
BMPER Improves Vascular Remodeling and the Contractile Vascular SMC Phenotype

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
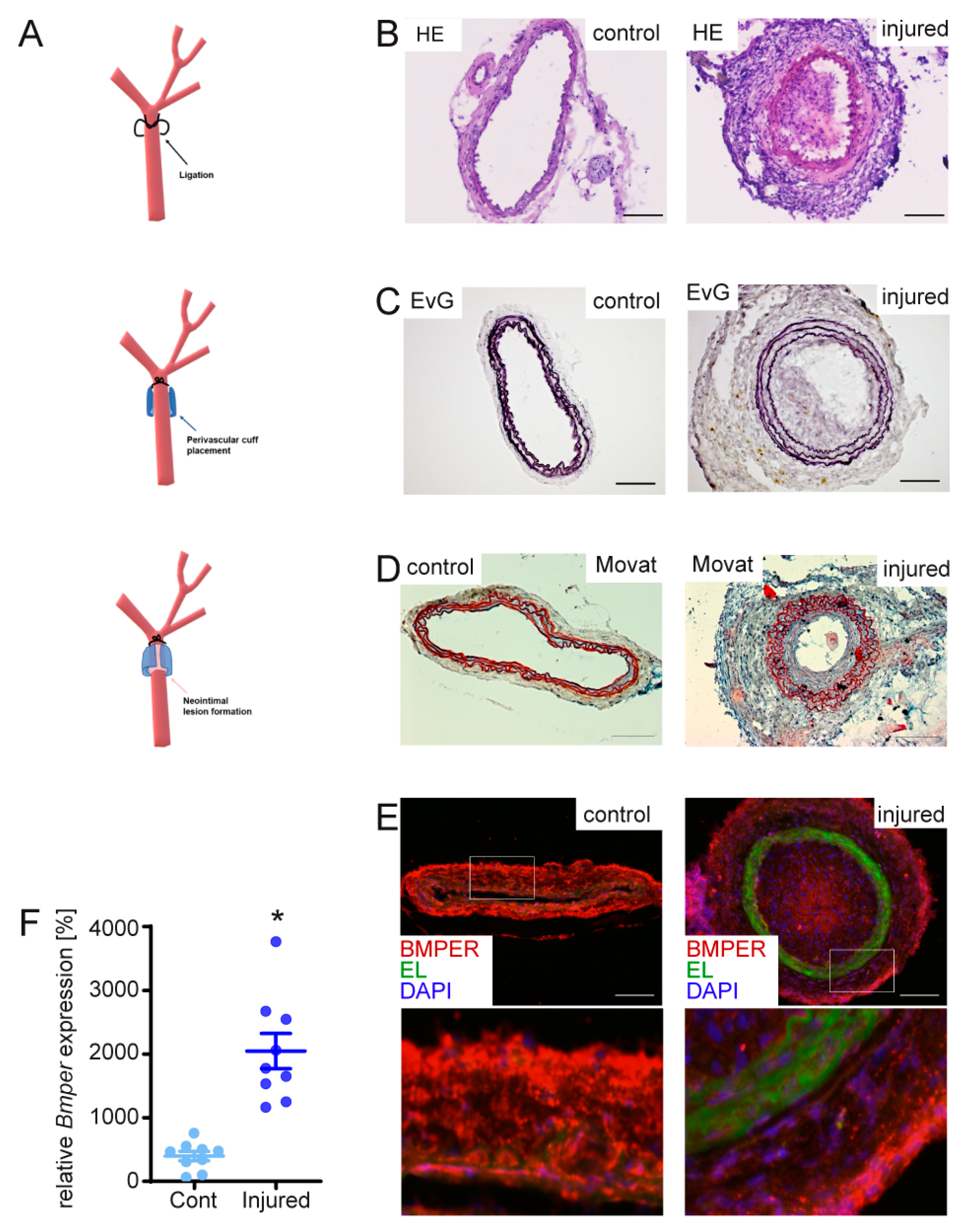
2.1. BMPER Expression in Healthy and Injured Carotid Artery
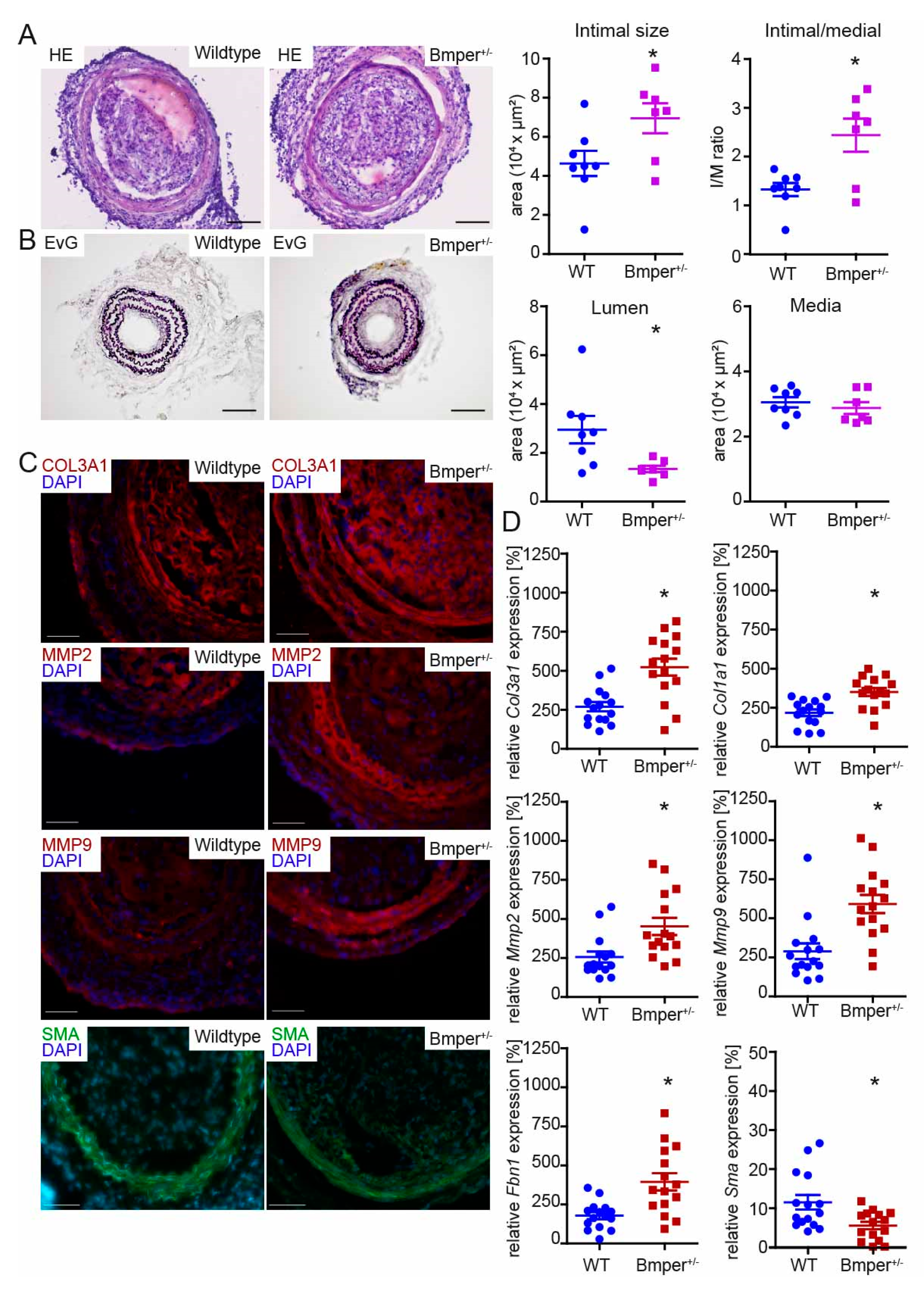
2.2. Loss of Bmper in C57BL/6N Mice Exacerbates Intimal Hyperplasia and ECM Remodeling
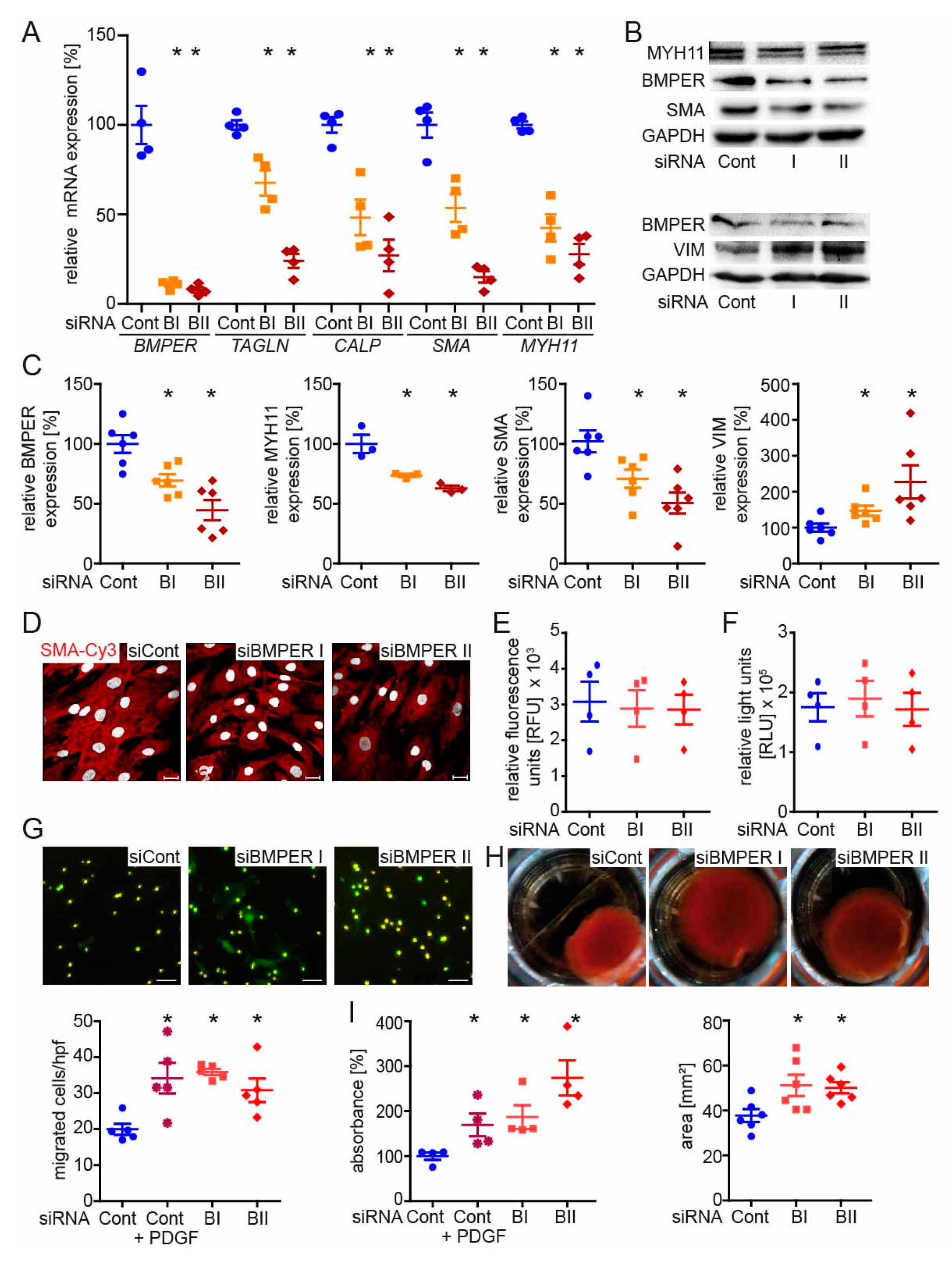
2.3. BMPER-Silencing in Human vSMCs Leads to a Synthetic vSMC Phenotype
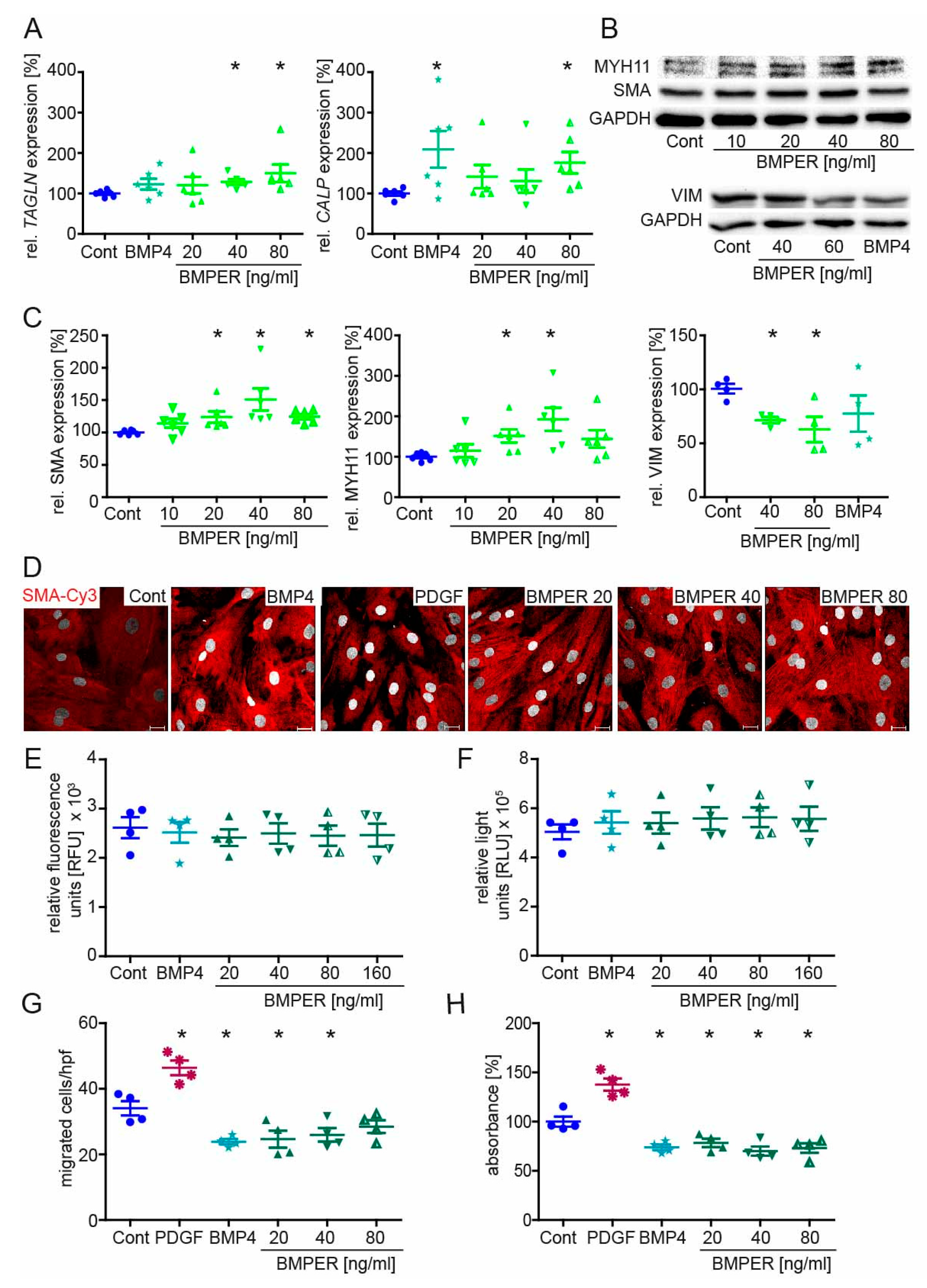
2.4. Recombinant BMPER Protein Stimulation of Human vSMC Induces a Contractile Phenotype
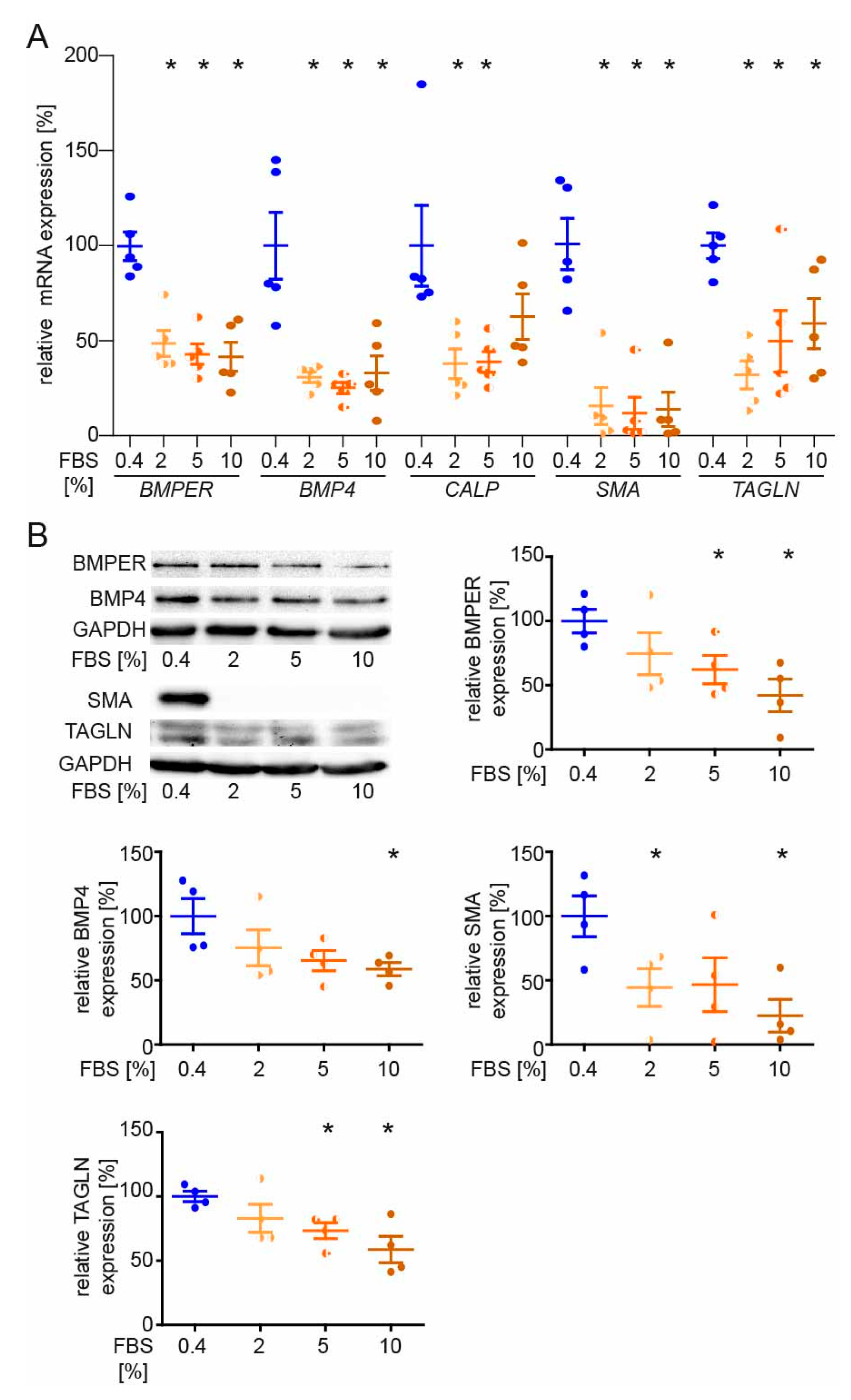
2.5. BMPER and BMP4 Expression Are Reduced in Serum-Stimulated Synthetic vSMCs
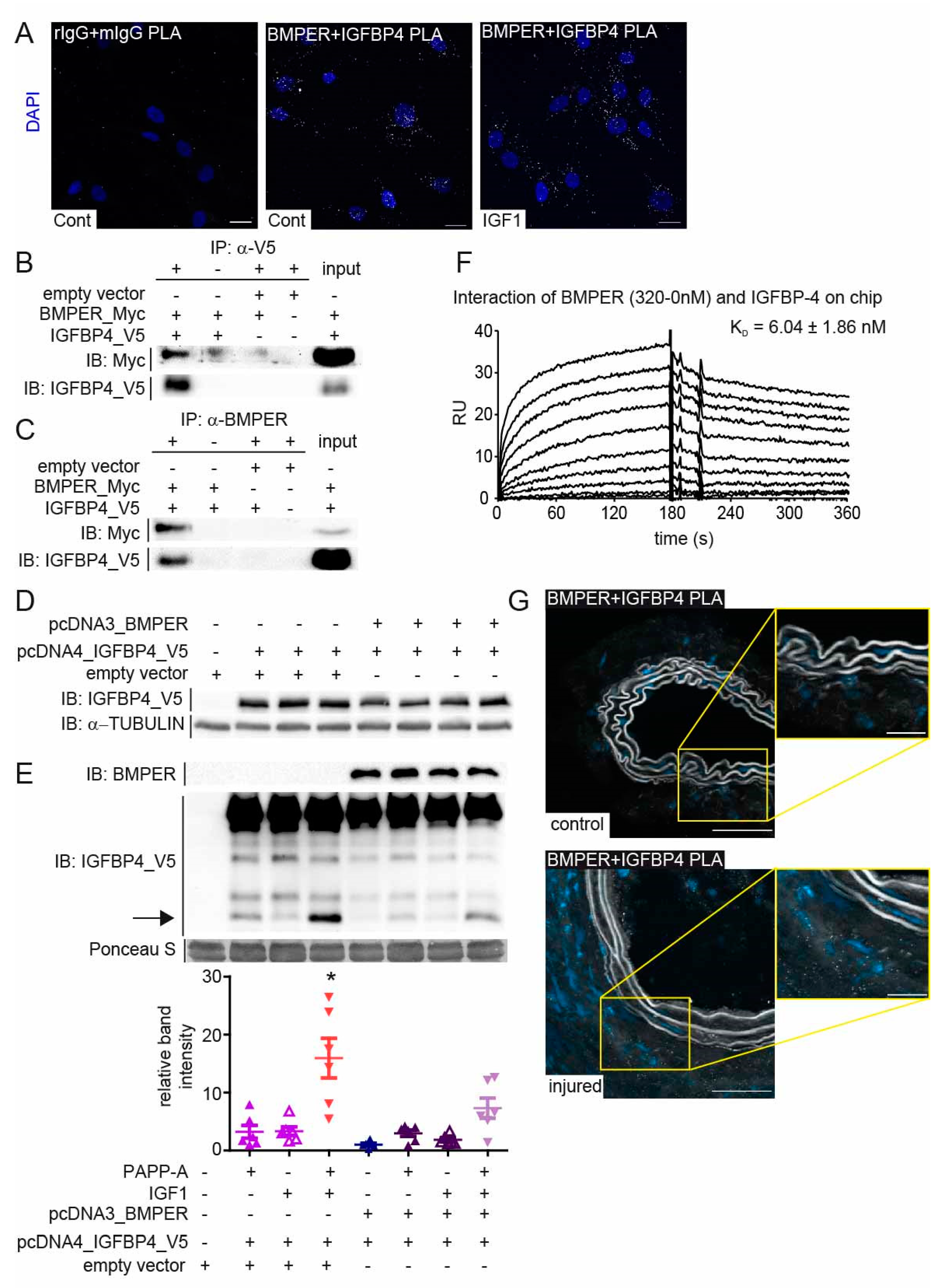
2.6. BMPER Physically Interacts with Insulin-like Growth Factor-Binding Protein 4 (IGFBP4)
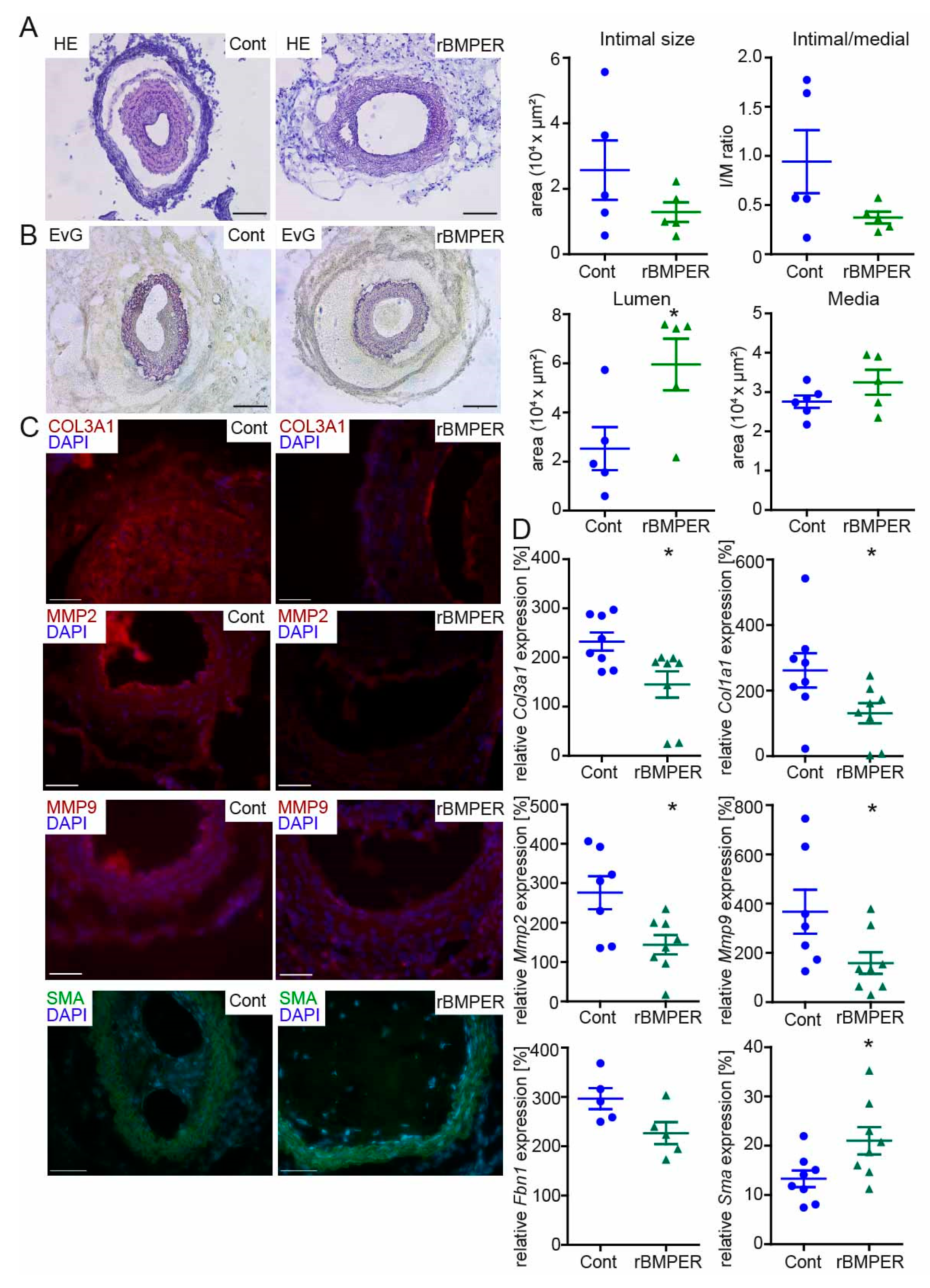
2.7. Recombinant BMPER Protein Prevents Intimal Hyperplasia and ECM Deposition in C57BL/6N Mice after Carotid Injury
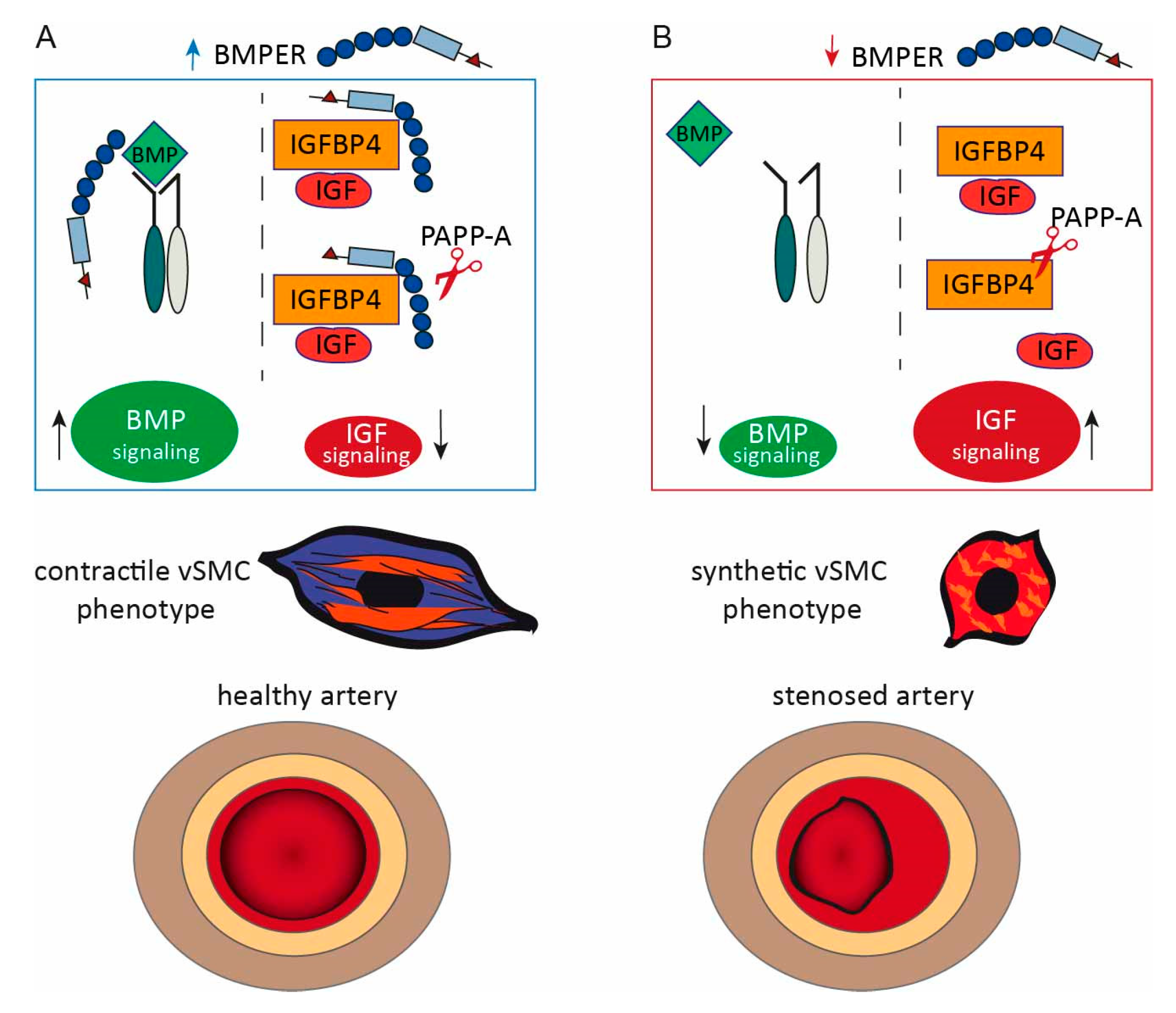
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Mouse Carotid Artery Ligation and Cuff-Induced Neointimal Formation Model
4.3. Histological Examination and Immunostaining
4.4. RNAscope In Situ Hybridization
4.5. Cell Culture
4.6. Transfection
4.7. Proliferation Assay
4.8. Migration Assay
4.9. Collagen-Gel Contraction Assay
4.10. Cell Viability Assay
4.11. Cell Apoptosis Assay
4.12. In Situ Proximity Ligation Assay (PLA)
4.13. RNA Extraction and Reverse Transcription
4.14. Quantitative Real-Time PCR
4.15. Western Blot Analysis
4.16. IGFBP4 Protease Assay
4.17. Immunoprecipitation (IP)
4.18. Surface Plasmon Resonance (SPR)
4.19. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ritchie, H. Causes of Death. Our World Data. 2018. Available online: https://ourworldindata.org/causes-of-death (accessed on 29 September 2022).
