
Neuroprotective Effects of the Neural-Induced Adipose-Derived Stem Cell Secretome against Rotenone-Induced Mitochondrial and Endoplasmic Reticulum Dysfunction

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
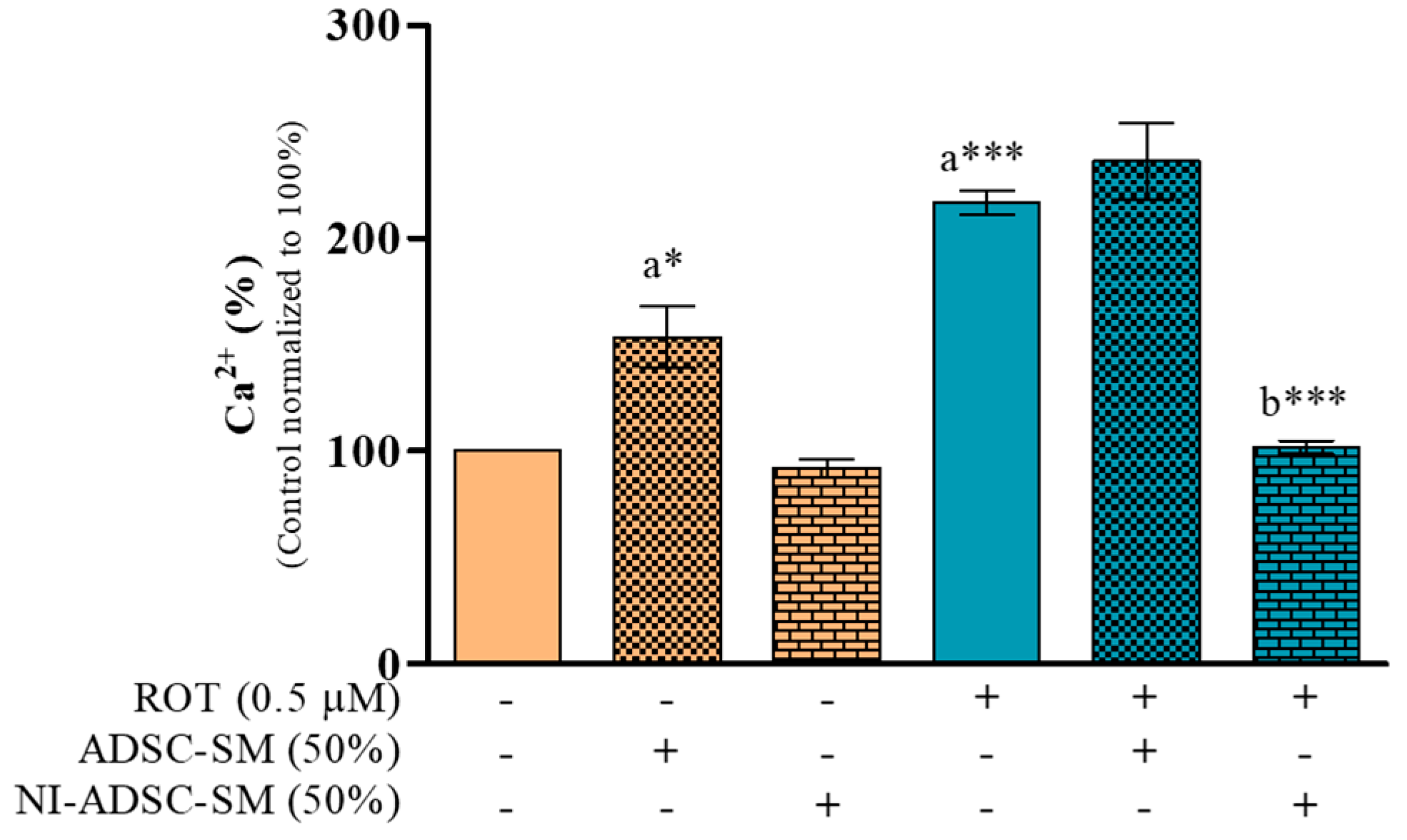
2.1. Effects of the NI-ADSC-SM on Intracellular Ca2+ Levels after ROT Exposure
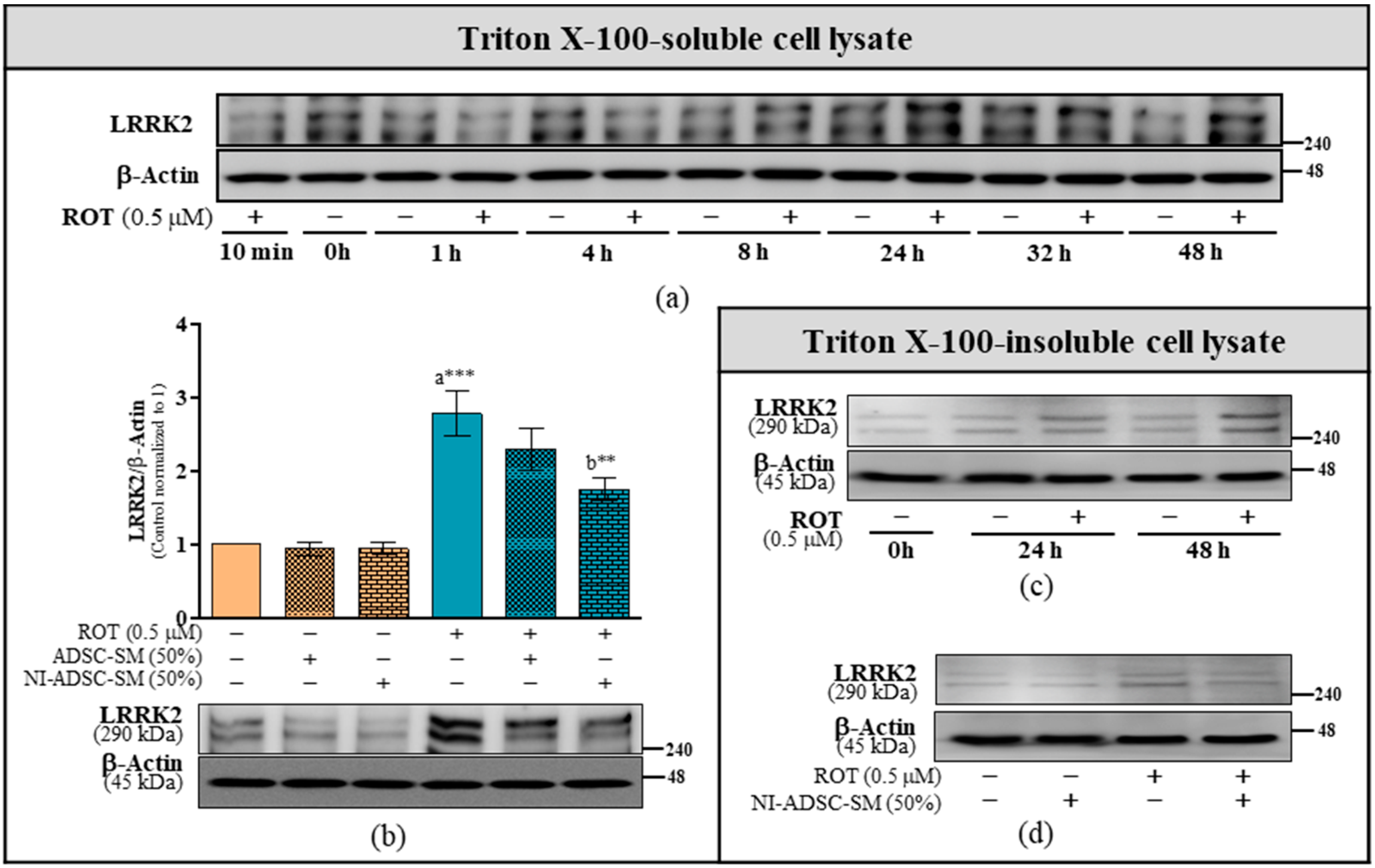
2.2. Effects of the NI-ADSC-SM on LRRK2 Protein Expression after ROT Exposure
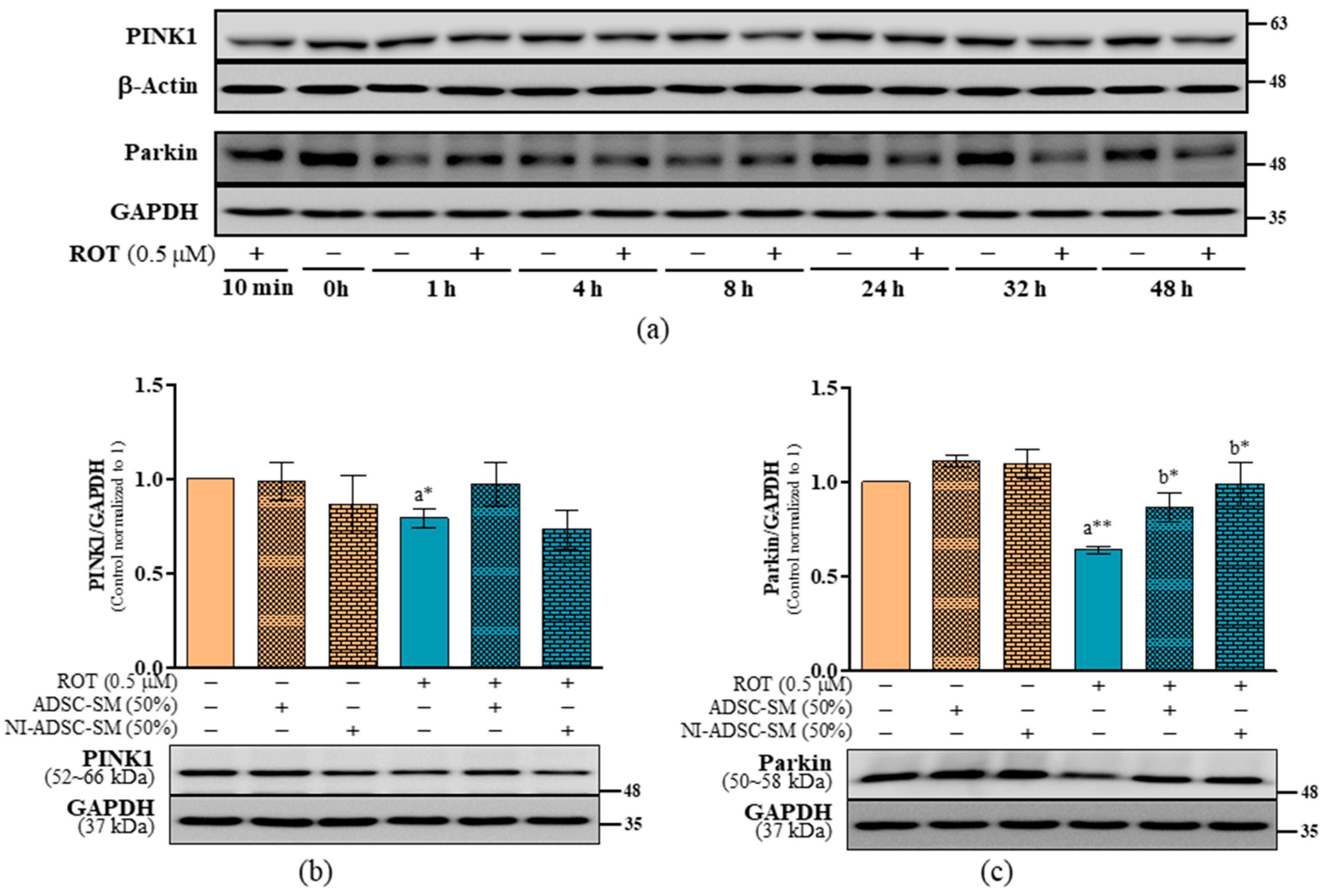
2.3. Effects of the NI-ADSC-SM on PINK1 and Parkin Protein Expression after ROT Exposure
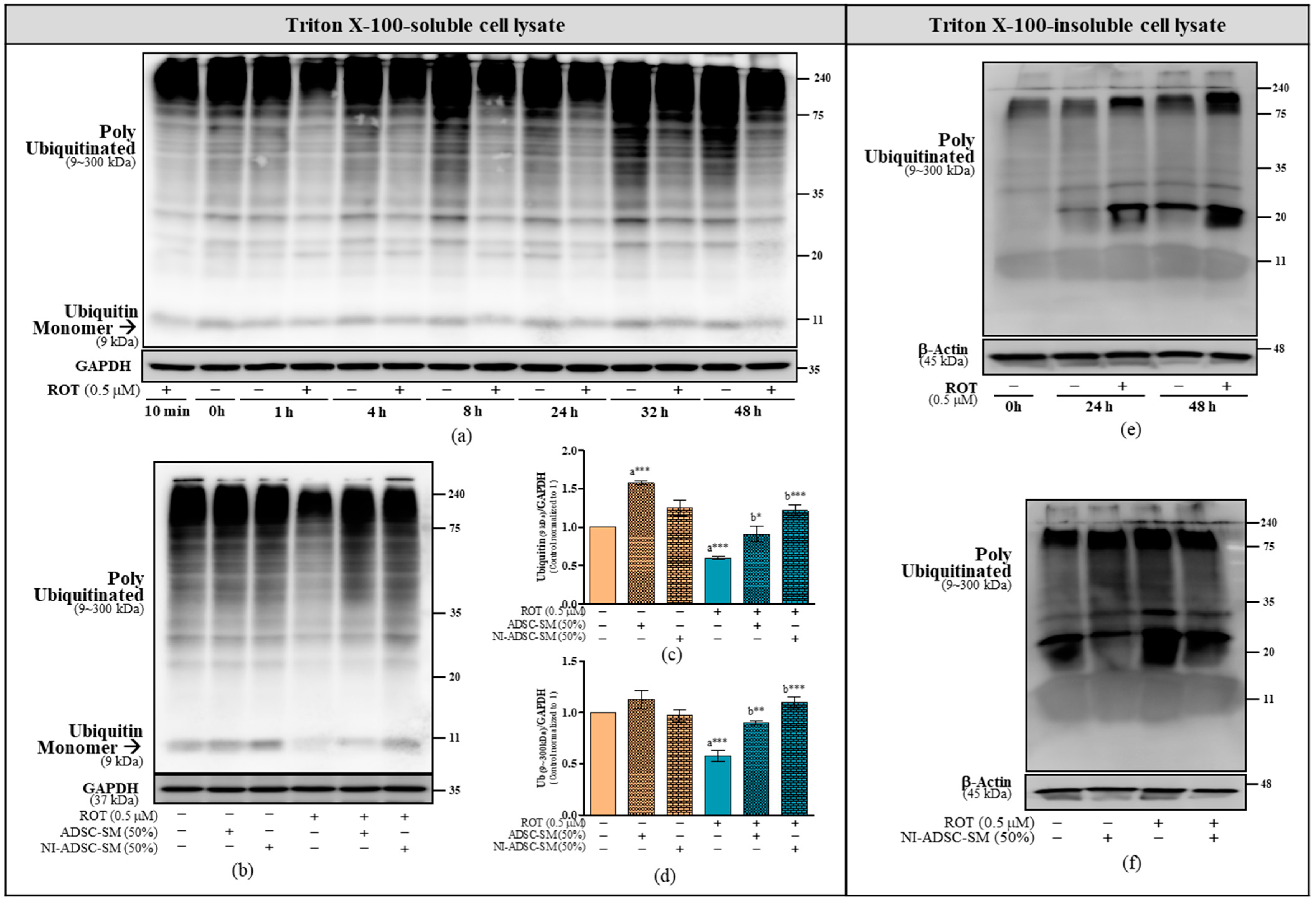
2.4. Effects of the NI-ADSC-SM on Ub Protein Expression after ROT Exposure
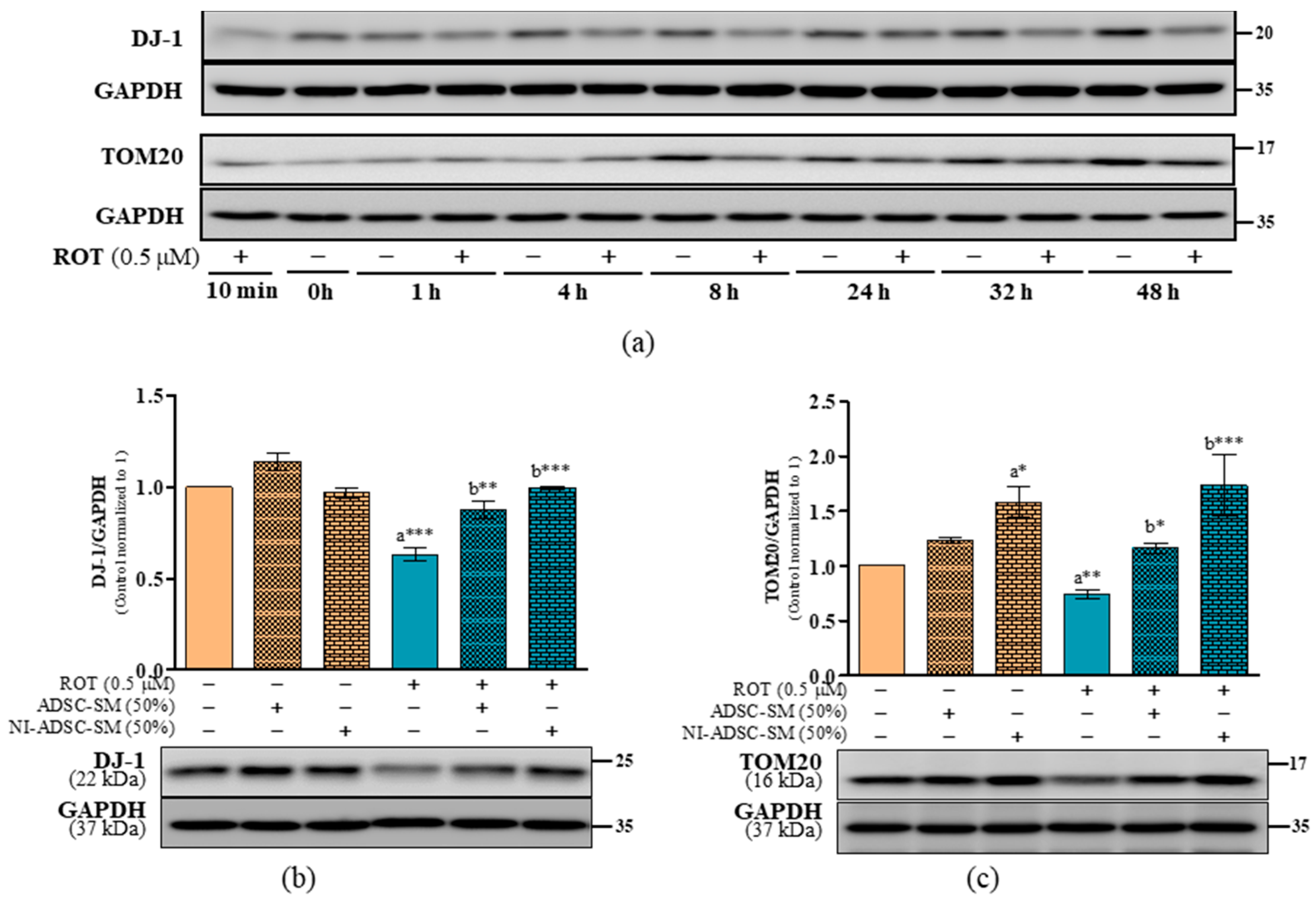
2.5. Effects of the NI-ADSC-SM on DJ-1 and TOM20 Protein Expression after ROT Exposure
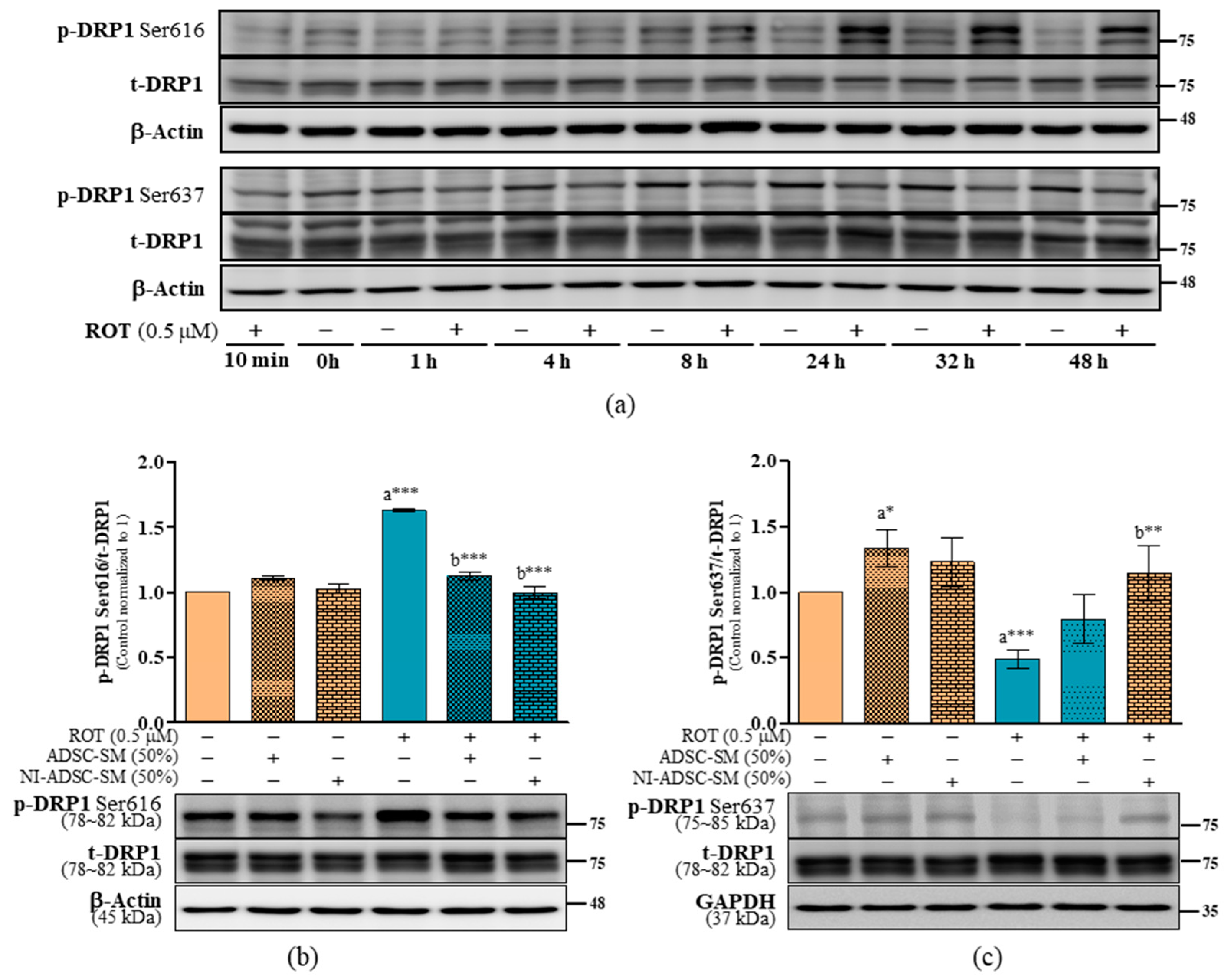
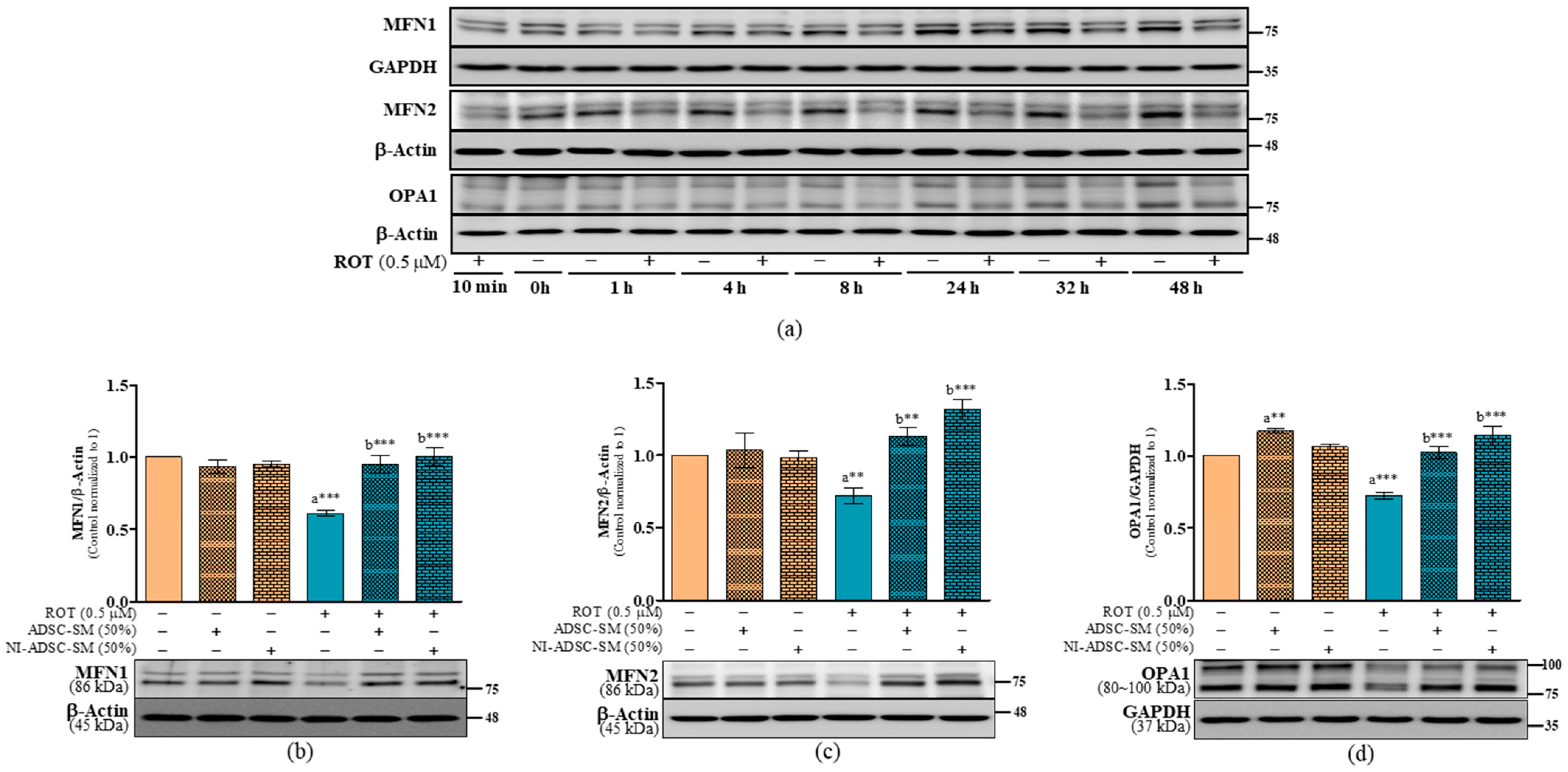
2.6. Effects of the NI-ADSC-SM on Mitochondrial Fission and Fusion Protein Expression after ROT Exposure
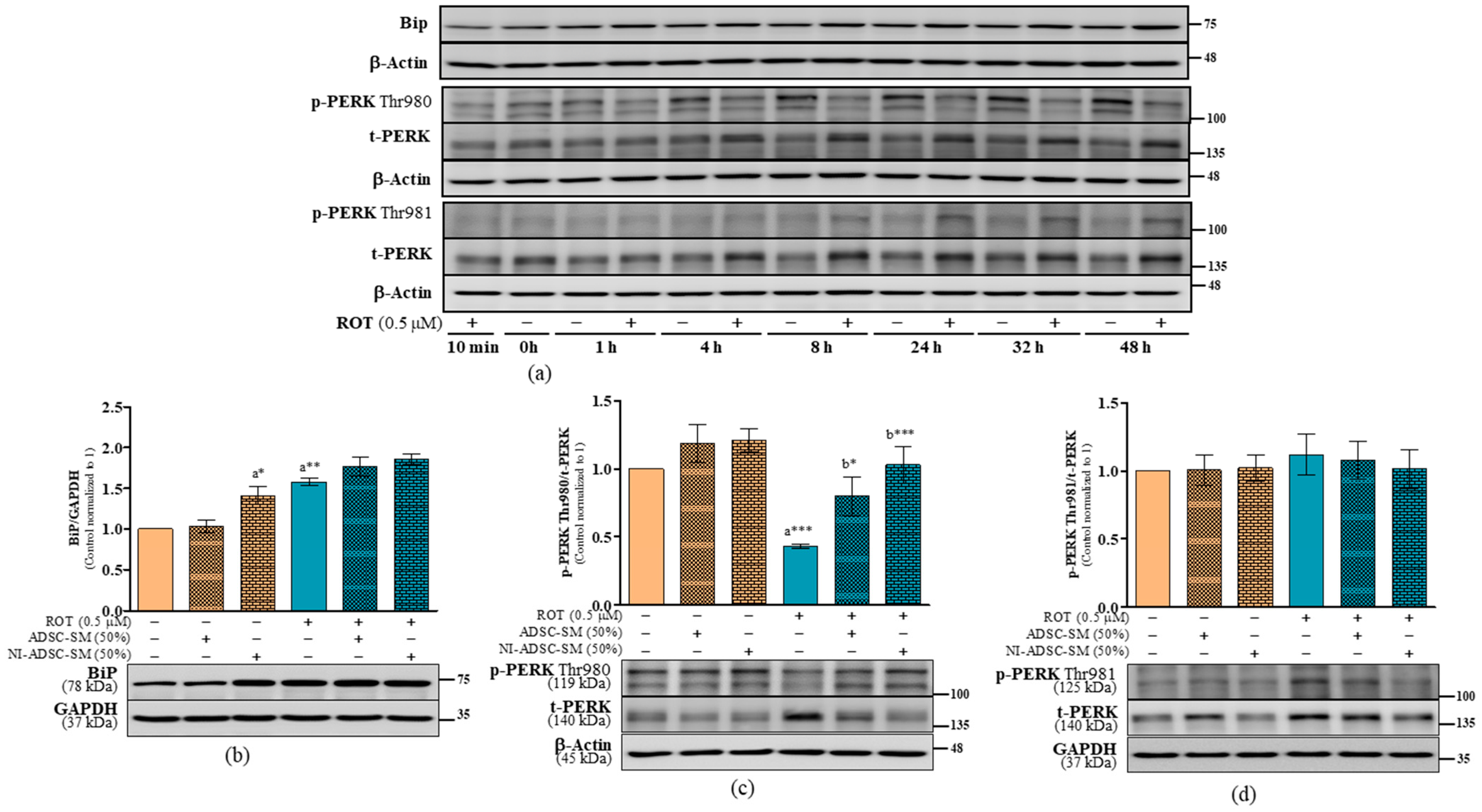
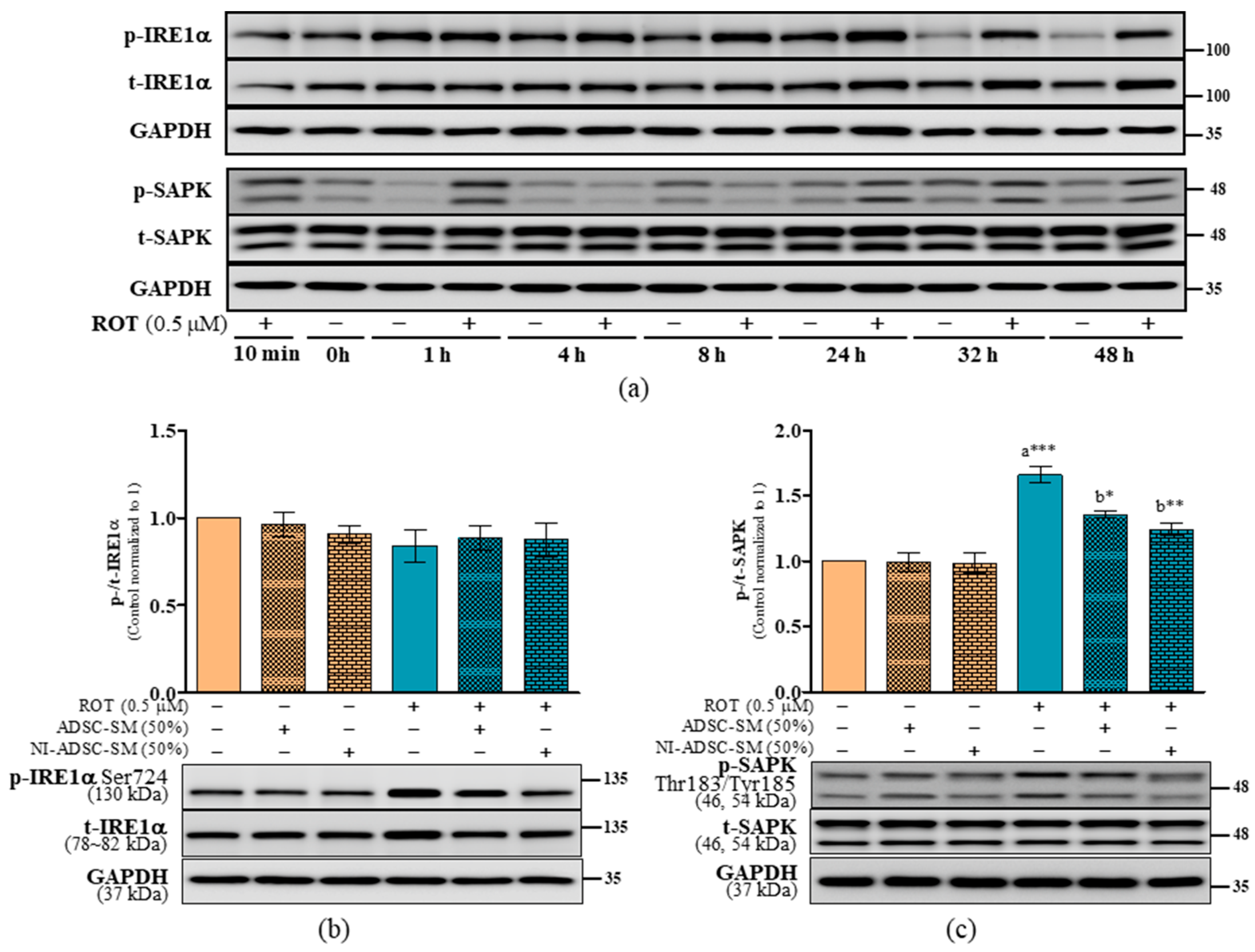
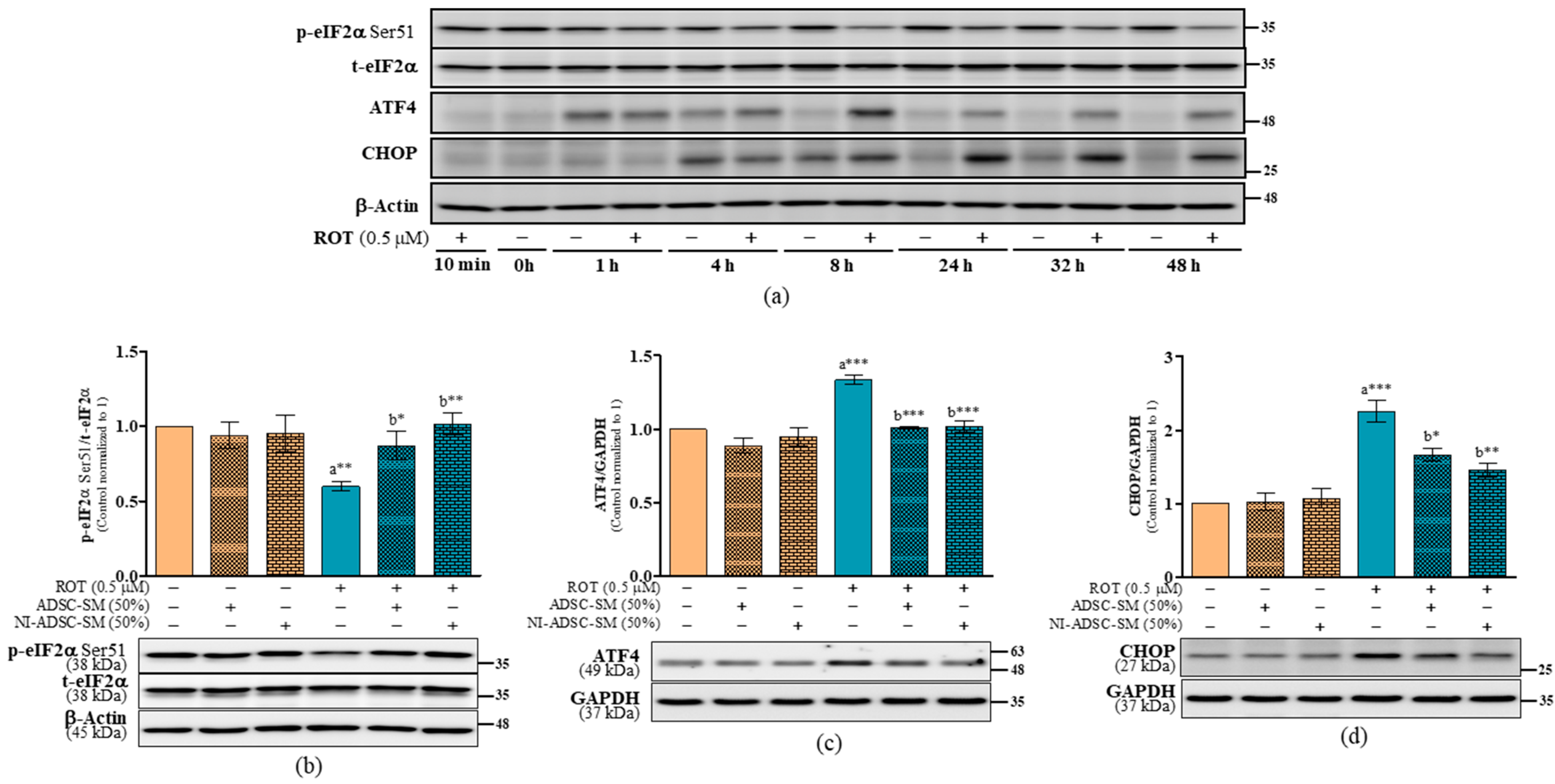
2.7. Effects of the NI-ADSC-SM on Endoplasmic Reticulum Stress Protein Expression after ROT Exposure
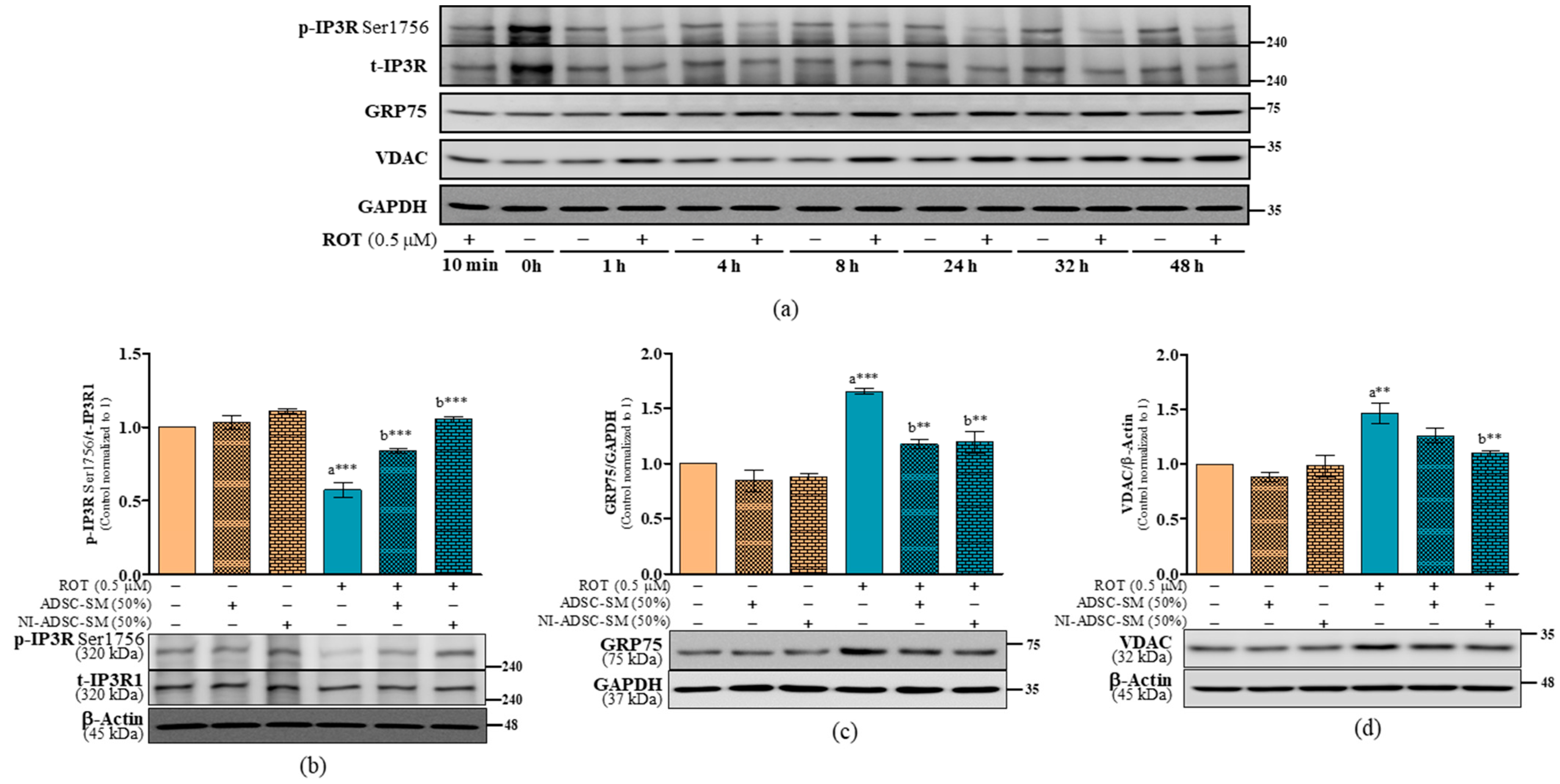
2.8. Effects of the NI-ADSC-SM on IP3R-GRP75-VDAC Tethering Protein Expression after ROT Exposure
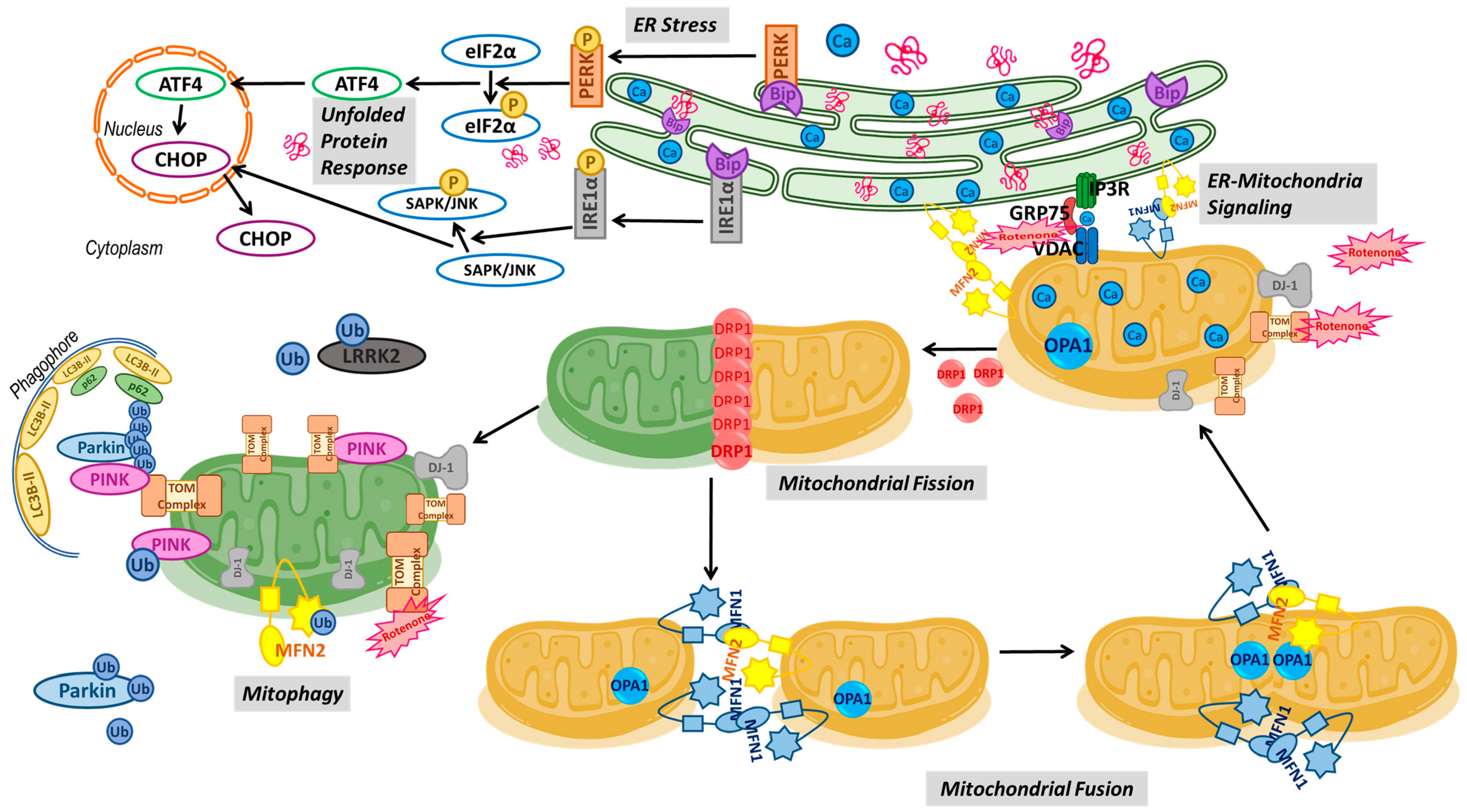
3. Discussion
4. Materials and Methods
4.1. Secretome-Containing Culture Medium Collection from Primary ADSCs and Neural-Induced ADSCs
4.2. Cell Culture
4.3. Rotenone Preparation
4.4. ROT Toxicity and ADSC-SM and NI-ADSC-SM Treatments
4.5. Estimation of Intracellular Calcium (Ca2+) by Fura-2AM
4.6. Preparation of Triton X-100-Soluble and -Insoluble Fractions and Western Blotting
