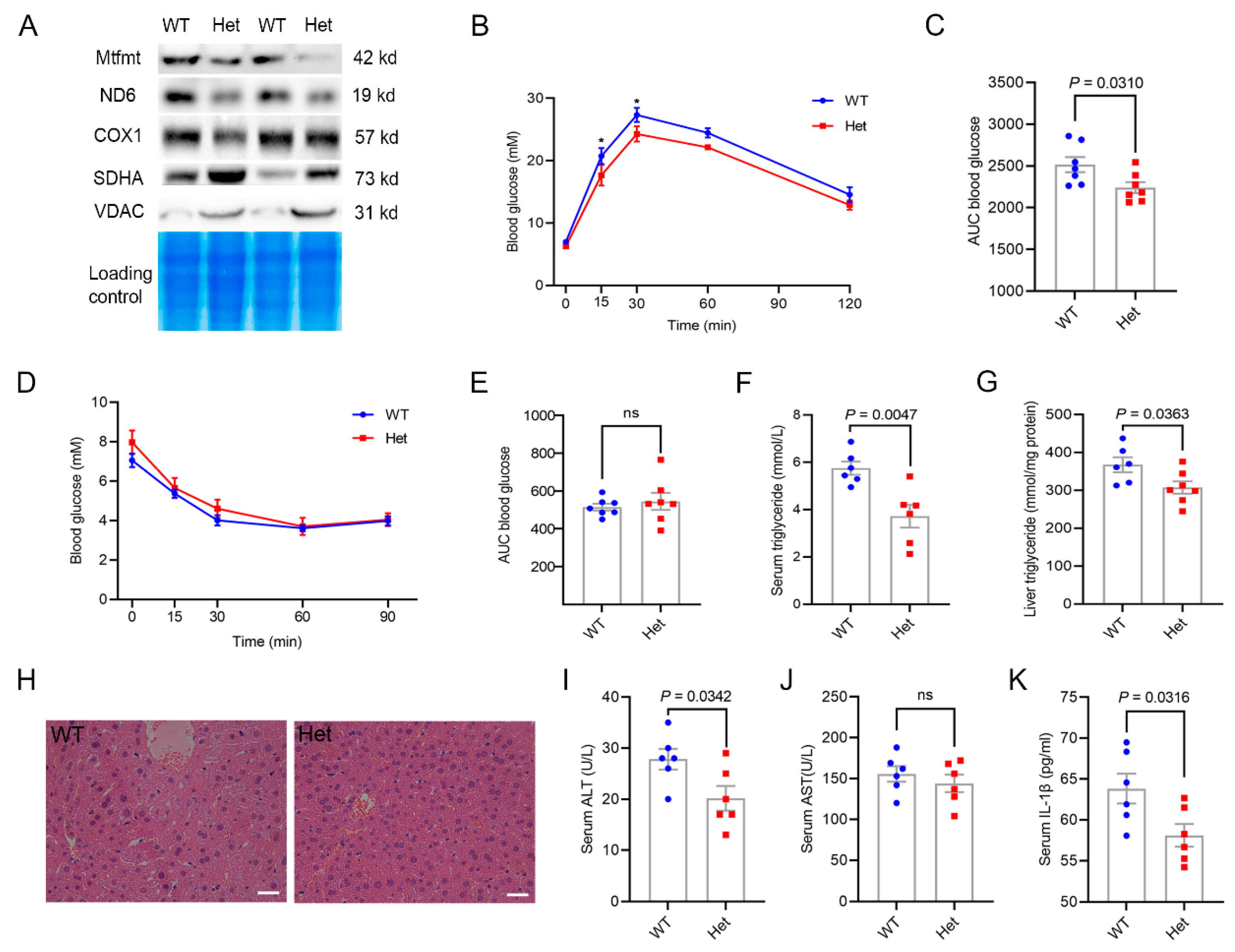
2.3. Mtfmt Haploinsufficiency Improved Hepatic Metabolic Health
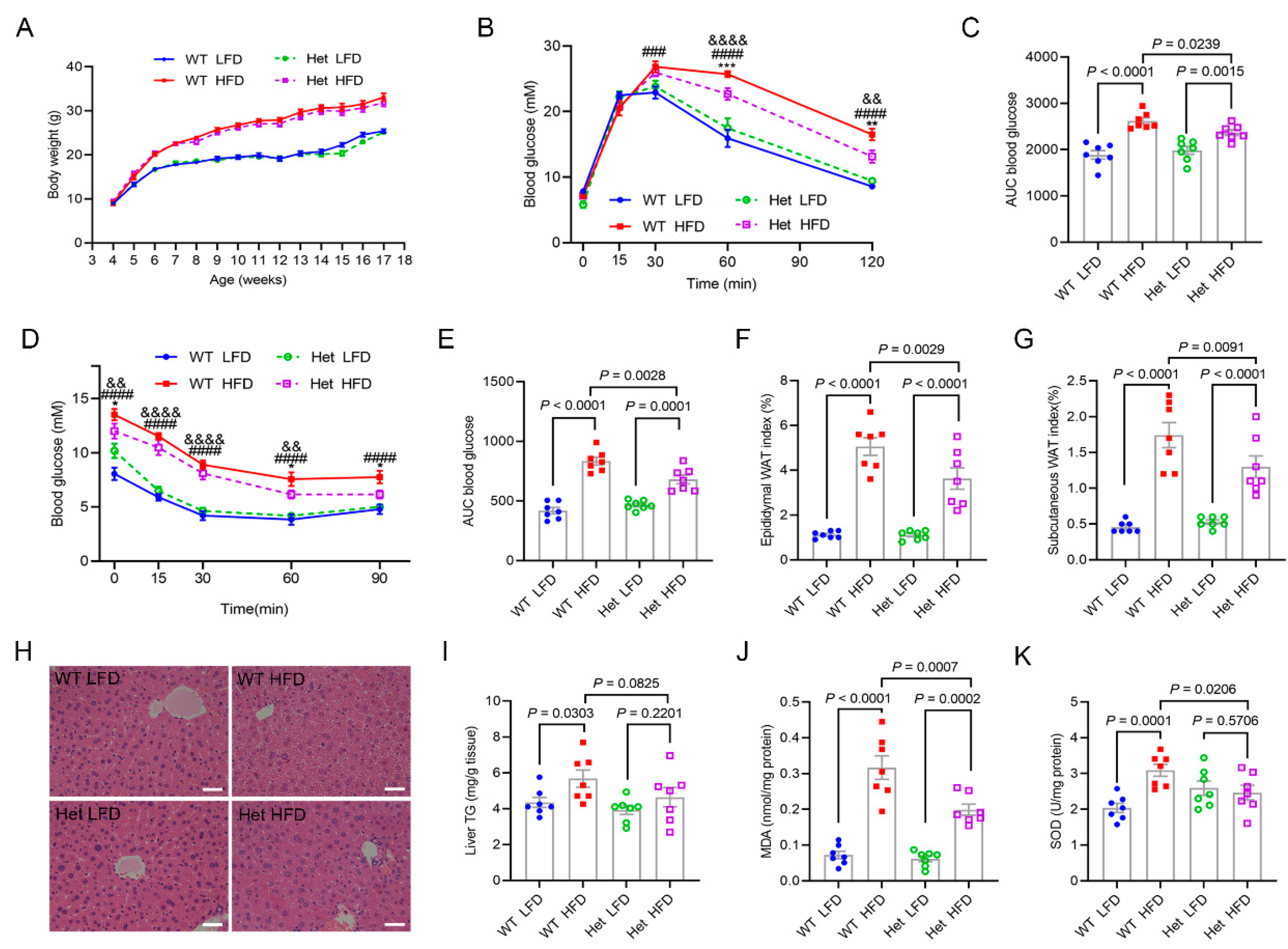
2.4. Mtfmt Haploinsufficiency Alleviated HFD-Induced Metabolic Disorders
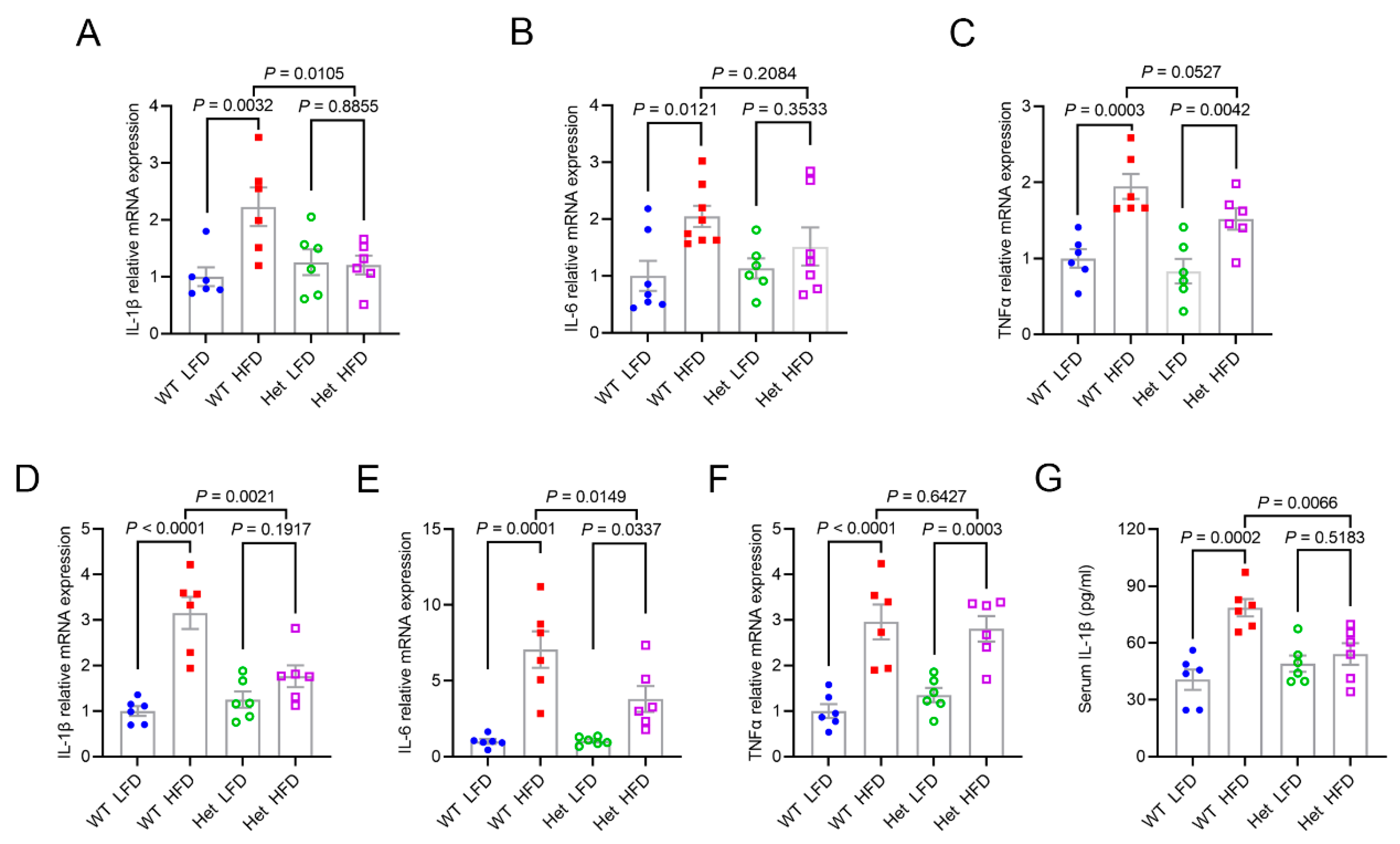
2.5. Mtfmt Haploinsufficiency Alleviated HFD-Induced Inflammation
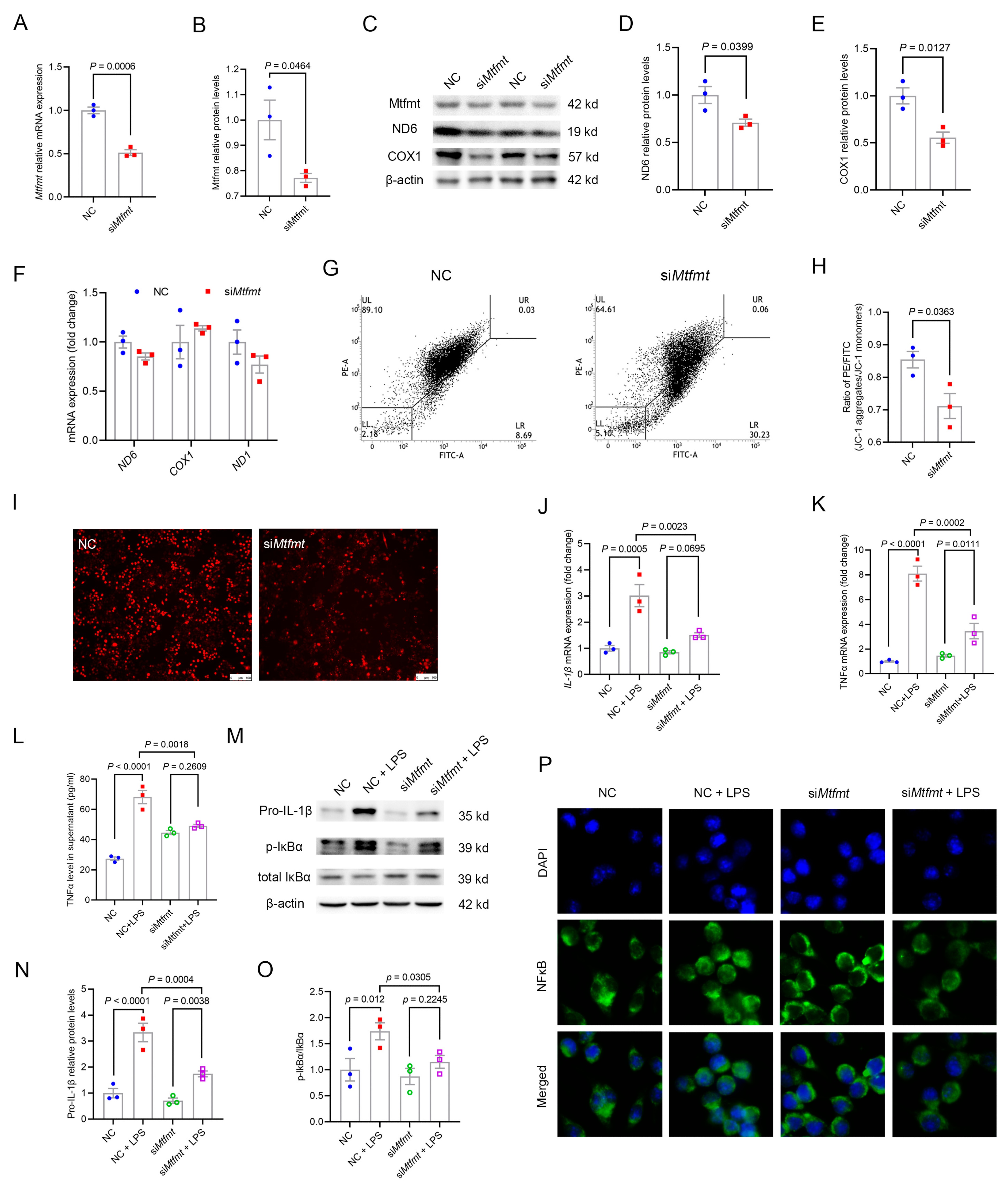
2.6. Mtfmt Knockdown in Macrophages Decreased Mitochondrial Activity and mROS Signaling and Blunted NFκB Signaling
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Real-Time Polymerase Chain Reaction (RT-PCR)
4.3. Western Blot Analysis
4.4. Transmission Electron Microscopy
4.5. Glucose Tolerance Test (GTT)
4.6. Insulin Tolerance Test (ITT)
4.7. Histological Analysis
4.8. Serum Biochemical Measurement
4.9. Detection of MDA, SOD, and TGs in Mouse Livers
4.10. Enzyme-Linked Immunosorbent Assay (ELISA)
4.11. Cell Culture and Cell Transfection
4.12. Flow Cytometry Analysis
4.13. Immunofluorescence
4.14. Mitochondrial ROS Determination
4.15. Isolation of Mitochondria
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kalyani, R.R.; Golden, S.H.; Cefalu, W.T. Diabetes and Aging: Unique Considerations and Goals of Care. Diabetes Care 2017, 40, 440–443. [Google Scholar] [CrossRef]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef]
