
Two Single Nucleotide Deletions in the ABCD1 Gene Causing Distinct Phenotypes of X-Linked Adrenoleukodystrophy
 and
and
Abstract
1. Introduction
2. Results
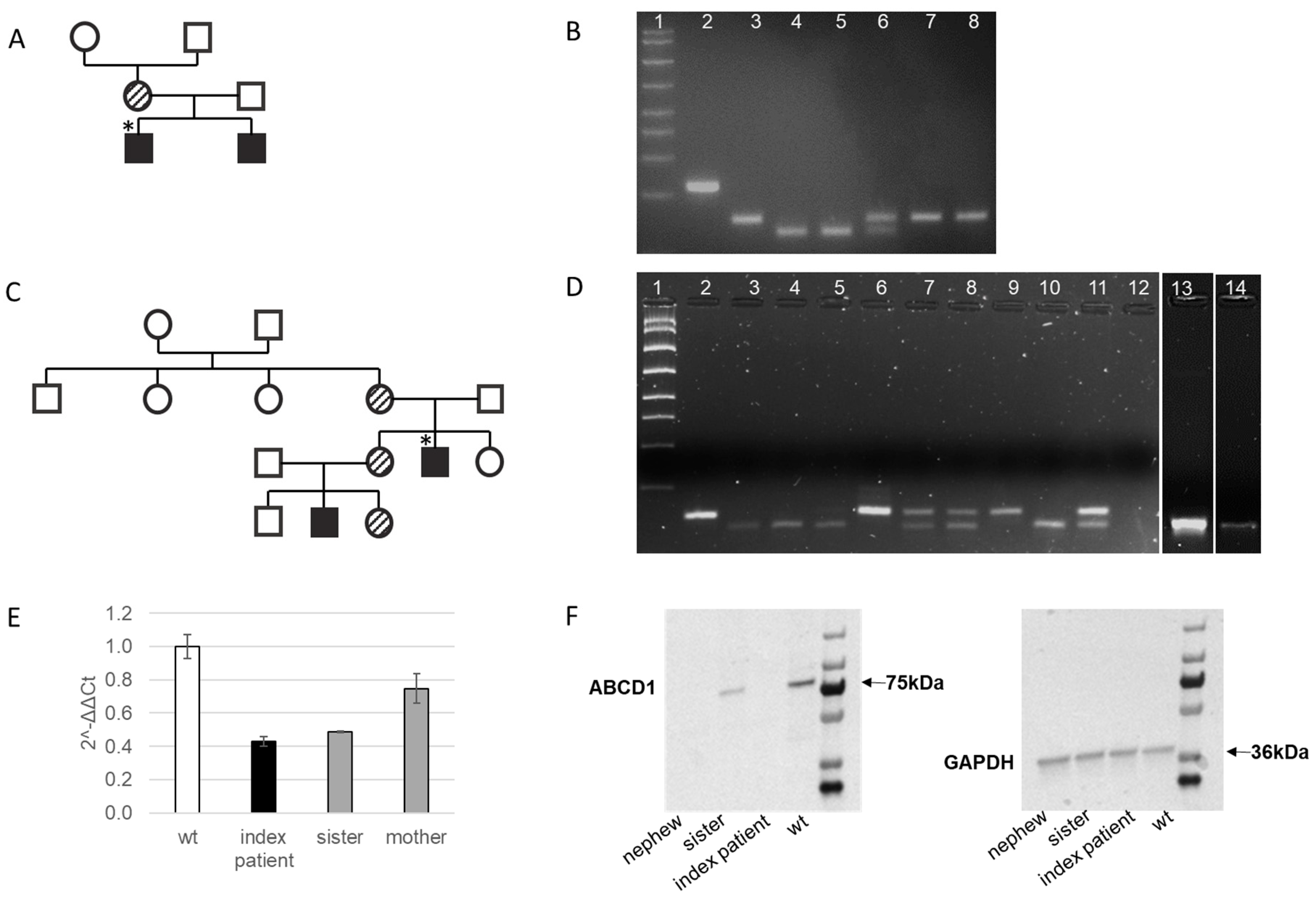
2.1. Clinical Characteristics of the Index Patient in Family 1 with Cerebral X-ALD and His Affected Older Brother with AMN
2.2. Genetic Confirmation of X-ALD in Family 1
2.3. Clinical Characteristics of the Index Patient in Family 2 with AMN and His Young Nephew with Addison’s Disease
2.4. Genetic Confirmation of AMN in the Index Patient in Familiy 2 and Family Members Evaluation
2.5. Molecular Characterization of the Novel Frameshift Variant c.1275delA [p.Phe426Leufs*15]
3. Discussion
4. Methods
4.1. Plasma VLFCA Measurements
4.2. DNA Isolation
4.3. Sequencing and Restriction Analysis
4.4. RT-qPCR
4.5. Western Blot
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roermund, C.W.T.; Visser, W.F.; IJlst, L.; Cruchten, A.; Boek, M.; Kulik, W.; Waterham, H.R.; Wanders, R.J.A. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. FASEB J. 2008, 22, 4201–4208. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Eichler, F.; Biffi, A.; Duncan, C.N.; Williams, D.A.; Majzoub, J.A. The Changing Face of Adrenoleukodystrophy. Endocr. Rev. 2020, 41, 577–593. [Google Scholar] [CrossRef]
- Moser, H.W.; Moser, A.B.; Frayer, K.K.; Chen, W.; Schulman, J.D.; O’Neill, B.P.; Kishimoto, Y. Adrenoleukodystrophy: Increased plasma content of saturated very long chain fatty acids. Neurology 1981, 31, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.A.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.R.; Theda, C.; Fatemi, A.; Moser, A.B. X-linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int. J. Dev. Neurosci. 2020, 80, 52–72. [Google Scholar] [CrossRef]
- Berger, J.; Molzer, B.; Fae, I.; Bernheimer, H. X-Linked Adrenoleukodystrophy (ALD): A Novel Mutation of the ALD Gene in 6 Members of a Family Presenting with 5 Different Phenotypes. Biochem. Biophys. Res. Commun. 1994, 205, 1638–1643. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Mallack, E.J.; Gao, K.; Engelen, M.; Kemp, S. Structure and Function of the ABCD1 Variant Database: 20 Years, 940 Pathogenic Variants, and 3400 Cases of Adrenoleukodystrophy. Cells 2022, 11, 283. [Google Scholar] [CrossRef]
- Mahmood, A.; Dubey, P.; Moser, H.W.; Moser, A. X-linked adrenoleukodystrophy: Therapeutic approaches to distinct phenotypes. Pediatr. Transplant. 2005, 9, 55–62. [Google Scholar] [CrossRef]