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenamyre, J.T.; Cannon, J.R.; Drolet, R.; Mastroberardino, P.G. Lessons from the rotenone model of Parkinson’s disease. Trends Pharmacol. Sci. 2010, 31, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Ryu, E.J.; Harding, H.P.; Angelastro, J.M.; Vitolo, O.V.; Ron, D.; Greene, L.A. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J. Neurosci. 2002, 22, 10690–10698. [Google Scholar] [CrossRef]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaar5429. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Keeney, M.T.; Di Maio, R.; De Miranda, B.R.; Greenamyre, J.T. LRRK2 and idiopathic Parkinson’s disease. Trends Neurosci. 2022, 45, 224–236. [Google Scholar] [CrossRef]
- Bakula, D.; Scheibye-Knudsen, M. MitophAging: Mitophagy in aging and disease. Front. Cell Dev. Biol. 2020, 8, 239. [Google Scholar] [CrossRef]
- McLelland, G.L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 2018, 7, e32866. [Google Scholar] [CrossRef] [PubMed]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lammermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef]
- Lee, S.; Min, K.T. The interface between ER and mitochondria: Molecular compositions and functions. Mol. Cells 2018, 41, 1000–1007. [Google Scholar]
- Pereira, C.M.F. Crosstalk between endoplasmic reticulum stress and protein misfolding in neurodegenerative diseases. ISRN Cell Biol. 2013, 2013, 256404. [Google Scholar] [CrossRef]
- DeGracia, D.J.; Montie, H.L. Cerebral ischemia and the unfolded protein response. J. Neurochem. 2004, 91, 1–8. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Thivolet, C.; Vial, G.; Cassel, R.; Rieusset, J.; Madec, A.M. Reduction of endoplasmic reticulum- mitochondria interactions in beta cells from patients with type 2 diabetes. PLoS ONE 2017, 12, e0182027. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, M.; Jang, S.; Jeong, H.S. Neural-induced human adipose tissue-derived stem cells conditioned medium ameliorates rotenone-induced toxicity in SH-SY5Y cells. Int. J. Mol. Sci. 2021, 22, 2322. [Google Scholar] [CrossRef]