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Dangas, G.D.; Claessen, B.E.; Caixeta, A.; Sanidas, E.A.; Mintz, G.S.; Mehran, R. In-stent restenosis in the drug-eluting stent era. J. Am. Coll. Cardiol. 2010, 56, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Lacolley, P.; Regnault, V.; Nicoletti, A.; Li, Z.; Michel, J.B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012, 95, 194–204. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Davis-Dusenbery, B.N.; Wu, C.; Hata, A. Micromanaging vascular smooth muscle cell differentiation and phenotypic modulation. Arter. Thromb. Vasc. Biol. 2011, 31, 2370–2377. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Arthur, H.M. Extracellular control of TGFbeta signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef]
- David, L.; Feige, J.J.; Bailly, S. Emerging role of bone morphogenetic proteins in angiogenesis. Cytokine Growth Factor Rev. 2009, 20, 203–212. [Google Scholar] [CrossRef]
- Goumans, M.J.; Zwijsen, A.; Ten Dijke, P.; Bailly, S. Bone Morphogenetic Proteins in Vascular Homeostasis and Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef] [PubMed]
- Lagna, G.; Ku, M.M.; Nguyen, P.H.; Neuman, N.A.; Davis, B.N.; Hata, A. Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors. J. Biol. Chem. 2007, 282, 37244–37255. [Google Scholar] [CrossRef]
- Morrell, N.W.; Bloch, D.B.; ten Dijke, P.; Goumans, M.J.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP signalling in cardiovascular disease and anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Horbelt, D.; Denkis, A.; Knaus, P. A portrait of Transforming Growth Factor beta superfamily signalling: Background matters. Int. J. Biochem. Cell Biol. 2012, 44, 469–474. [Google Scholar] [CrossRef]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef]
- Piccolo, S.; Sasai, Y.; Lu, B.; De Robertis, E.M. Dorsoventral patterning in Xenopus: Inhibition of ventral signals by direct binding of chordin to BMP-4. Cell 1996, 86, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.C.; Harland, R.M. Expression cloning of noggin, a new dorsalizing factor localized to the Spemann organizer in Xenopus embryos. Cell 1992, 70, 829–840. [Google Scholar] [CrossRef]