- Zemanova, M.; Chrastina, P.; Dvorakova, L.; Reboun, M.; Vlaskova, H.; Jahnova, H.; El-Lababidi, N.; Cepova, J.; Honzik, T.; Zeman, J. X-linked adrenoleukodystrophy: Phenotype-genotype correlation in hemizygous males and heterozygous females with ABCD1 mutations. Neuroendocrinol. Lett. 2021, 42, 359–367. [Google Scholar]
- Huffnagel, I.C.; Laheji, F.K.; Aziz-Bose, R.; Tritos, N.A.; Marino, R.; Linthorst, G.E.; Kemp, S.; Engelen, M.; Eichler, F. The Natural History of Adrenal Insufficiency in X-Linked Adrenoleukodystrophy: An International Collaboration. J. Clin. Endocrinol. Metab. 2019, 104, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Shimozawa, N.; Kashiwayama, Y.; Suzuki, Y.; Imanaka, T. ABC subfamily D proteins and very long chain fatty acid metabolism as novel targets in adrenoleukodystrophy. Curr. Drug Targets 2011, 12, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Priestley, J.R.C.; Adang, L.A.; Drewes Williams, S.; Lichter-Konecki, U.; Menello, C.; Engelhardt, N.M.; DiPerna, J.C.; DiBoscio, B.; Ahrens-Nicklas, R.C.; Edmondson, A.C.; et al. Newborn Screening for X-Linked Adrenoleukodystrophy: Review of Data and Outcomes in Pennsylvania. Int. J. Neonatal Screen. 2022, 8, 24. [Google Scholar] [CrossRef]
- Zierfuss, B.; Buda, A.; Villoria-González, A.; Logist, M.; Fabjan, J.; Parzer, P.; Battin, C.; Vandersteene, S.; Dijkstra, I.M.E.; Waidhofer-Söllner, P.; et al. Saturated very long-chain fatty acids regulate macrophage plasticity and invasiveness. J. Neuroinflammation 2022, 19, 305. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Pujol, A. Pathomechanisms Underlying X-Adrenoleukodystrophy: A Three-Hit Hypothesis: X-Adrenoleukodystrophy. Brain Pathol. 2009, 20, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Fode, P.; Jespersgaard, C.; Hardwick, R.J.; Bogle, H.; Theisen, M.; Dodoo, D.; Lenicek, M.; Vitek, L.; Vieira, A.; Freitas, J.; et al. Determination of beta-defensin genomic copy number in different populations: A comparison of three methods. PLoS ONE 2011, 6, e16768. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, T.; Mitsui, J.; Ishiura, H.; Takahashi, Y.; Tanaka, M.; Hayashi, S.; Goto, J.; Inazawa, J.; Tsuji, S. Mutational analysis of abcd1 in 86 adrenoleukodystrophy patients, including identification of complex mutations. J. Neurol. Sci. 2017, 381, 159. [Google Scholar] [CrossRef]
- Moser, A.B.; Kreiter, N.; Bezman, L.; Lu, S.; Raymond, G.V.; Naidu, S.; Moser, H.W. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann. Neurol. 1999, 45, 100–110. [Google Scholar] [CrossRef]
- Kosaki, A.; Pillay, T.S.; Xu, L.; Webster, N.J.G. The B Isoform of the Insulin Receptor Signals More Efficiently Than the A Isoform in HepG2 Cells. J. Biol. Chem. 1995, 270, 20816–20823. [Google Scholar] [CrossRef]