- Lee, Y.S.; Olefsky, J. Chronic tissue inflammation and metabolic disease. Genes Dev. 2021, 35, 307–328. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Embgenbroich, M.; van der Zande, H.J.P.; Hussaarts, L.; Schulte-Schrepping, J.; Pelgrom, L.R.; Garcia-Tardon, N.; Schlautmann, L.; Stoetzel, I.; Handler, K.; Lambooij, J.M.; et al. Soluble mannose receptor induces proinflammatory macrophage activation and metaflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2103304118. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; O’Doherty, R.M. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef]
- Pospisilik, J.A.; Knauf, C.; Joza, N.; Benit, P.; Orthofer, M.; Cani, P.D.; Ebersberger, I.; Nakashima, T.; Sarao, R.; Neely, G.; et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 2007, 131, 476–491. [Google Scholar] [CrossRef]
- Masand, R.; Paulo, E.; Wu, D.; Wang, Y.; Swaney, D.L.; Jimenez-Morales, D.; Krogan, N.J.; Wang, B. Proteome Imbalance of Mitochondrial Electron Transport Chain in Brown Adipocytes Leads to Metabolic Benefits. Cell Metab. 2018, 27, 616–629.e4. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Zarse, K.; Schmeisser, S.; Groth, M.; Priebe, S.; Beuster, G.; Kuhlow, D.; Guthke, R.; Platzer, M.; Kahn, C.R.; Ristow, M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012, 15, 451–465. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- He, F.; Huang, Y.; Song, Z.; Zhou, H.J.; Zhang, H.; Perry, R.J.; Shulman, G.I.; Min, W. Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance. J. Exp. Med. 2021, 218, e20201416. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, J.; Deng, H.; Ma, R.; Liao, J.Y.; Liang, H.; Hu, J.; Li, J.; Guo, Z.; Cai, J.; et al. Targeting Mitochondria-Located circRNA SCAR Alleviates NASH via Reducing mROS Output. Cell 2020, 183, 76–93.e22. [Google Scholar] [CrossRef]