- Oelgeschlager, M.; Larrain, J.; Geissert, D.; De Robertis, E.M. The evolutionarily conserved BMP-binding protein Twisted gastrulation promotes BMP signalling. Nature 2000, 405, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Binder, O.; Wu, Y.; Aitsebaomo, J.; Ren, R.; Bode, C.; Bautch, V.L.; Conlon, F.L.; Patterson, C. BMPER, a novel endothelial cell precursor-derived protein, antagonizes bone morphogenetic protein signaling and endothelial cell differentiation. Mol. Cell Biol. 2003, 23, 5664–5679. [Google Scholar] [CrossRef] [PubMed]
- Heinke, J.; Wehofsits, L.; Zhou, Q.; Zoeller, C.; Baar, K.M.; Helbing, T.; Laib, A.; Augustin, H.; Bode, C.; Patterson, C.; et al. BMPER Is an Endothelial Cell Regulator and Controls Bone Morphogenetic Protein-4-Dependent Angiogenesis. Circ. Res. 2008, 103, 804–812. [Google Scholar] [CrossRef]
- Serpe, M.; Umulis, D.; Ralston, A.; Chen, J.; Olson, D.J.; Avanesov, A.; Othmer, H.; O’Connor, M.B.; Blair, S.S. The BMP-binding protein Crossveinless 2 is a short-range, concentration-dependent, biphasic modulator of BMP signaling in Drosophila. Dev. Cell 2008, 14, 940–953. [Google Scholar] [CrossRef]
- Kelley, R.; Ren, R.; Pi, X.; Wu, Y.; Moreno, I.; Willis, M.; Moser, M.; Ross, M.; Podkowa, M.; Attisano, L.; et al. A concentration-dependent endocytic trap and sink mechanism converts Bmper from an activator to an inhibitor of Bmp signaling. J. Cell Biol. 2009, 184, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Jumabay, M.; Ly, A.; Radparvar, M.; Wang, A.H.; Abdmaulen, R.; Bostrom, K.I. Crossveinless 2 regulates bone morphogenetic protein 9 in human and mouse vascular endothelium. Blood 2012, 119, 5037–5047. [Google Scholar] [CrossRef]
