


Enhanced Apoptosis and Loss of Cell Viability in Melanoma Cells by Combined Inhibition of ERK and Mcl-1 Is Related to Loss of Mitochondrial Membrane Potential, Caspase Activation and Upregulation of Proapoptotic Bcl-2 Proteins
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Strongly Decreased Cell Viability by Combination of MAPK and Mcl-1 Inhibitors
2.2. Loss of Cell Proliferation in Parallel with Decreased Cell Viability
2.3. Decreased Cell Viability Correlates with Induction of Apoptosis
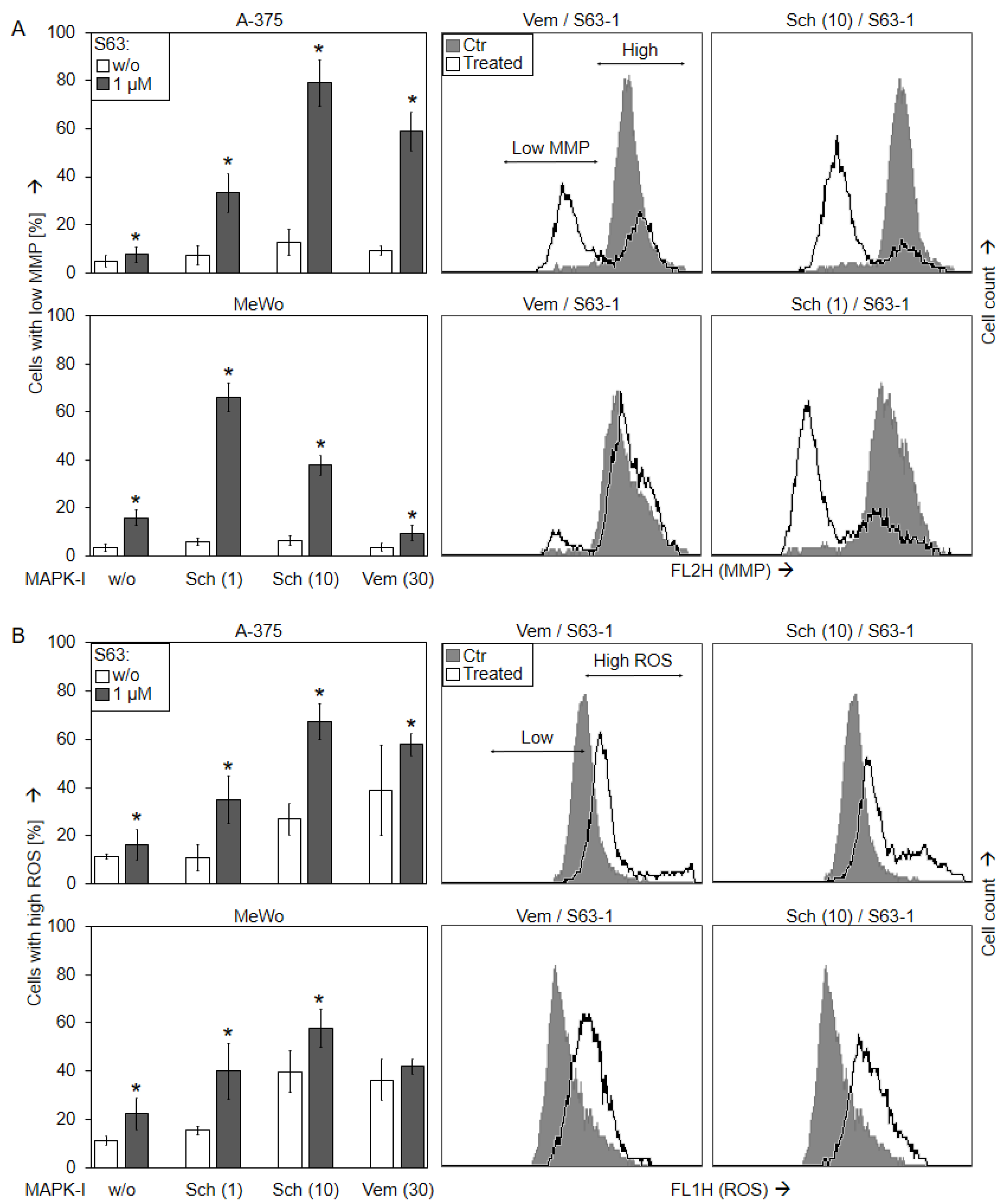
2.4. Loss of mMP and Production of ROS
2.5. Significant Role of Caspase Activation
2.6. Upregulation of Proapoptotic BH3-Only Proteins
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Quantification of Cell Viability and Apoptosis
4.3. Cell Proliferation Assays
4.4. Determination of Mitochondrial Membrane Potential and Reactive Oxygen Species (ROS)
4.5. Caspase Activation Assay and Cytochrome C Release
4.6. Western Blotting
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olsen, C.M. Global trends in melanoma mortality differ by sex and age. Br. J. Derm. 2020, 183, 985–986. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dréno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics: Update 2022. Eur. J. Cancer 2022, 170, 236–255. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Long, G.V. Systemic treatment for BRAF-mutant melanoma: Where do we go next? Lancet Oncol. 2014, 15, e371–e381. [Google Scholar] [CrossRef]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dreno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: Treatment—Update 2022. Eur. J. Cancer 2022, 170, 256–284. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.; Klairmont, M.; Sharfman, W.H.; Kaufman, H.L. Interleukin-2, Ipilimumab, and Anti-PD-1: Clinical management and the evolving role of immunotherapy for the treatment of patients with metastatic melanoma. Cancer Biol. 2021, 22, 513–526. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Menzies, A.M.; Amaria, R.N.; Rozeman, E.A.; Huang, A.C.; Tetzlaff, M.T.; van de Wiel, B.A.; Lo, S.; Tarhini, A.A.; Burton, E.M.; Pennington, T.E.; et al. Pathological response and survival with neoadjuvant therapy in melanoma: A pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Nat. Med. 2021, 27, 301–309. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.J.; Eroglu, Z.; Brockstein, B.; Poklepovic, A.S.; Bajaj, M.; Babu, S.; Hallmeyer, S.; Velasco, M.; Lutzky, J.; Higgs, E.; et al. Pembrolizumab Plus Ipilimumab Following Anti-PD-1/L1 Failure in Melanoma. J. Clin. Oncol. 2021, 39, 2647–2655. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Krammer, P.H.; Arnold, R.; Lavrik, I.N. Life and death in peripheral T cells. Nat. Rev. Immunol. 