


FapydG in the Shadow of OXOdG—A Theoretical Study of Clustered DNA Lesions


Abstract
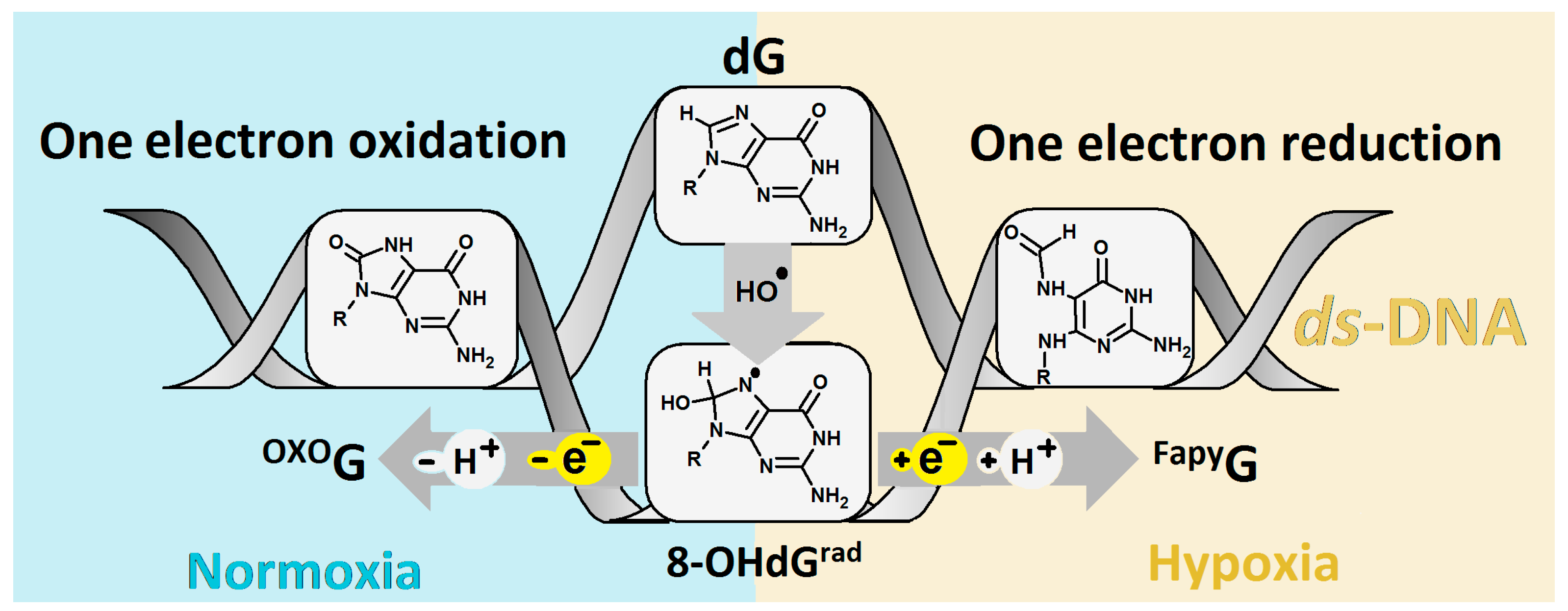
1. Introduction
2. Results
2.1. Electronic Properties and Geometry of oligo-FapyG
2.2. The Charge and Spin Distribution Oligo-FapyG Structure
2.3. Electronic Properties of a Single Base-Pair Extracted from Oligo-FapyG
2.4. The Rate Constant of Charge Transfer through oligo-FapyG
3. Discussion
4. Materials and Methods
5. Conclusions
- The vertical/adiabatic ionization potential (VIPEQ, AIP) and electron affinity (VEA, AEA) of the investigated ds-oligo were noted in [eV] as 5.87/5.39 and −1.41/−2.09, respectively;
- The analysis of four optimized oligo-FapyG spatial geometries contained in the FapydG structure in the two cis and two trans-rotameric forms showed the trans-2 to be energetically privileged. Subsequently, the influence of a multi-damage site including FapyG and OXOG as subunits on the discussed ds-oligo structure was noted as negligible according to the standard DNA reference frame for the analysis of structural parameters. Furthermore, neither the adoption of an excess electron or the loss of an electron by the oligo-FapyG forced a significant macromolecule structural distortion;
- Additionally, for the FapyGC base-pair isolated from the discussed ds-oligo, the ionization potential and electron affinity values were observed to be higher than those assigned for OXOGC;
- These results imply that FapyGC does not have a significant effect on electron–hole or excess–electron charge transfer. In parallel to this, the OXOGC base-pair becomes the radical cation/anion sink in the oligo-FapyG structure, as expected.