- Karousis, E.D.; Nasif, S.; Mühlemann, O. Nonsense-mediated mRNA decay: Novel mechanistic insights and biological impact. Wiley Interdiscip. Rev. RNA 2016, 7, 661–682. [Google Scholar] [CrossRef]
- Chapman, J.H.; Craig, J.M.; Wang, C.D.; Gundlach, J.H.; Neuman, K.C.; Hogg, J.R. UPF1 mutants with intact ATPase but deficient helicase activities promote efficient nonsense-mediated mRNA decay. Nucleic Acids Res. 2022, 50, 11876–11894. [Google Scholar] [CrossRef]
- Volmrich, A.M.; Cuénant, L.M.; Forghani, I.; Hsieh, S.L.; Shapiro, L.T. ABCD1 Gene Mutations: Mechanisms and Management of Adrenomyeloneuropathy. Appl. Clin. Genet. 2022, 15, 111–123. [Google Scholar] [CrossRef]
- Mehrpour, M.; Gohari, F.; Dizaji, M.Z.; Ahani, A.; Malicdan, M.C.V.; Behnam, B. An ABCD1 Mutation (c.253dupC) Caused Diverse Phenotypes of Adrenoleukodystrophy in an Iranian Consanguineous Pedigree. J. Mol. Genet. Med. 2016, 10, 222. [Google Scholar] [CrossRef] [PubMed]
- Sobue, G.; Ueno-Natsukari, I.; Okamoto, H.; Connell, T.A.; Aizawa, I.; Mizoguchi, K.; Honma, M.; Ishikawa, G.; Mitsuma, T.; Natsukari, N. Phenotypic heterogeneity of an adult form of adrenoleukodystrophy in monozygotic twins. Ann. Neurol. 1994, 36, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Heinzer, A.K.; McGuinness, M.C.; Lu, J.-F.; Stine, O.C.; Wei, H.; Van der Vlies, M.; Dong, G.-X.; Powers, J.; Watkins, P.A.; Smith, K.D. Mouse models and genetic modifiers in X-linked adrenoleukodystrophy. Adv. Exp. Med. Biol. 2003, 544, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Palakuzhiyil, S.V.; Christopher, R.; Chandra, S.R. Deciphering the modifiers for phenotypic variability of X-linked adrenoleukodystrophy. World J. Biol. Chem. 2020, 11, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Brand, B.A.; Blesson, A.E.; Smith-Hicks, C.L. The Impact of X-Chromosome Inactivation on Phenotypic Expression of X-Linked Neurodevelopmental Disorders. Brain Sci. 2021, 11, 904. [Google Scholar] [CrossRef] [PubMed]
- Maier, E.M.; Kammerer, S.; Muntau, A.C.; Wichers, M.; Braun, A.; Roscher, A.A. Symptoms in carriers of adrenoleukodystrophy relate to skewed X inactivation. Ann. Neurol. 2002, 52, 683–688. [Google Scholar] [CrossRef]
- Dumic, M.; Gubarev, N.; Sikic, N.; Roscher, A.; Plavsic, V.; Filipovic-Grcic, B. Sparse hair and multiple endocrine disorders in two women heterozygous for adrenoleukodystrophy. Am. J. Med. Genet. 1992, 43, 829–832. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | C22 | C24 | C26 | C24/C22 | C26/C22 | |