2007, 7, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Eberle, J. Countering TRAIL Resistance in Melanoma. Cancers 2019, 11, 656. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell. 2010, 37, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell. Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Zhivkova, V.; Kiecker, F.; Langer, P.; Eberle, J. Crucial role of reactive oxygen species (ROS) for the proapoptotic effects of indirubin derivative DKP-073 in melanoma cells. Mol. Carcinog. 2019, 58, 258–269. [Google Scholar] [CrossRef]
- Quast, S.A.; Berger, A.; Eberle, J. ROS-dependent phosphorylation of Bax by wortmannin sensitizes melanoma cells for TRAIL-induced apoptosis. Cell. Death Dis. 2013, 4, e839. [Google Scholar] [CrossRef]
- Plotz, M.; Eberle, J. BH3-only proteins: Possible proapoptotic triggers for melanoma therapy. Exp. Derm. 2014, 23, 375–378. [Google Scholar] [CrossRef]
- Inamdar, G.S.; Madhunapantula, S.V.; Robertson, G.P. Targeting the MAPK pathway in melanoma: Why some approaches succeed and other fail. Biochem. Pharm. 2010, 80, 624–637. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef]
- Morris, E.J.; Jha, S.; Restaino, C.R.; Dayananth, P.; Zhu, H.; Cooper, A.; Carr, D.; Deng, Y.; Jin, W.; Black, S.; et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013, 3, 742–750. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.F.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Nieves-Neira, W.; Boon, C.; Pommier, Y.; Bonner, W.M. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000, 275, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Townsend, P.A.; Kozhevnikova, M.V.; Cexus, O.N.F.; Zamyatnin, A.A., Jr.; Soond, S.M. BH3-mimetics: Recent developments in cancer therapy. J. Exp. Clin. Cancer Res. 2021, 40, 355. [Google Scholar] [CrossRef]
- Brennan, M.S.; Chang, C.; Tai, L.; Lessene, G.; Strasser, A.; Dewson, G.; Kelly, G.L.; Herold, M.J. Humanized Mcl-1 mice enable accurate preclinical evaluation of MCL-1 inhibitors destined for clinical use. Blood 2018, 132, 1573–1583. [Google Scholar] [CrossRef]
- Senichkin, V.V.; Streletskaia, A.Y.; Gorbunova, A.S.; Zhivotovsky, B.; Kopeina, G.S. Saga of Mcl-1: Regulation from transcription to degradation. Cell. Death Differ. 2020, 27, 405–419. [Google Scholar] [CrossRef]
- Tseng, H.Y.; Dreyer, J.; Emran, A.A.; Gunatilake, D.; Pirozyan, M.; Cullinane, C.; Dutton-Regester, K.; Rizos, H.; Hayward, N.K.; McArthur, G.; et al. Co-targeting bromodomain and extra-terminal proteins and MCL1 induces synergistic cell death in melanoma. Int. J. Cancer 2020, 147, 2176–2189. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Grob, J.J.; Amonkar, M.M.; Karaszewska, B.; Schachter, J.; Dummer, R.; Mackiewicz, A.; Stroyakovskiy, D.; Drucis, K.; Grange, F.; Chiarion-Sileni, V.; et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): Results of a phase 3, open-label, randomised trial. Lancet Oncol. 2015, 16, 1389–1398. [Google Scholar] [CrossRef]