- Ramalingam, M.; Jeong, H.S.; Hwang, J.; Cho, H.H.; Kim, B.C.; Kim, E.; Jang, S. Autophagy signaling by neural-induced human adipose tissue-derived stem cell-conditioned medium during rotenone-induced toxicity in SH-SY5Y cells. Int. J. Mol. Sci. 2022, 23, 4193. [Google Scholar] [CrossRef]
- Napoli, E.; Borlongan, C.V. Cell Therapy in Parkinson’s Disease: Host Brain Repair Machinery Gets a Boost from Stem Cell Grafts. Stem Cells 2017, 35, 1443–1445. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Salgado, A.J. Mesenchymal stem cells secretome: Current trends and future challenges. Neural Regen. Res. 2020, 15, 75–77. [Google Scholar] [CrossRef]
- Jang, S.; Cho, H.H.; Cho, Y.B.; Park, J.S.; Jeong, H.S. Functional neural differentiation of human adipose tissue-derived stem cells using bFGF and forskolin. BMC Cell Biol. 2010, 11, 25. [Google Scholar] [CrossRef]
- Jang, S.; Cho, H.H.; Kim, S.H.; Lee, K.H.; Cho, Y.B.; Park, J.S.; Jeong, H.S. Transplantation of human adipose tissue-derived stem cells for repair of injured spiral ganglion neurons in deaf guinea pigs. Neural Regen. Res. 2016, 11, 994–1000. [Google Scholar] [CrossRef]
- Jang, S.; Park, J.S.; Jeong, H.S. Neural differentiation of human adipose tissue-derived stem cells involves activation of the Wnt5a/JNK signalling. Stem Cells Int. 2015, 2015, 178618. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet. 2007, 16, R183–R194. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.; Flinkman, D.; Hong, Y.; Goltseva, E.; Siino, V.; Sun, L.H.; Peltonen, S.; Elo, L.L.; Kaasinen, V.; James, P.; et al. Protein synthesis is suppressed in sporadic and familial Parkinson’s disease by LRRK2. FASEB J. 2020, 34, 14217–14233. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Kim, J.W.; Lee, B.D.; Kang, H.C.; Xu, J.C.; Jia, H.; Stankowski, J.; Kim, M.S.; Zhong, J.; Kumar, M.; et al. