
Molecular Mechanisms of Neutrophil Extracellular Trap (NETs) Degradation

{kind=link}
Abstract
1. NETs Generation and Their Biological Role
2. NETs in Clinical Pathology
2.1. NETs in Autoimmune Diseases
2.2. NETs—Coagulation
2.3. NETs in COVID-19 and in Other Severe Infections
2.4. NETs in Cancer
2.5. NETs in Ischemic Stroke
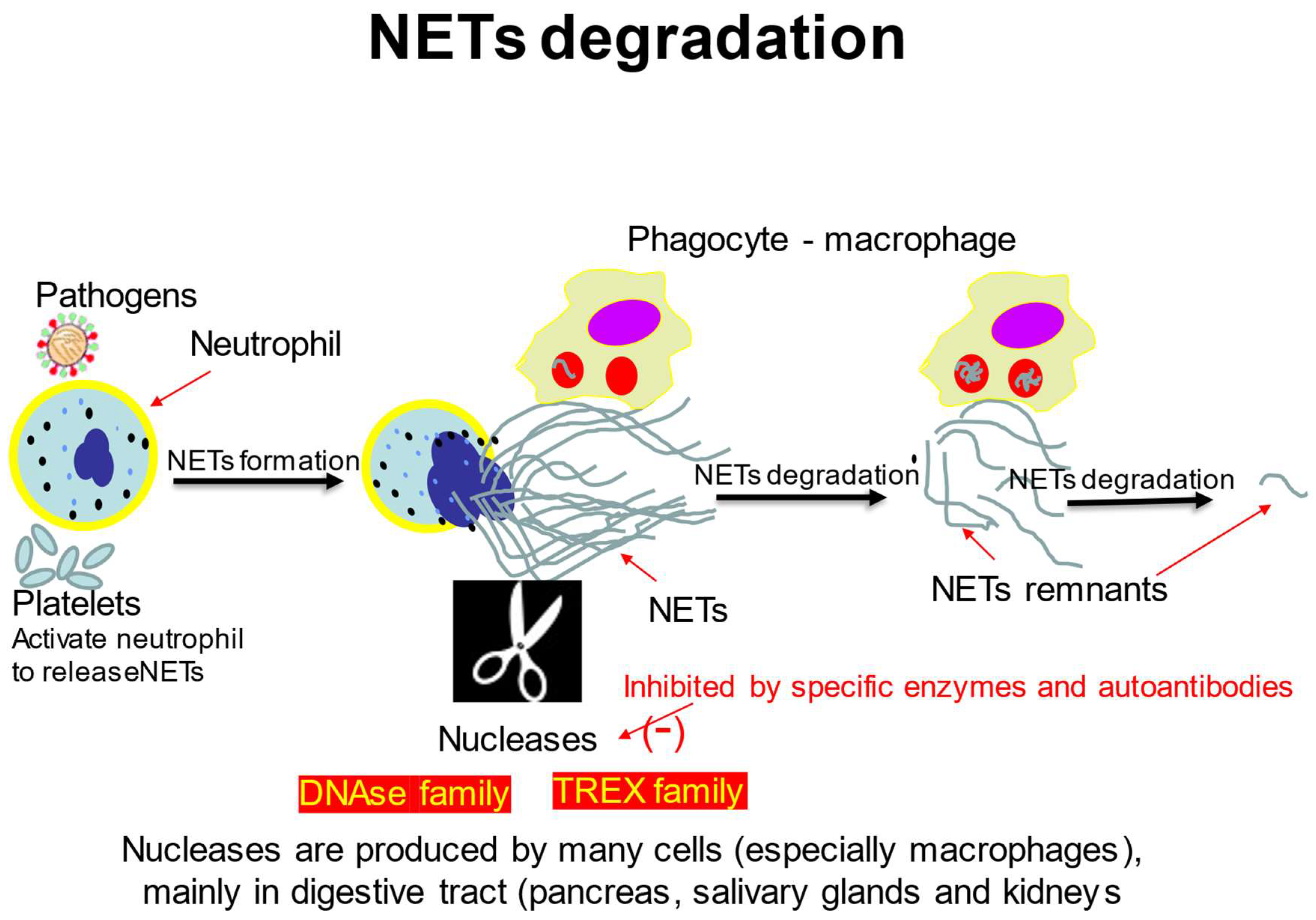
3. The Mechanisms of NETs Degradation
3.1. DNA Degrading Enzymes
3.2. NETs Degradation by Macrophages
4. NETs Degradation Defects
4.1. NETs Degradation Defects in SLE
4.2. TREX Defects
4.3. Other Clinical Consequences of DNase Mutations
5. The Potential Applications of NETs-Inhibiting Drugs
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Manda-Handzlik, A.; Demkow, U. Neutrophils: The role of oxidative and nitrosative stress in health and disease. Adv. Exp. Med. Biol. 2015, 857, 51–60. [Google Scholar] [CrossRef]
- Gierlikowska, B.; Stachura, A.; Gierlikowski, W.; Demkow, U. The Impact of Cytokines on Neutrophils’ Phagocytosis and NET Formation during Sepsis—A Review. Int. J. Mol. Sci. 2022, 23, 5076. [Google Scholar] [CrossRef]
- Demkow, U. Neutrophil Extracellular Traps (NETs) in Cancer Invasion, Evasion and Metastasis. Cancers 2021, 13, 4495. [Google Scholar] [CrossRef]
- Szturmowicz, M.; Demkow, U. Neutrophil Extracellular Traps (NETs) in Severe SARS-CoV-2 Lung Disease. Int. J. Mol. Sci. 2021, 22, 8854. [Google Scholar] [CrossRef]
- Manda-Handzlik, A.; Demkow, U. The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System. Cells 2019, 8, 1477. [Google Scholar] [CrossRef]
- Szturmowicz, M.; Barańska, I.; Skoczylas, A.; Jędrych, M.; Demkow, U. Correlation of bronchoalveolar lavage lymphocyte count with the extend of lung fibrosis and with plethysmographic lung volumes in patients with newly recognized hypersensitivity pneumonitis. Cent. Eur. J. Immunol. 2020, 45, 276–282. [Google Scholar] [CrossRef]
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil extracellular traps and thrombosis in COVID-19. J. Thromb. Thrombolysis 2020, 51, 446–453. [Google Scholar] [CrossRef]
- Berthelot, J.-M.; Le Goff, B.; Neel, A.; Maugars, Y.; Hamidou, M. NETosis: At the crossroads of rheumatoid arthritis, lupus, and vasculitis. Jt. Bone Spine 2017, 84, 255–262. [Google Scholar] [CrossRef]
- Block, H.; Rossaint, J.; Zarbock, A. The Fatal Circle of NETs and NET-Associated DAMPs Contributing to Organ Dysfunction. Cells 2022, 11, 1919. [Google Scholar] [CrossRef]
- Ciesielski, O.; Biesiekierska, M.; Panthu, B.; Soszyński, M.; Pirola, L.; Balcerczyk, A. Citrullination in the pathology of inflammatory and autoimmune disorders: Recent advances and future perspectives. Cell. Mol. Life Sci. 2022, 79, 94. [Google Scholar] [CrossRef]