- Dummer, R.; Brase, J.C.; Garrett, J.; Campbell, C.D.; Gasal, E.; Squires, M.; Gusenleitner, D.; Santinami, M.; Atkinson, V.; Mandala, M.; et al. Adjuvant dabrafenib plus trametinib versus placebo in patients with resected, BRAF(V600)-mutant, stage III melanoma (COMBI-AD): Exploratory biomarker analyses from a randomised, phase 3 trial. Lancet Oncol. 2020, 21, 358–372. [Google Scholar] [CrossRef]
- Bhagwat, S.V.; McMillen, W.T.; Cai, S.; Zhao, B.; Whitesell, M.; Shen, W.; Kindler, L.; Flack, R.S.; Wu, W.; Anderson, B.; et al. ERK Inhibitor LY3214996 Targets ERK Pathway-Driven Cancers: A Therapeutic Approach Toward Precision Medicine. Mol. Cancer Ther. 2020, 19, 325–336. [Google Scholar] [CrossRef]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer 2017, 16, 2351–2363. [Google Scholar] [CrossRef]
- Moschos, S.J.; Sullivan, R.J.; Hwu, W.J.; Ramanathan, R.K.; Adjei, A.A.; Fong, P.C.; Shapira-Frommer, R.; Tawbi, H.A.; Rubino, J.; Rush, T.S., 3rd; et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 2018, 3, e92352. [Google Scholar] [CrossRef]
- Varga, A.; Soria, J.C.; Hollebecque, A.; LoRusso, P.; Bendell, J.; Huang, S.A.; Wagle, M.C.; Okrah, K.; Liu, L.; Murray, E.; et al. A First-in-Human Phase I Study to Evaluate the ERK1/2 Inhibitor GDC-0994 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 1229–1236. [Google Scholar] [CrossRef]
- Goodwin, C.M.; Waters, A.M.; Klomp, J.E.; Javaid, S.; Bryant, K.L.; Stalnecker, C.A.; Drizyte-Miller, K.; Papke, B.; Yang, R.; Amparo, A.M.; et al. Combination Therapies with CDK4/6 Inhibitors to Treat KRAS-mutant Pancreatic Cancer. Cancer Res. 2022, 83, 141–157. [Google Scholar] [CrossRef]
- Wong, D.J.L.; Robert, L.; Atefi, M.S.; Lassen, A.; Avarappatt, G.; Cerniglia, M.; Avramis, E.; Tsoi, J.; Foulad, D.; Graeber, T.G.; et al. Antitumor activity of the ERK inhibitor SCH722984 against BRAF mutant, NRAS mutant and wild-type melanoma. Mol. Cancer 2014, 13, 194. [Google Scholar] [CrossRef]
- Dummer, R.; Lebbe, C.; Atkinson, V.; Mandala, M.; Nathan, P.D.; Arance, A.; Richtig, E.; Yamazaki, N.; Robert, C.; Schadendorf, D.; et al. Combined PD-1, BRAF and MEK inhibition in advanced BRAF-mutant melanoma: Safety run-in and biomarker cohorts of COMBI-i. Nat. Med. 2020, 26, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Tarhini, A.A.; Cohen, G.I.; Truong, T.G.; Moon, H.H.; Davar, D.; O’Rourke, M.; Stephenson, J.J.; et al. Combination Dabrafenib and Trametinib Versus Combination Nivolumab and Ipilimumab for Patients with Advanced BRAF-Mutant Melanoma: The DREAMseq Trial-ECOG-ACRIN EA6134. J. Clin. Oncol. 