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA Damage and Disease: Induction, Repair and Significance; Elsevier: Amsterdam, The Netherlands, 2004; Volume 567, ISBN 1301975850. [Google Scholar]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Steenken, S.; Jovanovic, S.V. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Seidel, C.A.M.; Schulz, A.; Sauer, M.H.M. Nucleobase-Specific Quenching of Fluorescent Dyes. 1. Nucleobase One-Electron Redox.pdf. Society 1996, 100, 5541–5553. [Google Scholar]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef]
- Keith, B.; Simon, M.C. Hypoxia-Inducible Factors, Stem Cells, and Cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Kupan, A.; Saulière, A.; Broussy, S.; Seguy, C.; Pratviel, G.; Meunier, B. Guanine oxidation by electron transfer: One-versus two-electron oxidation mechanism. ChemBioChem 2006, 7, 125–133. [Google Scholar] [CrossRef]
- Shukla, L.I.; Adhikary, A.; Pazdro, R.; Becker, D.; Sevilla, M.D. Formation of 8-oxo-7,8-dihydroguanine-radicals in γ-irradiated DNA by multiple one-electron oxidations. Nucleic Acids Res. 2004, 32, 6565–6574. [Google Scholar] [CrossRef]
- Shafirovich, V.; Mock, S.; Kolbanovskiy, A.; Geacintov, N.E. Photochemically catalyzed generation of site-specific 8-nitroguanine adducts in DNA by the reaction of long-lived neutral guanine radicals with nitrogen dioxide. Chem. Res. Toxicol. 2002, 15, 591–597. [Google Scholar] [CrossRef]
- Shukla, P.K.; Mishra, P.C. Catalytic involvement of CO2 in the mutagenesis caused by reactions of ONOO- with guanine. J. Phys. Chem. B 2008, 112, 4779–4789. [Google Scholar] [CrossRef]
- Scanlan, L.D.; Coskun, S.H.; Jaruga, P.; Hanna, S.K.; Sims, C.M.; Almeida, J.L.; Catoe, D.; Coskun, E.; Golan, R.; Dizdaroglu, M.; et al. Measurement of Oxidatively Induced DNA Damage in Caenorhabditis elegans with High-Salt DNA Extraction and Isotope-Dilution Mass Spectrometry. Anal. Chem. 2019, 91, 12149–12155. [Google Scholar] [CrossRef]
- Greenberg, M.M. The formamidopyrimidines: Purine lesions formed in competition with 8-oxopurines from oxidative stress. Acc. Chem. Res. 2012, 45, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Candeias, S.S.L. Reaction of HO* with guanine derivatives in aqueous solution: Formation of two different redox-active OH-adduct radicals and their unimolecular transformation reactions. Properties of G(-H)*. Chem.—A Eur. J. 2000, 475–484. [Google Scholar] [CrossRef]
- Nyaga, S.G.; Jaruga, P.; Lohani, A.; Dizdaroglu, M.; Evans, M.K. Accumulation of oxidatively induced DNA damage in human breast cancer cell lines following treatment with hydrogen peroxide. Cell Cycle 2007, 6, 1471–1477. [Google Scholar] [CrossRef]
- Arczewska, K.D.; Tomazella, G.G.; Lindvall, J.M.; Kassahun, H.; Maglioni, S.; Torgovnick, A.; Henriksson, J.; Matilainen, O.; Marquis, B.J.; Nelson, B.C.; et al. Active transcriptomic and proteomic reprogramming in the C. elegans nucleotide excision repair mutant xpa-1. Nucleic Acids Res. 2013, 41, 5368–5381. [Google Scholar] [CrossRef]