- Rosen, P.; Wiernsperger, N.F. Metformin delays the manifestation of diabetes and vascular dysfunction in Goto-Kakizaki rats by reduction of mitochondrial oxidative stress. Diabetes Metab. Res. Rev. 2006, 22, 323–330. [Google Scholar] [CrossRef]
- Christian, B.E.; Spremulli, L.L. Mechanism of protein biosynthesis in mammalian mitochondria. Biochim. Biophys. Acta 2012, 1819, 1035–1054. [Google Scholar] [CrossRef]
- Pearce, S.; Nezich, C.L.; Spinazzola, A. Mitochondrial diseases: Translation matters. Mol. Cell. Neurosci. 2013, 55, 1–12. [Google Scholar] [CrossRef]
- Tucker, E.J.; Hershman, S.G.; Kohrer, C.; Belcher-Timme, C.A.; Patel, J.; Goldberger, O.A.; Christodoulou, J.; Silberstein, J.M.; McKenzie, M.; Ryan, M.T.; et al. Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 2011, 14, 428–434. [Google Scholar] [CrossRef]
- Hayhurst, H.; de Coo, I.F.M.; Piekutowska-Abramczuk, D.; Alston, C.L.; Sharma, S.; Thompson, K.; Rius, R.; He, L.; Hopton, S.; Ploski, R.; et al. Leigh syndrome caused by mutations in MTFMT is associated with a better prognosis. Ann. Clin. Transl. Neurol. 2019, 6, 515–524. [Google Scholar] [CrossRef]
- Hemelsoet, D.M.; Vanlander, A.V.; Smet, J.; Vantroys, E.; Acou, M.; Goethals, I.; Sante, T.; Seneca, S.; Menten, B.; Van Coster, R. Leigh syndrome followed by parkinsonism in an adult with homozygous c.626C>T mutation in MTFMT. Neurol. Genet. 2018, 4, e298. [Google Scholar] [CrossRef]