2023, 41, 186–197. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Overall survival with first-line atezolizumab in combination with vemurafenib and cobimetinib in BRAF(V600) mutation-positive advanced melanoma (IMspire150): Second interim analysis of a multicentre, randomised, phase 3 study. Lancet Oncol. 2023, 24, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Missert, D.J.; Asciolla, J.J.; Podgrabinska, S.; Wieder, S.Y.; Izadmehr, S.; Belbin, G.; Skobe, M.; Chipuk, J.E. Anti-apoptotic BCL-2 proteins govern cellular outcome following B-RAF(V600E) inhibition and can be targeted to reduce resistance. Oncogene 2015, 34, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Frederick, D.T.; Salas Fragomeni, R.A.; Schalck, A.; Ferreiro-Neira, I.; Hoff, T.; Cooper, Z.A.; Haq, R.; Panka, D.J.; Kwong, L.N.; Davies, M.A.; et al. Clinical profiling of BCL-2 family members in the setting of BRAF inhibition offers a rationale for targeting de novo resistance using BH3 mimetics. PLoS One 2014, 9, e101286. [Google Scholar] [CrossRef]
- Karakas, B.; Ozmay, Y.; Basaga, H.; Gul, O.; Kutuk, O. Distinct apoptotic blocks mediate resistance to panHER inhibitors in HER2+ breast cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1073–1087. [Google Scholar] [CrossRef]
- Su, Y.; Li, X.; Ma, J.; Zhao, J.; Liu, S.; Wang, G.; Edwards, H.; Taub, J.W.; Lin, H.; Ge, Y. Targeting PI3K, mTOR, ERK, and Bcl-2 signaling network shows superior antileukemic activity against AML ex vivo. Biochem. Pharm. 2018, 148, 13–26. [Google Scholar] [CrossRef]
- Lee, E.F.; Harris, T.J.; Tran, S.; Evangelista, M.; Arulananda, S.; John, T.; Ramnac, C.; Hobbs, C.; Zhu, H.; Gunasingh, G.; et al. BCL-XL and MCL-1 are the key BCL-2 family proteins in melanoma cell survival. Cell. Death Dis. 2019, 10, 342. [Google Scholar] [CrossRef]
- Winkler, M.; Friedrich, J.; Boedicker, C.; Dolgikh, N. Co-targeting MCL-1 and ERK1/2 kinase induces mitochondrial apoptosis in rhabdomyosarcoma cells. Transl. Oncol. 2022, 16, 101313. [Google Scholar] [CrossRef]
- Szlávik, Z.; Ondi, L.; Csékei, M.; Paczal, A.; Szabó, Z.B.; Radics, G.; Murray, J.; Davidson, J.; Chen, I.; Davis, B.; et al. Structure-Guided Discovery of a Selective Mcl-1 Inhibitor with Cellular Activity. J. Med. Chem. 2019, 62, 6913–6924. [Google Scholar] [CrossRef]
- Hartman, M.L.; Gajos-Michniewicz, A.; Talaj, J.A.; Mielczarek-Lewandowska, A.; Czyz, M. BH3 mimetics potentiate pro-apoptotic activity of encorafenib in BRAF(V600E) melanoma cells. Cancer Lett. 2021, 499, 122–136. [Google Scholar] [CrossRef]