- Dyer, L.; Wu, Y.; Moser, M.; Patterson, C. BMPER-induced BMP signaling promotes coronary artery remodeling. Dev. Biol. 2014, 386, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Helbing, T.; Rothweiler, R.; Heinke, J.; Goetz, L.; Diehl, P.; Zirlik, A.; Patterson, C.; Bode, C.; Moser, M. BMPER is upregulated by statins and modulates endothelial inflammation by intercellular adhesion molecule-1. Arter. Thromb. Vasc. Biol. 2010, 30, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Helbing, T.; Rothweiler, R.; Ketterer, E.; Goetz, L.; Heinke, J.; Grundmann, S.; Duerschmied, D.; Patterson, C.; Bode, C.; Moser, M. BMP activity controlled by BMPER regulates the proinflammatory phenotype of endothelium. Blood 2011, 118, 5040–5049. [Google Scholar] [CrossRef] [PubMed]
- Pi, X.; Lockyer, P.; Dyer, L.A.; Schisler, J.C.; Russell, B.; Carey, S.; Sweet, D.T.; Chen, Z.; Tzima, E.; Willis, M.S.; et al. Bmper inhibits endothelial expression of inflammatory adhesion molecules and protects against atherosclerosis. Arter. Thromb. Vasc. Biol. 2012, 32, 2214–2222. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Miralles, I.; Ren, R.; Moser, M.; Hartnett, M.E.; Patterson, C. Bone morphogenetic protein endothelial cell precursor-derived regulator regulates retinal angiogenesis in vivo in a mouse model of oxygen-induced retinopathy. Arter. Thromb. Vasc. Biol. 2011, 31, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Ikeya, M.; Kawada, M.; Kiyonari, H.; Sasai, N.; Nakao, K.; Furuta, Y.; Sasai, Y. Essential pro-Bmp roles of crossveinless 2 in mouse organogenesis. Development 2006, 133, 4463–4473. [Google Scholar] [CrossRef]
- Wasteson, P.; Johansson, B.R.; Jukkola, T.; Breuer, S.; Akyurek, L.M.; Partanen, J.; Lindahl, P. Developmental origin of smooth muscle cells in the descending aorta in mice. Development 2008, 135, 1823–1832. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Lindner, V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arter. Thromb. Vasc. Biol. 1997, 17, 2238–2244. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Sata, M.; Hirata, Y.; Nagai, R. Diverse contribution of bone marrow cells to neointimal hyperplasia after mechanical vascular injuries. Circ. Res. 2003, 93, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar]