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell 2014, 157, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Callio, J.; Otero, P.A.; Sekler, I.; Wills, Z.P.; Chu, C.T. Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-associated LRRK2 mutants. J. Neurosci. 2017, 37, 11151–11165. [Google Scholar] [CrossRef]
- Dodson, M.W.; Zhang, T.; Jiang, C.; Chen, S.; Guo, M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012, 21, 1350–1363. [Google Scholar] [CrossRef]
- Dwyer, Z.; Rudyk, C.; Thompson, A.; Farmer, K.; Fenner, B.; Fortin, T.; Derksen, A.; Sun, H.; Hayley, S. Clint, Leucine-rich repeat kinase-2 (LRRK2) modulates microglial phenotype and dopaminergic neurodegeneration. Neurobiol. Aging 2020, 91, 45–55. [Google Scholar] [CrossRef]
- Xu, L.; Wang, X.; Tong, C. Endoplasmic reticulum-mitochondria contact sites and neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 428. [Google Scholar] [CrossRef]
- Van Laar, V.S.; Otero, P.A.; Hastings, T.G.; Berman, S.B. Potential role of Mic60/mitofilin in Parkinson’s disease. Front. Neurosci. 2018, 12, 898. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.L.; Metzakopian, E. ER-mitochondria contact sites in neurodegeneration: Genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ. 2021, 28, 1804–1821. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.K.; Cortese, G.P.; Amodeo, K.D.; Weihofen, A.; Letai, A.; LaVoie, M.J. Parkin selectively alters the intrinsic threshold for mitochondrial cytochrome c release. Hum. Mol. Genet. 2009, 18, 4317–4328. [Google Scholar] [CrossRef]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef]
- Han, S.W.; Choi, J.; Ryu, K.Y. Stress response of mouse embryonic fibroblasts exposed to polystyrene nanoplastics. Int. J. Mol. Sci. 2021, 22, 2094. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef]