- Boiteux, S.; Laval, J. Imidazole open ring 7-methylguanine: An inhibitor of DNA synthesis. Biochem. Biophys. Res. Commun. 1983, 110, 552–558. [Google Scholar] [CrossRef]
- Tudek, B.; Gra̧ziewicz, M.; Kazanova, O.; Zastawny, T.H.; Obtułowicz, T.; Laval, J. Mutagenic specificity of imidazole ring-opened 7-methylpurines in M13mp18 phage DNA. Acta Biochim. Pol. 1999, 46, 785–799. [Google Scholar] [CrossRef]
- Burgdorf, L.T.; Carell, T. Synthesis, Stability, and Conformation of the Formamidopyrimidine G DNA Lesion. Chem.—A Eur. J. 2002, 8, 293–301. [Google Scholar] [CrossRef]
- Wiederholt, C.J.; Greenberg, M.M. Fapy·dG instructs Klenow Exo- to misincorporate deoxyadenosine. J. Am. Chem. Soc. 2002, 124, 7278–7279. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Boiteux, S.; O’Connor, T.R.; Laval, J. Formamidopyrimidine-DNA glycosylase of Escherichia coli: Cloning and sequencing of the fpg structural gene and overproduction of the protein. EMBO J. 1987, 6, 3177–3183. [Google Scholar] [CrossRef]
- Jaruga, P.; Birincioglu, M.; Rosenquist, T.A.; Dizdaroglu, M. Mouse NEIL1 protein is specific for excision of 2,6-diamino-4-hydroxy-5- formamidopyrimidine and 4,6-diamino-5-formamidopyrimidine from oxidatively damaged DNA. Biochemistry 2004, 43, 15909–15914. [Google Scholar] [CrossRef] [PubMed]
- Wiederholt, C.J.; Delaney, M.O.; Pope, M.A.; David, S.S.; Greenberg, M.M. Repair of DNA containing Fapy·dG and its β-C-nucleoside analogue by formamidopyrimidine DNA glycosylase and MutY. Biochemistry 2003, 42, 9755–9760. [Google Scholar] [CrossRef] [PubMed]
- Jena, N.R.; Mark, A.E.; Mishra, P.C. Does tautomerization of FapyG influence its mutagenicity? ChemPhysChem 2014, 15, 1779–1784. [Google Scholar] [CrossRef]
- Lin, J.C.; Singh, R.R.P.; Cox, D.L. Theoretical study of DNA damage recognition via electron transfer from the [4Fe-4S] complex of MutY. Biophys. J. 2008, 95, 3259–3268. [Google Scholar] [CrossRef]
- Boal, A.K.; Yavin, E.; Barton, J.K. DNA repair glycosylases with a [4Fe-4S] cluster: A redox cofactor for DNA-mediated charge transport? J. Inorg. Biochem. 2007, 101, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Munk, B.H.; Burrows, C.J.; Schlegel, H.B. Exploration of mechanisms for the transformation of 8-hydroxy guanine radical to FAPyG by density functional theory. Chem. Res. Toxicol. 2007, 20, 432–444. [Google Scholar] [CrossRef]
- Kumar, A.; Adhikary, A.; Sevilla, M.D.; Close, D.M. One-electron oxidation of ds(5′-GGG-3′) and ds(5′-G(8OG)G-3′) and the nature of hole distribution: A density functional theory (DFT) study. Phys. Chem. Chem. Phys. 2020, 22, 5078–5089. [Google Scholar] [CrossRef]