- Zakin, L.; Metzinger, C.A.; Chang, E.Y.; Coffinier, C.; De Robertis, E.M. Development of the vertebral morphogenetic field in the mouse: Interactions between Crossveinless-2 and Twisted Gastrulation. Dev. Biol. 2008, 323, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Beamish, J.A.; He, P.; Kottke-Marchant, K.; Marchant, R.E. Molecular regulation of contractile smooth muscle cell phenotype: Implications for vascular tissue engineering. Tissue Eng. Part B Rev. 2010, 16, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef]
- Dyer, L.A.; Pi, X.; Patterson, C. The role of BMPs in endothelial cell function and dysfunction. Trends Endocrinol. Metab. 2014, 25, 472–480. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Conover, C.A.; Schwartz, R.S. The insulin-like growth factor axis: A review of atherosclerosis and restenosis. Circ. Res. 2000, 86, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Smith, Y.E.; Toomey, S.; Napoletano, S.; Kirwan, G.; Schadow, C.; Chubb, A.J.; Mikkelsen, J.H.; Oxvig, C.; Harmey, J.H. Recombinant PAPP-A resistant insulin-like growth factor binding protein 4 (dBP4) inhibits angiogenesis and metastasis in a murine model of breast cancer. BMC Cancer 2018, 18, 1016. [Google Scholar] [CrossRef]
- Esser, J.S.; Rahner, S.; Deckler, M.; Bode, C.; Patterson, C.; Moser, M. Fibroblast growth factor signaling pathway in endothelial cells is activated by BMPER to promote angiogenesis. Arter. Thromb. Vasc. Biol. 2015, 35, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Cidade, M.T.; Ramos, D.J.; Santos, J.; Carrelo, H.; Calero, N.; Borges, J.P. Injectable Hydrogels Based on Pluronic/Water Systems Filled with Alginate Microparticles for Biomedical Applications. Materials 2019, 12, 1083. [Google Scholar] [CrossRef]
- Rzucidlo, E.M.; Martin, K.A.; Powell, R.J. Regulation of vascular smooth muscle cell differentiation. J. Vasc. Surg. 2007, 45, A25–A32. [Google Scholar] [CrossRef]