- Sampathu, D.M.; Giasson, B.I.; Pawlyk, A.C.; Trojanowski, J.Q.; Lee, V.M. Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am. J. Pathol. 2003, 163, 91–100. [Google Scholar] [CrossRef]
- Lo Bianco, C.; Schneider, B.L.; Bauer, M.; Sajadi, A.; Brice, A.; Iwatsubo, T.; Aebischer, P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 17510–17515. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Usami, Y.; Hatano, T.; Imai, S.; Kubo, S.; Sato, S.; Saiki, S.; Fujioka, Y.; Ohba, Y.; Sato, F.; Funayama, M.; et al. DJ-1 associates with synaptic membranes. Neurobiol. Dis. 2011, 43, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Wang, D.; Chen, L.; Choo, Y.S.; Ma, H.; Tang, C.; Xia, K.; Jiang, W.; Ronai, Z.; Zhuang, X.; et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Investig. 2009, 119, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef]
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S.D. DJ-1 Inhibits alpha-synuclein aggregation by regulating chaperone-mediated autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef]
- Huang, M.; Chen, S. DJ-1 in neurodegenerative diseases: Pathogenesis and clinical application. Prog. Neurobiol. 2021, 204, 102114. [Google Scholar] [CrossRef] [PubMed]
- Shiota, T.; Imai, K.; Qiu, J.; Hewitt, V.L.; Tan, K.; Shen, H.H.; Sakiyama, N.; Fukasawa, Y.; Hayat, S.; Kamiya, M.; et al. Molecular architecture of the active mitochondrial protein gate. Science 2015, 349, 1544–1548. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef]
- Koo, J.H.; Cho, J.Y.; Lee, U.B. Treadmill exercise alleviates motor deficits and improves mitochondrial import machinery in an MPTP-induced mouse model of Parkinson’s disease. Exp. Gerontol. 2017, 89, 20–29. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Suleiman, J.; Almannai, M.; Scaglia, F. Mitochondrial dynamics: Biological roles, molecular machinery, and related diseases. Mol. Genet. Metab. 2018, 125, 315–321. [Google Scholar] [CrossRef]