- Olson, W.K.; Bansal, M.; Burley, S.K.; Dickerson, R.E.; Gerstein, M.; Harvey, S.C.; Heinemann, U.; Lu, X.; Neidle, S.; Shakked, Z.; et al. N OMENCLATURE A Standard Reference Frame for the Description of Nucleic Acid Base-pair Geometry. J. Mol. Biol. 2001, 313, 229–237. [Google Scholar] [CrossRef]
- Karwowski, B.T. How Clustered DNA Damage Can Change the Electronic Properties of ds-DNA. Differences between GAG, GAOXOG, and OXOGAOXOG. Biomolecules 2023. (Accepted for publication). [Google Scholar]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Sanii, L.; Schuster, G.B.; June, R. V Long-Distance Charge Transport in DNA: Sequence-Dependent Radical Cation Injection Efficiency. J. Am. Chem. Soc. 2000, 122, 11545–11546. [Google Scholar] [CrossRef]
- Karwowsk, B.T. The influence of single, tandem, and clustered DNA damage on the electronic properties of the double helix: A theoretical study. Molecules 2020, 25, 3126. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. Electron Transfer Reactions in Chemistry: Theory and Experiment (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1993, 32, 1111–1121. [Google Scholar] [CrossRef]
- Karwowski, B.T. The AT Interstrand Cross-Link: Structure, Electronic Properties, and Influence on Charge Transfer in dsDNA. Mol. Ther.—Nucleic Acids 2018, 13, 665–685. [Google Scholar] [CrossRef] [PubMed]
- Mignon, P.; Loverix, S.; Steyaert, J.; Geerlings, P. Influence of the π-π interaction on the hydrogen bonding capacity of stacked DNA/RNA bases. Nucleic Acids Res. 2005, 33, 1779–1789. [Google Scholar] [CrossRef]
- Arnold, A.R.; Grodick, M.A.; Barton, J.K. Review DNA Charge Transport: From Chemical Principles to the Cell. Cell Chem. Biol. 2016, 23, 183–197. [Google Scholar] [CrossRef]
- Boal, A.K.; Yavin, E.; Lukianova, O.A.; O’Shea, V.L.; David, S.S.; Barton, J.K. DNA-bound redox activity of DNA repair glycosylases containing [4Fe-4S] clusters. Biochemistry 2005, 44, 8397–8407. [Google Scholar] [CrossRef]
- Cammack, R. Iron-Sulfur Proteins, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; Volume 2, ISBN 9780123786319. [Google Scholar]
- Lomax, M.E.; Folkes, L.K.; Neill, P.O. Biological Consequences of Radiation-induced DNA Damage: Relevance to Radiotherapy Statement of Search Strategies Used and Sources of Information Why Radiation Damage is More Effective than Endogenous Damage at Killing Cells Ionising Radiation-induced Do. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 2011, 711, 123–133. [Google Scholar] [CrossRef]
- Jena, N.R.; Mishra, P.C. Is FapyG mutagenic?: Evidence from the DFT study. ChemPhysChem 2013, 14, 3263–3270. [Google Scholar] [CrossRef]
- Reynisson, J.; Steenken, S. DFT calculations on the electrophilic reaction with water of the guanine and adenine radical cations. A model for the situation in DNA. Phys. Chem. Chem. Phys. 2002, 4, 527–532. [Google Scholar] [CrossRef]
- Elias, B.; Shao, F.; Barton, J.K. Charge migration along the DNA duplex: Hole versus electron transport. J. Am. Chem. Soc. 2008, 130, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Voityuk, A.A.; Jortner, J.; Bixon, M.; Rösch, N. Energetics of hole transfer in DNA. Chem. Phys. Lett. 2000, 324, 430–434. [Google Scholar] [CrossRef]