- Newby, A.C.; Zaltsman, A.B. Molecular mechanisms in intimal hyperplasia. J. Pathol. 2000, 190, 300–309. [Google Scholar] [CrossRef]
- Sedlmeier, G.; Sleeman, J.P. Extracellular regulation of BMP signaling: Welcome to the matrix. Biochem. Soc. Trans. 2017, 45, 173–181. [Google Scholar] [CrossRef]
- Corriere, M.A.; Rogers, C.M.; Eliason, J.L.; Faulk, J.; Kume, T.; Hogan, B.L.; Guzman, R.J. Endothelial Bmp4 is induced during arterial remodeling: Effects on smooth muscle cell migration and proliferation. J. Surg. Res. 2008, 145, 142–149. [Google Scholar] [CrossRef]
- Chang, K.; Weiss, D.; Suo, J.; Vega, J.D.; Giddens, D.; Taylor, W.R.; Jo, H. Bone morphogenic protein antagonists are coexpressed with bone morphogenic protein 4 in endothelial cells exposed to unstable flow in vitro in mouse aortas and in human coronary arteries: Role of bone morphogenic protein antagonists in inflammation and atherosclerosis. Circulation 2007, 116, 1258–1266. [Google Scholar] [PubMed]
- Huang, J.; Liu, Y.; Filas, B.; Gunhaga, L.; Beebe, D.C. Negative and positive auto-regulation of BMP expression in early eye development. Dev. Biol. 2015, 407, 256–264. [Google Scholar] [CrossRef]
- Zinski, J.; Tajer, B.; Mullins, M.C. TGF-beta Family Signaling in Early Vertebrate Development. Cold Spring Harb. Perspect. Biol. 2018, 10, a033274. [Google Scholar] [CrossRef] [PubMed]
- Balemans, W.; Van Hul, W. Extracellular regulation of BMP signaling in vertebrates: A cocktail of modulators. Dev. Biol. 2002, 250, 231–250. [Google Scholar] [CrossRef]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Pi, X.; Schmitt, C.E.; Xie, L.; Portbury, A.L.; Wu, Y.; Lockyer, P.; Dyer, L.A.; Moser, M.; Bu, G.; Flynn, E.J., 3rd; et al. LRP1-dependent endocytic mechanism governs the signaling output of the bmp system in endothelial cells and in angiogenesis. Circ. Res. 2012, 111, 564–574. [Google Scholar] [CrossRef]
- Boldt, H.B.; Conover, C.A. Pregnancy-associated plasma protein-A (PAPP-A): A local regulator of IGF bioavailability through cleavage of IGFBPs. Growth Horm. IGF Res. 2007, 17, 10–18. [Google Scholar] [CrossRef]