- Ko, H.J.; Tsai, C.Y.; Chiou, S.J.; Lai, Y.L.; Wang, C.H.; Cheng, J.T.; Chuang, T.H.; Huang, C.F.; Kwan, A.L.; Loh, J.K.; et al. The phosphorylation status of Drp1-ser637 by PKA in mitochondrial fission modulates mitophagy via PINK1/parkin to exert multipolar spindles assembly during mitosis. Biomolecules 2021, 11, 424. [Google Scholar] [CrossRef]
- Chang, C.R.; Blackstone, C. Drp1 phosphorylation and mitochondrial regulation. EMBO Rep. 2007, 8, 1088–1089. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial quality control in neurodegenerative diseases: Focus on Parkinson’s disease and Huntington’s disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Monaco, G.; Missiaen, L.; De Smedt, H.; Parys, J.B.; Bultynck, G. IP(3) receptors, mitochondria, and Ca2+ signaling: Implications for aging. J. Aging Res. 2011, 2011, 920178. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-mitochondria contacts by parkin via mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernandez-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Chen, G.; Fan, Z.; Luo, J.; Ke, Z.J. GSK3beta and endoplasmic reticulum stress mediate rotenone-induced death of SK-N-MC neuroblastoma cells. Biochem. Pharmacol. 2008, 76, 128–138. [Google Scholar] [CrossRef]
- Lima, N.C.R.; Melo, T.Q.; Sakugawa, A.Y.S.; Melo, K.P.; Ferrari, M.F.R. Restoration of Rab1 levels prevents endoplasmic reticulum stress in hippocampal cells during protein aggregation triggered by rotenone. Neuroscience 2019, 419, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 2018, 9, 875. [Google Scholar] [CrossRef] [PubMed]