- Senthilkumar, K.; Grozema, F.C.; Guerra, C.F.; Bickelhaupt, F.M.; Siebbeles, L.D.A. Mapping the Sites for Selective Oxidation of Guanines in DNA. J. Am. Chem. Soc. 2003, 125, 13658–13659. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Majima, T. Hole transfer kinetics of DNA. Acc. Chem. Res. 2013, 46, 2616–2625. [Google Scholar] [CrossRef]
- Lewis, F.D.; Liu, J.; Weigel, W.; Rettig, W.; Kurnikov, I.V.; Beratan, D.N. Donor-bridge-acceptor energetics determine the distance dependence of electron tunneling in DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 12536–12541. [Google Scholar] [CrossRef]
- Aust, A.E.; Eveleigh, J.F. Mechanisms of DNA oxidation. Proc. Soc. Exp. Biol. Med. 1999, 222, 246–252. [Google Scholar] [CrossRef]
- Schuster, G.B.; Landman, U. The Mechanism of Long-Distance Radical Cation Transport in Duplex DNA: Ion-Gated Hopping of Polaron-Like Distortions. In Long-Range Charge Transfer in DNA I; Springer: Berlin/Heidelberg, Germany, 2012; pp. 139–161. [Google Scholar] [CrossRef]
- BIOVIA. Discovery Studio Visualizer; v16.1.0.15350; BIOVIA: San Diego, CA, USA, 2015. [Google Scholar]
- Dapprich, S.; Komáromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. THEOCHEM 1999, 461–462, 1–21. [Google Scholar] [CrossRef]
- Zhao, Y.; Pu, J.; Lynch, B.J.; Truhlar, D.G. Tests of second-generation and third-generation density functionals for thermochemical kineticsElectronic supplementary information (ESI) available: Mean errors for pure and hybrid DFT methods. See http://www.rsc.org/suppdata/cp/b3/b316260e/. Phys. Chem. Chem. Phys. 2004, 6, 673. [Google Scholar] [CrossRef]
- Karwowski, B.T. Ionisation potential and electron affinity of free 5′,8-cyclopurine-2′-deoxynucleosides. DFT study in gaseous and aqueous phase. Cent. Eur. J. Chem. 2010, 8, 70–76. [Google Scholar] [CrossRef]
- Kumar, A.; Sevilla, M.D. Photoexcitation of dinucleoside radical cations: A time-dependent density functional study. J. Phys. Chem. B 2006, 110, 24181–24188. [Google Scholar] [CrossRef] [PubMed]
- Cave, R.J.; Newton, M.D. Generalization of the Mulliken-Hush treatment for the calculation of electron transfer matrix elements. Chem. Phys. Lett. 1996, 249, 15–19. [Google Scholar] [CrossRef]
- Miertus̃, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2019; Volume 2019, p. 2019. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Base Pair | HB1 | HB2 | HB3 | λ1 | λ2 | dC1′.C1′ | Base Pair Dimmer | Rise |
|---|---|---|---|---|---|---|---|---|
| A1T5 | 3.03 | 2.82 | 55.4 | 57.9 | 10.4 | A1T5|FG2C4 | 2.87 | |
| FG2C4 | 2.84 | 2.90 | 2.92 | 55.6 | 20.4 | 10.5 | G2C4|A3T3 | 2.95 |
| A3T3 | 2.97 | 2.88 | 54.6 | 53.1 | 10.5 | A3T3|OG2C4 | 3.25 | |
| OG2C4 | 2.90 | 2.89 | 2.88 | 55.0 | 51.3 | 10.7 | OG2C4|A5T1 | 3.15 |