- Duan, C.; Clemmons, D.R. Differential expression and biological effects of insulin-like growth factor-binding protein-4 and -5 in vascular smooth muscle cells. J. Biol. Chem. 1998, 273, 16836–16842. [Google Scholar] [CrossRef]
- Resch, Z.T.; Simari, R.D.; Conover, C.A. Targeted disruption of the pregnancy-associated plasma protein-A gene is associated with diminished smooth muscle cell response to insulin-like growth factor-I and resistance to neointimal hyperplasia after vascular injury. Endocrinology 2006, 147, 5634–5640. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Schwartz, R.S.; Lewis, D.A.; Overgaard, M.T.; Christiansen, M.; Oxvig, C.; Ashai, K.; Holmes, D.R., Jr.; Conover, C.A. Insulin-like growth factor binding protein-4 protease produced by smooth muscle cells increases in the coronary artery after angioplasty. Arter. Thromb. Vasc. Biol. 2001, 21, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Nichols, T.C.; Busby, W.H., Jr.; Merricks, E.; Sipos, J.; Rowland, M.; Sitko, K.; Clemmons, D.R. Protease-resistant insulin-like growth factor (IGF)-binding protein-4 inhibits IGF-I actions and neointimal expansion in a porcine model of neointimal hyperplasia. Endocrinology 2007, 148, 5002–5010. [Google Scholar] [CrossRef]
- Lockhart-Cairns, M.P.; Lim, K.T.W.; Zuk, A.; Godwin, A.R.F.; Cain, S.A.; Sengle, G.; Baldock, C. Internal cleavage and synergy with twisted gastrulation enhance BMP inhibition by BMPER. Matrix Biol. 2019, 77, 73–86. [Google Scholar] [CrossRef]
- Roberts, D.D. Emerging functions of matricellular proteins. Cell Mol. Life Sci. 2011, 68, 3133–3136. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Sessa, W.C. Endothelial NOS: Perspective and recent developments. Br. J. Pharmacol. 2019, 176, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Sano, M. God gives IL-19 with both hands: Anti-inflammatory but pro-angiogenic. J. Mol. Cell Cardiol. 2015, 80, 20–22. [Google Scholar] [CrossRef]