- Morano, K.A. New tricks for an old dog: The evolving world of Hsp70. Ann. N. Y. Acad. Sci. 2007, 1113, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.W.; Coen, D.M.; Ron, D.; et al. A selective inhibitor-of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Lu, P.D.; Jousse, C.; Marciniak, S.J.; Zhang, Y.H.; Novoa, I.; Scheuner, D.; Kaufman, R.J.; Ron, D.; Harding, H.P. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004, 23, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.H.; Zeng, H.Q.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef]
- Mercado, G.; Castillo, V.; Soto, P.; Lopez, N.; Axten, J.M.; Sardi, S.P.; Hoozemans, J.J.M.; Hetz, C. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis. 2018, 112, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Baleriola, J.; Walker, C.A.; Jean, Y.Y.; Crary, J.F.; Troy, C.M.; Nagy, P.L.; Hengst, U. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 2014, 158, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Hoppins, S.; Nunnari, J. Cell Biology. Mitochondrial dynamics and apoptosis--the ER connection. Science 2012, 337, 1052–1054. [Google Scholar] [CrossRef]
- Kim, J.W.; Yin, X.L.; Jhaldiyal, A.; Khan, M.R.; Martin, I.; Xie, Z.; Perez-Rosello, T.; Kumar, M.; Abalde-Atristain, L.; Xu, J.C.; et al. Defects in mRNA translation in LRRK2-mutant hiPSC-derived dopaminergic neurons lead to dysregulated calcium homeostasis. Cell Stem Cell 2020, 27, 633–645. [Google Scholar] [CrossRef]
- Finch, E.A.; Turner, T.J.; Goldin, S.M. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science 1991, 252, 443–446. [Google Scholar] [CrossRef]
- Higo, T.; Hamada, K.; Hisatsune, C.; Nukina, N.; Hashikawa, T.; Hattori, M.; Nakamura, T.; Mikoshiba, K. Mechanism of ER stress-induced brain damage by IP3 receptor. Neuron 2010, 68, 865–878. [Google Scholar] [CrossRef]
- Cardenas, C.; Muller, M.; McNeal, A.; Lovy, A.; Jana, F.; Bustos, G.; Urra, F.; Smith, N.; Molgo, J.; Diehl, J.A.; et al. Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep. 2016, 14, 2313–2324. [Google Scholar] [CrossRef]