- La Piana, R.; Weraarpachai, W.; Ospina, L.H.; Tetreault, M.; Majewski, J.; Care4Rare Canada, C.; Bruce Pike, G.; Decarie, J.C.; Tampieri, D.; Brais, B.; et al. Identification and functional characterization of a novel MTFMT mutation associated with selective vulnerability of the visual pathway and a mild neurological phenotype. Neurogenetics 2017, 18, 97–103. [Google Scholar] [CrossRef]
- Bi, X.; Ai, H.; Wu, Q.; Fan, Q.; Ding, F.; Hu, C.; Ding, W. Insulin Resistance Is Associated with Interleukin 1beta (IL-1beta) in Non-Diabetic Hemodialysis Patients. Med. Sci. Monit. 2018, 24, 897–902. [Google Scholar] [CrossRef]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- Cerritelli, S.M.; Frolova, E.G.; Feng, C.; Grinberg, A.; Love, P.E.; Crouch, R.J. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell 2003, 11, 807–815. [Google Scholar] [CrossRef]
- Yagi, M.; Uchiumi, T.; Takazaki, S.; Okuno, B.; Nomura, M.; Yoshida, S.; Kanki, T.; Kang, D. p32/gC1qR is indispensable for fetal development and mitochondrial translation: Importance of its RNA-binding ability. Nucleic Acids Res. 2012, 40, 9717–9737. [Google Scholar] [CrossRef]
- Piruat, J.I.; Pintado, C.O.; Ortega-Saenz, P.; Roche, M.; Lopez-Barneo, J. The mitochondrial SDHD gene is required for early embryogenesis, and its partial deficiency results in persistent carotid body glomus cell activation with full responsiveness to hypoxia. Mol. Cell. Biol. 2004, 24, 10933–10940. [Google Scholar] [CrossRef] [PubMed]
- Nonn, L.; Williams, R.R.; Erickson, R.P.; Powis, G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 2003, 23, 916–922. [Google Scholar] [CrossRef]