- Fofaria, N.M.; Frederick, D.T.; Sullivan, R.J.; Flaherty, K.T.; Srivastava, S.K. Overexpression of Mcl-1 confers resistance to BRAFV600E inhibitors alone and in combination with MEK1/2 inhibitors in melanoma. Oncotarget 2015, 6, 40535–40556. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.; Werth, F.; Nguyen, H.A.; Kiecker, F.; Eberle, J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell. Death Dis. 2017, 8, e2594. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, M.; Bauer, J.A.; Martín de la Vega, C.; Wang, G.; Wolter, K.G.; Brenner, J.C.; Nikolovska-Coleska, Z.; Bengtson, A.; Nair, R.; Elder, J.T.; et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 2006, 66, 11348–11359. [Google Scholar] [CrossRef] [PubMed]
- Franke, J.C.; Plötz, M.; Prokop, A.; Geilen, C.C.; Schmalz, H.G.; Eberle, J. New caspase-independent but ROS-dependent apoptosis pathways are targeted in melanoma cells by an iron-containing cytosine analogue. Biochem. Pharm. 2010, 79, 575–586. [Google Scholar] [CrossRef]
- Li, Z.; He, S.; Look, A.T. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia 2019, 33, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Alcon, C.; Gómez Tejeda Zañudo, J.; Albert, R.; Wagle, N.; Scaltriti, M.; Letai, A.; Samitier, J.; Montero, J. ER+ Breast Cancer Strongly Depends on MCL-1 and BCL-xL Anti-Apoptotic Proteins. Cells 2021, 10, 1659. [Google Scholar] [CrossRef] [PubMed]
- Hird, A.W.; Tron, A.E. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol. Ther. 2019, 198, 59–67. [Google Scholar] [CrossRef]
- Qin, J.; Xin, H.; Nickoloff, B.J. Specifically targeting ERK1 or ERK2 kills melanoma cells. J. Transl. Med. 2012, 10, 15. [Google Scholar] [CrossRef]
- van Delft, M.F.; Huang, D.C. How the Bcl-2 family of proteins interact to regulate apoptosis. Cell. Res. 2006, 16, 203–213. [Google Scholar] [CrossRef]
- Wakeyama, H.; Akiyama, T.; Takahashi, K.; Amano, H.; Kadono, Y.; Nakamura, M.; Oshima, Y.; Itabe, H.; Nakayama, K.I.; Nakayama, K.; et al. Negative feedback loop in the Bim-caspase-3 axis regulating apoptosis and activity of osteoclasts. J. Bone Min. Res. 2007, 22, 1631–1639. [Google Scholar] [CrossRef]
- Hadji, A.; Clybouw, C.; Auffredou, M.T.; Alexia, C.; Poalas, K.; Burlion, A.; Feraud, O.; Leca, G.; Vazquez, A. Caspase-3 triggers a TPCK-sensitive protease pathway leading to degradation of the BH3-only protein puma. Apoptosis 2010, 15, 1529–1539. [Google Scholar] [CrossRef]
- Berger, A.; Quast, S.A.; Plötz, M.; Kuhn, N.F.; Trefzer, U.; Eberle, J. RAF inhibition overcomes resistance to TRAIL-induced apoptosis in melanoma cells. J. Investig. Dermatol. 2014, 134, 430–440. [Google Scholar] [CrossRef]
- Montero, J.; Gstalder, C.; Kim, D.J.; Sadowicz, D.; Miles, W.; Manos, M.; Cidado, J.R.; Paul Secrist, J.; Tron, A.E.; Flaherty, K.; et al. Destabilization of NOXA mRNA as a common resistance mechanism to targeted therapies. Nat. Commun. 2019, 10, 5157. [Google Scholar] [CrossRef]