- Liang, T.; Hang, W.J.; Chen, J.H.; Wu, Y.; Wen, B.; Xu, K.; Ding, B.B.; Chen, J. ApoE4 (Delta 272–299) induces mitochondrial-associated membrane formation and mitochondrial impairment by enhancing GRP75-modulated mitochondrial calcium overload in neuron. Cell Biosci. 2021, 11, 50. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Scorrano, L.; Opferman, J.T.; Bassik, M.C.; Nishino, M.; Pozzan, T.; Korsmeyer, S.J. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Bartok, A.; Weaver, D.; Golenar, T.; Nichtova, Z.; Katona, M.; Bansaghi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramalingam, M.; Jang, S.; Hwang, J.; Kim, B.; Cho, H.-H.; Kim, E.; Jeong, H.-S. Neuroprotective Effects of the Neural-Induced Adipose-Derived Stem Cell Secretome against Rotenone-Induced Mitochondrial and Endoplasmic Reticulum Dysfunction. Int. J. Mol. Sci. 2023, 24, 5622. https://doi.org/10.3390/ijms24065622
Ramalingam M, Jang S, Hwang J, Kim B, Cho H-H, Kim E, Jeong H-S. Neuroprotective Effects of the Neural-Induced Adipose-Derived Stem Cell Secretome against Rotenone-Induced Mitochondrial and Endoplasmic Reticulum Dysfunction. International Journal of Molecular Sciences. 2023; 24(6):5622. https://doi.org/10.3390/ijms24065622
Chicago/Turabian StyleRamalingam, Mahesh, Sujeong Jang, Jinsu Hwang, Boeun Kim, Hyong-Ho Cho, Eungpil Kim, and Han-Seong Jeong. 2023. "Neuroprotective Effects of the Neural-Induced Adipose-Derived Stem Cell Secretome against Rotenone-Induced Mitochondrial and Endoplasmic Reticulum Dysfunction" International Journal of Molecular Sciences 24, no. 6: 5622. https://doi.org/10.3390/ijms24065622
APA StyleRamalingam, M., Jang, S., Hwang, J., Kim, B., Cho, H.-H., Kim, E., & Jeong, H.-S. (2023). Neuroprotective Effects of the Neural-Induced Adipose-Derived Stem Cell Secretome against Rotenone-Induced Mitochondrial and Endoplasmic Reticulum Dysfunction. International Journal of Molecular Sciences, 24(6), 5622. https://doi.org/10.3390/ijms24065622