| A5T1 | 2.82 | 3.01 | 55.4 | 57.9 | 10.4 | |||
| Reference parameters calculated for native ds-oligo structure [30] | ||||||||
| AT | 2.79 | 2.95 | 52.2 | 48.1 | 10.8 | AT|GC | 3.29 | |
| GC | 2.87 | 2.87 | 2.88 | 54.4 | 49.6 | 10.7 | GC|AT | 2.96 |
| oligo-FapyG | |||||
|---|---|---|---|---|---|
| VIPNE | VIPEQ | AIP | VEANE | VEAEQ | AEA |
| (a) 6.50 | 5.87 | 5.39 | −0.90 | −1.41 | −2.09 |
| (b) 6.32 | 5.80 | 5.38 | −0.65 | −1.35 | −1.94 |
| *(c) 6.72 | 6.08 | 5.65 | −0.84 | −1.58 | −2.09 |
| *(d) 6.48 | 5.98 | 5.58 | −0.60 | −1.34 | −1.90 |
| RMSD: Anion versus Neutral | RMSD: Cation versus Neutral | ||||
| ds-DNA | BP | PS-Frame | ds-DNA | BP | PS-Frame |
| 0.19 | 0.16 | 0.22 | 0.34 | 0.28 | 0.39 |
| * 0.17 | 0.16 | 0.17 | 0.36 | 0.29 | 0.42 |
| Electronic Properties in [eV] | ||||
|---|---|---|---|---|
| Base-Pair | VIP | AIP | VEA | AEA |
| A1T5 | 6.65 | 6.65 | −1.39 | −1.39 |
| FapyG2C4 | 6.17 | 6.16 | −1.46 | −1.47 |
| A3T3 | 6.63 | 6.63 | −1.38 | −1.31 |
| OXOG4C2 | 5.90 | 5.56 | −1.53 | −1.97 |
| A5T1 | 6.73 | 6.69 | −1.43 | −1.39 |
| GC32 | 6.13 | 5.83 | −1.52 | −1.95 |
| AT32 | 6.65 | 6.60 | −1.40 | −1.40 |
| Electron–Hole Transfer | |||||
|---|---|---|---|---|---|
| System | λ | ΔG | Ea | V12 | kHT(s−1) |
| A1T1→FapyG2C2 | 0.02 | −0.49 | 3.78 | 0.24 | ~0.0 |
| FapyG2C2←A3T3 | 0.01 | −0.47 | 6.99 | 0.23 | ~0.0 |
| A3T3→OG4C4 | 0.34 | −1.07 | 0.40 | 0.40 | 7.53 × 108 |
| OXOG4C4←A5T5 | 0.39 | −1.13 | 0.34 | 0.41 | 6.90 × 109 |
| A1T1→A3T3 | 0.00 | −0.02 | −0.12 | 0.01 | ND |
| FapyG2C2→OG4C4 | 0.35 | −0.60 | 0.05 | 0.17 | 1.4 × 1014 |
| A3T5←A5T5 | 0.00 | −0.06 | −1.70 | 0.05 | ND |
| Excess Electron Transfer | |||||
| A1T1→FapyG 2C2 | 0.02 | −0.09 | 0.07 | 0.01 | 1.62 × 1012 |
| FapyG 2C2←A3T3 | 0.06 | −0.17 | 0.04 | 0.04 | 2.20 × 1013 |
| A3T3→OG 4C4 | 0.49 | −0.66 | 0.02 | 0.07 | 5.65 × 1013 |
| OXOG 4C4←A5T5 | 0.50 | −0.58 | 0.00 | 0.05 | 5.18 × 1013 |
| A1T1←A3T3 | −0.06 | −0.08 | −0.08 | 0.09 | ND |
| FapyG2C2→OG4C4 | 0.44 | −0.49 | 0.002 | 0.05 | 6.9 × 1013 |
| A3T5→A5T5 | 0.02 | −0.08 | 0.08 | 0.08 | 4.7 × 1013 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karwowski, B.T. FapydG in the Shadow of OXOdG—A Theoretical Study of Clustered DNA Lesions. Int. J. Mol. Sci. 2023, 24, 5361. https://doi.org/10.3390/ijms24065361
Karwowski BT. FapydG in the Shadow of OXOdG—A Theoretical Study of Clustered DNA Lesions. International Journal of Molecular Sciences. 2023; 24(6):5361. https://doi.org/10.3390/ijms24065361
Chicago/Turabian StyleKarwowski, Bolesław T. 2023. "FapydG in the Shadow of OXOdG—A Theoretical Study of Clustered DNA Lesions" International Journal of Molecular Sciences 24, no. 6: 5361. https://doi.org/10.3390/ijms24065361
APA StyleKarwowski, B. T. (2023). FapydG in the Shadow of OXOdG—A Theoretical Study of Clustered DNA Lesions. International Journal of Molecular Sciences, 24(6), 5361. https://doi.org/10.3390/ijms24065361