- Tani, H.; Ohnishi, S.; Shitara, H.; Mito, T.; Yamaguchi, M.; Yonekawa, H.; Hashizume, O.; Ishikawa, K.; Nakada, K.; Hayashi, J.I. Mice deficient in the Shmt2 gene have mitochondrial respiration defects and are embryonic lethal. Sci. Rep. 2018, 8, 425. [Google Scholar] [CrossRef]
- Vrbacky, M.; Kovalcikova, J.; Chawengsaksophak, K.; Beck, I.M.; Mracek, T.; Nuskova, H.; Sedmera, D.; Papousek, F.; Kolar, F.; Sobol, M.; et al. Knockout of Tmem70 alters biogenesis of ATP synthase and leads to embryonal lethality in mice. Hum. Mol. Genet. 2016, 25, 4674–4685. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Lane, N. Mitochondrial disease: Powerhouse of disease. Nature 2006, 440, 600–602. [Google Scholar] [CrossRef]
- Zhao, Q.; Luo, T.; Gao, F.; Fu, Y.; Li, B.; Shao, X.; Chen, H.; Zhou, Z.; Guo, S.; Shen, L.; et al. GRP75 Regulates Mitochondrial-Supercomplex Turnover to Modulate Insulin Sensitivity. Diabetes 2022, 71, 233–248. [Google Scholar] [CrossRef]
- Keller, M.P.; Attie, A.D. Physiological insights gained from gene expression analysis in obesity and diabetes. Annu. Rev. Nutr. 2010, 30, 341–364. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef]
- Rabol, R.; Boushel, R.; Dela, F. Mitochondrial oxidative function and type 2 diabetes. Appl. Physiol. Nutr. Metab. 2006, 31, 675–683. [Google Scholar] [CrossRef]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Wredenberg, A.; Freyer, C.; Sandstrom, M.E.; Katz, A.; Wibom, R.; Westerblad, H.; Larsson, N.G. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem. Biophys. Res. Commun. 2006, 350, 202–207. [Google Scholar] [CrossRef]