|---|---|---|---|---|---|---|
| Reference Range: | 11.2–48.7 | 7.10–28.9 | 0.18–0.48 | 0.68–0.98 | 0.007–0.03 | |
| Family 1. (c.253delC) | ||||||
| Index Case (hemizygous) | X-ALD | 26.1 | 44.70 | 1.97 | 1.70 | 0.070 |
| Brother (hemizygous) | AMN | 14.4 | 26.40 | 1.06 | 1.80 | 0.070 |
| Family 2. (c.1275delA) | ||||||
| Index Case (hemizygous) | AMN | 25.70 | 49.30 | 1.68 | 1.92 | 0.065 |
| Mother (heterozygous) | asymptomatic | 35.4 | 39.00 | 0.74 | 1.10 | 0.021 |
| Father (wild type) | 20.6 | 15.9 | 0.14 | 0.77 | 0.007 | |
| Sister (wild type) | 27 | 25.5 | 0.19 | 0.94 | 0.007 | |
| Sister (heterozygous) | asymptomatic | 24.6 | 29.4 | 0.56 | 1.2 | 0.023 |
| Nephew (hemizygous) | Addison´s | 20.8 | 36.6 | 1.10 | 1.76 | 0.053 |
| Nephew (wild type) | 19.8 | 21.2 | 0.16 | 1.07 | 0.008 | |
| Niece (heterozygous) | asymptomatic | 26.2 | 36.9 | 0.65 | 1.41 | 0.025 |
| Physical Location | Variant | Type | Mutation Taster | SIFT | Protein Length |
|---|---|---|---|---|---|
| chr23:152990974delC | c.253delC | Frameshift | damaging (LOF: 0.98; tree vote: 190|10) | damaging (score: 0.858) | NMD |
| chr23:153001849delA | c.1275delA | Frameshift | damaging (LOF: 0.98; tree vote: 186|14) | damaging (score: 0.858) | NMD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dohr, K.A.; Tokic, S.; Gastager-Ehgartner, M.; Stojakovic, T.; Dumic, M.; Plecko, B.; Dumic, K.K. Two Single Nucleotide Deletions in the ABCD1 Gene Causing Distinct Phenotypes of X-Linked Adrenoleukodystrophy. Int. J. Mol. Sci. 2023, 24, 5957. https://doi.org/10.3390/ijms24065957
Dohr KA, Tokic S, Gastager-Ehgartner M, Stojakovic T, Dumic M, Plecko B, Dumic KK. Two Single Nucleotide Deletions in the ABCD1 Gene Causing Distinct Phenotypes of X-Linked Adrenoleukodystrophy. International Journal of Molecular Sciences. 2023; 24(6):5957. https://doi.org/10.3390/ijms24065957
Chicago/Turabian StyleDohr, Katrin A., Silvija Tokic, Magdalena Gastager-Ehgartner, Tatjana Stojakovic, Miroslav Dumic, Barbara Plecko, and Katja K. Dumic. 2023. "Two Single Nucleotide Deletions in the ABCD1 Gene Causing Distinct Phenotypes of X-Linked Adrenoleukodystrophy" International Journal of Molecular Sciences 24, no. 6: 5957. https://doi.org/10.3390/ijms24065957
APA StyleDohr, K. A., Tokic, S., Gastager-Ehgartner, M., Stojakovic, T., Dumic, M., Plecko, B., & Dumic, K. K. (2023). Two Single Nucleotide Deletions in the ABCD1 Gene Causing Distinct Phenotypes of X-Linked Adrenoleukodystrophy. International Journal of Molecular Sciences, 24(6), 5957. https://doi.org/10.3390/ijms24065957