- Robinet, P.; Milewicz, D.M.; Cassis, L.A.; Leeper, N.J.; Lu, H.S.; Smith, J.D. Consideration of Sex Differences in Design and Reporting of Experimental Arterial Pathology Studies-Statement From ATVB Council. Arter. Thromb. Vasc. Biol. 2018, 38, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Moroi, M.; Zhang, L.; Yasuda, T.; Virmani, R.; Gold, H.K.; Fishman, M.C.; Huang, P.L. Interaction of genetic deficiency of endothelial nitric oxide, gender, and pregnancy in vascular response to injury in mice. J. Clin. Invest. 1998, 101, 1225–1232. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pankratz, F.; Maksudova, A.; Goesele, R.; Meier, L.; Proelss, K.; Marenne, K.; Thut, A.-K.; Sengle, G.; Correns, A.; Begelspacher, J.; et al. BMPER Improves Vascular Remodeling and the Contractile Vascular SMC Phenotype. Int. J. Mol. Sci. 2023, 24, 4950. https://doi.org/10.3390/ijms24054950
Pankratz F, Maksudova A, Goesele R, Meier L, Proelss K, Marenne K, Thut A-K, Sengle G, Correns A, Begelspacher J, et al. BMPER Improves Vascular Remodeling and the Contractile Vascular SMC Phenotype. International Journal of Molecular Sciences. 2023; 24(5):4950. https://doi.org/10.3390/ijms24054950
Chicago/Turabian StylePankratz, Franziska, Aziza Maksudova, Roman Goesele, Lena Meier, Kora Proelss, Katia Marenne, Ann-Kathrin Thut, Gerhard Sengle, Annkatrin Correns, Jeanina Begelspacher, and et al. 2023. "BMPER Improves Vascular Remodeling and the Contractile Vascular SMC Phenotype" International Journal of Molecular Sciences 24, no. 5: 4950. https://doi.org/10.3390/ijms24054950
APA StylePankratz, F., Maksudova, A., Goesele, R., Meier, L., Proelss, K., Marenne, K., Thut, A.-K., Sengle, G., Correns, A., Begelspacher, J., Alkis, D., Siegel, P. M., Smolka, C., Grundmann, S., Moser, M., Zhou, Q., & Esser, J. S. (2023). BMPER Improves Vascular Remodeling and the Contractile Vascular SMC Phenotype. International Journal of Molecular Sciences, 24(5), 4950. https://doi.org/10.3390/ijms24054950