- Vernochet, C.; Mourier, A.; Bezy, O.; Macotela, Y.; Boucher, J.; Rardin, M.J.; An, D.; Lee, K.Y.; Ilkayeva, O.R.; Zingaretti, C.M.; et al. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab. 2012, 16, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lv, W.; Xu, J.; Zheng, A.; Zeng, M.; Cao, K.; Wang, X.; Cui, Y.; Li, H.; Yang, M.; et al. Hepatic Suppression of Mitochondrial Complex II Assembly Drives Systemic Metabolic Benefits. Adv. Sci. 2022, 9, e2105587. [Google Scholar] [CrossRef] [PubMed]
- Colaco, H.G.; Barros, A.; Neves-Costa, A.; Seixas, E.; Pedroso, D.; Velho, T.; Willmann, K.L.; Faisca, P.; Grabmann, G.; Yi, H.S.; et al. Tetracycline Antibiotics Induce Host-Dependent Disease Tolerance to Infection. Immunity 2021, 54, 53–67.e7. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Hwang, C.S.; Yoo, J.Y. MTFMT deficiency correlates with reduced mitochondrial integrity and enhanced host susceptibility to intracellular infection. Sci. Rep. 2020, 10, 11183. [Google Scholar] [CrossRef]
- Jais, A.; Einwallner, E.; Sharif, O.; Gossens, K.; Lu, T.T.; Soyal, S.M.; Medgyesi, D.; Neureiter, D.; Paier-Pourani, J.; Dalgaard, K.; et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell 2014, 158, 25–40. [Google Scholar] [CrossRef]
- Fan, G.; Li, Y.; Chen, J.; Zong, Y.; Yang, X. DHA/AA alleviates LPS-induced Kupffer cells pyroptosis via GPR120 interaction with NLRP3 to inhibit inflammasome complexes assembly. Cell Death Dis. 2021, 12, 73. [Google Scholar] [CrossRef]