- Corsiero, E.; Bombardieri, M.; Carlotti, E.; Pratesi, F.; Robinson, W.; Migliorini, P.; Pitzalis, C. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann. Rheum. Dis. 2016, 75, 1866–1875. [Google Scholar] [CrossRef]
- Chang, H.-H.; Dwivedi, N.; Nicholas, A.P.; Ho, I.-C. The W620 Polymorphism in PTPN22 Disrupts Its Interaction With Peptidylarginine Deiminase Type 4 and Enhances Citrullination and NETosis. Arthritis Rheumatol. 2015, 67, 2323–2334. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Z.; Liu, Z. Impact of Neutrophil Extracellular Traps on Thrombosis Formation: New Findings and Future Perspective. Front. Cell. Infect. Microbiol. 2022, 12, 910908. [Google Scholar] [CrossRef]
- Seo, J.D.; Gu, J.-Y.; Jung, H.S.; Kim, Y.J.; Kim, H.K. Contact System Activation and Neutrophil Extracellular Trap Markers: Risk Factors for Portal Vein Thrombosis in Patients With Hepatocellular Carcinoma. Clin. Appl. Thromb. 2019, 25, 1076029618825310. [Google Scholar] [CrossRef]
- Becker, R.C. COVID-19-associated vasculitis and vasculopathy. J. Thromb. Thrombolysis 2020, 50, 499–511. [Google Scholar] [CrossRef]
- Vassiliou, A.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- El Amki, M.; Glück, C.; Binder, N.; Middleham, W.; Wyss, M.T.; Weiss, T.; Meister, H.; Luft, A.; Weller, M.; Weber, B.; et al. Neutrophils Obstructing Brain Capillaries Are a Major Cause of No-Reflow in Ischemic Stroke. Cell Rep. 2020, 33, 108260. [Google Scholar] [CrossRef]
- Jin, J.; Qiao, S.; Liu, J.; Li, W.; Wang, F.; Gao, X.; Tian, J.; Wang, N.; Zhang, J.; Dong, J.; et al. Neutrophil extracellular traps promote thrombogenicity in cerebral venous sinus thrombosis. Cell Biosci. 2022, 12, 1–16. [Google Scholar] [CrossRef]
- Haider, P.; Kral-Pointner, J.B.; Mayer, J.; Richter, M.; Kaun, C.; Brostjan, C.; Eilenberg, W.; Fischer, M.B.; Speidl, W.S.; Hengstenberg, C.; et al. Neutrophil Extracellular Trap Degradation by Differently Polarized Macrophage Subsets. Arter. Thromb. Vasc. Biol. 2020, 40, 2265–2278. [Google Scholar] [CrossRef]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef]
- Jimenez-Alcazar, M.; Napirei, M.; Panda, R.; Köhler, E.C.; Hovinga, J.K.; Mannherz, H.G.; Peine, S.; Renné, T.; Lämmle, B.; Fuchs, T.A. Impaired DNase1-mediated degradation of neutrophil extracellular traps is associated with acute thrombotic microangiopathies. J. Thromb. Haemost. 2015, 13, 732–742. [Google Scholar] [CrossRef]
- Lauková, L.; Konečná, B.; Janovičová, Ľ.; Vlková, B.; Celec, P. Deoxyribonucleases and Their Applications in Biomedicine. Biomolecules 2020, 10, 1036. [Google Scholar] [CrossRef]
- Evans, C.J.; Aguilera, R.J. DNase II: Genes, enzymes and function. Gene 2003, 322, 1–15. [Google Scholar] [CrossRef]
- Nagata, S.; Nagase, H.; Kawane, K.; Mukae, N.; Fukuyama, H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 2003, 10, 108–116. [Google Scholar] [CrossRef]