- Basile, K.J.; Aplin, A.E. Downregulation of Noxa by RAF/MEK inhibition counteracts cell death response in mutant B-RAF melanoma cells. Am. J. Cancer Res. 2012, 2, 726–735. [Google Scholar]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, B.; Lehmann, J.M.; Zieglerheitbrock, H.W.L.; Funke, I.; Riethmuller, G. Glycoprotein P3.58, Associated with Tumor Progression in Malignant-Melanoma, Is a Novel Leukocyte Activation Antigen. Int. J. Cancer 1988, 41, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Bean, M.A.; Bloom, B.R.; Herberman, R.B.; Old, L.J.; Oettgen, H.F.; Klein, G.; Terry, W.D. Cell-mediated cytotoxicity for bladder carcinoma: Evaluation of a workshop. Cancer Res. 1975, 35, 2902–2913. [Google Scholar] [PubMed]
- Carey, T.E.; Takahashi, T.; Resnick, L.A.; Oettgen, H.F.; Old, L.J. Cell surface antigens of human malignant melanoma: Mixed hemadsorption assays for humoral immunity to cultured autologous melanoma cells. Proc. Natl. Acad. Sci. USA 1976, 73, 3278–3282. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Z.; Gillissen, B.; Richter, A.; Sinnberg, T.; Schlaak, M.S.; Eberle, J. Enhanced Apoptosis and Loss of Cell Viability in Melanoma Cells by Combined Inhibition of ERK and Mcl-1 Is Related to Loss of Mitochondrial Membrane Potential, Caspase Activation and Upregulation of Proapoptotic Bcl-2 Proteins. Int. J. Mol. Sci. 2023, 24, 4961. https://doi.org/10.3390/ijms24054961
Peng Z, Gillissen B, Richter A, Sinnberg T, Schlaak MS, Eberle J. Enhanced Apoptosis and Loss of Cell Viability in Melanoma Cells by Combined Inhibition of ERK and Mcl-1 Is Related to Loss of Mitochondrial Membrane Potential, Caspase Activation and Upregulation of Proapoptotic Bcl-2 Proteins. International Journal of Molecular Sciences. 2023; 24(5):4961. https://doi.org/10.3390/ijms24054961
Chicago/Turabian StylePeng, Zhe, Bernhard Gillissen, Antje Richter, Tobias Sinnberg, Max S. Schlaak, and Jürgen Eberle. 2023. "Enhanced Apoptosis and Loss of Cell Viability in Melanoma Cells by Combined Inhibition of ERK and Mcl-1 Is Related to Loss of Mitochondrial Membrane Potential, Caspase Activation and Upregulation of Proapoptotic Bcl-2 Proteins" International Journal of Molecular Sciences 24, no. 5: 4961. https://doi.org/10.3390/ijms24054961
APA StylePeng, Z., Gillissen, B., Richter, A., Sinnberg, T., Schlaak, M. S., & Eberle, J. (2023). Enhanced Apoptosis and Loss of Cell Viability in Melanoma Cells by Combined Inhibition of ERK and Mcl-1 Is Related to Loss of Mitochondrial Membrane Potential, Caspase Activation and Upregulation of Proapoptotic Bcl-2 Proteins. International Journal of Molecular Sciences, 24(5), 4961. https://doi.org/10.3390/ijms24054961