- Kang, J.; Tae, N.; Min, B.S.; Choe, J.; Lee, J.H. Malabaricone C suppresses lipopolysaccharide-induced inflammatory responses via inhibiting ROS-mediated Akt/IKK/NF-kappaB signaling in murine macrophages. Int. Immunopharmacol. 2012, 14, 302–310. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Liu, S.; Cai, J.; Yang, M.; Li, C.; Tan, M.; He, B. Mitochondrial Methionyl-tRNA Formyltransferase Deficiency Alleviates Metaflammation by Modulating Mitochondrial Activity in Mice. Int. J. Mol. Sci. 2023, 24, 5999. https://doi.org/10.3390/ijms24065999
Sun X, Liu S, Cai J, Yang M, Li C, Tan M, He B. Mitochondrial Methionyl-tRNA Formyltransferase Deficiency Alleviates Metaflammation by Modulating Mitochondrial Activity in Mice. International Journal of Molecular Sciences. 2023; 24(6):5999. https://doi.org/10.3390/ijms24065999
Chicago/Turabian StyleSun, Xiaoxiao, Suyuan Liu, Jiangxue Cai, Miaoxin Yang, Chenxuan Li, Meiling Tan, and Bin He. 2023. "Mitochondrial Methionyl-tRNA Formyltransferase Deficiency Alleviates Metaflammation by Modulating Mitochondrial Activity in Mice" International Journal of Molecular Sciences 24, no. 6: 5999. https://doi.org/10.3390/ijms24065999
APA StyleSun, X., Liu, S., Cai, J., Yang, M., Li, C., Tan, M., & He, B. (2023). Mitochondrial Methionyl-tRNA Formyltransferase Deficiency Alleviates Metaflammation by Modulating Mitochondrial Activity in Mice. International Journal of Molecular Sciences, 24(6), 5999. https://doi.org/10.3390/ijms24065999