- Kawane, K.; Nagata, S. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Inflamm. Regen. 2009, 29, 204–208. [Google Scholar] [CrossRef]
- Farrera, C.; Fadeel, B. Macrophage Clearance of Neutrophil Extracellular Traps Is a Silent Process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef]
- Mazur, D.J.; Perrino, F.W. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′-->5′ exonucleases. J. Biol. Chem. 1999, 274, 19655–19660. [Google Scholar] [CrossRef]
- Morita, M.; Stamp, G.; Robins, P.; Dulic, A.; Rosewell, I.; Hrivnak, G.; Daly, G.; Lindahl, T.; Barnes, D.E. Gene-targeted mice lacking the Trex1 (DNase III) 3′-->5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol. 2004, 24, 6719–6727. [Google Scholar] [CrossRef]
- Huang, K.-W.; Liu, T.-C.; Liang, R.-Y.; Chu, L.-Y.; Cheng, H.-L.; Chu, J.-W.; Hsiao, Y.-Y. Structural basis for overhang excision and terminal unwinding of DNA duplexes by TREX1. PLoS Biol. 2018, 16, e2005653. [Google Scholar] [CrossRef]
- Lazzaretto, B.; Fadeel, B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef]
- Jani, D.; Lutz, S.; Hurt, E.; Laskey, R.A.; Stewart, M.; Wickramasinghe, V.O. Functional and structural characterization of the mammalian TREX-2 complex that links transcription with nuclear messenger RNA export. Nucleic Acids Res. 2012, 40, 4562–4573. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, Y.; Lai, D.; Zhang, P.; Yang, Y.; Li, Y.; Fei, K.; Jiang, G.; Fan, J. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. 2018, 9, 597. [Google Scholar] [CrossRef]
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef]
- Hu, Z.; Murakami, T.; Tamura, H.; Reich, J.; Kuwahara-Arai, K.; Iba, T.; Tabe, Y.; Nagaoka, I. Neutrophil extracellular traps induce IL-1β production by macrophages in combination with lipopolysaccharide. Int. J. Mol. Med. 2017, 39, 549–558. [Google Scholar] [CrossRef]
- Nakazawa, D.; Shida, H.; Kusunoki, Y.; Miyoshi, A.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun. 2016, 67, 19–28. [Google Scholar] [CrossRef]
- Luo, L.; Wall, A.; Tong, S.; Hung, Y.; Xiao, Z.; Tarique, A.A.; Sly, P.D.; Fantino, E.; Marzolo, M.-P.; Stow, J.L. TLR Crosstalk Activates LRP1 to Recruit Rab8a and PI3Kγ for Suppression of Inflammatory Responses. Cell Rep. 2018, 24, 3033–3044. [Google Scholar] [CrossRef]
- Doodnauth, S.A.; Grinstein, S.; Maxson, M.E. Constitutive and stimulated macropinocytosis in macrophages: Roles in immunity and in the pathogenesis of atherosclerosis. Philos. Trans. R. Soc. B Biol. Sci. 2018, 374, 20180147. [Google Scholar] [CrossRef]
- Li, N.; Zheng, X.; Chen, M.; Huang, L.; Chen, L.; Huo, R.; Li, X.; Huang, Y.; Sun, M.; Mai, S.; et al. Deficient DNASE1L3 facilitates neutrophil extracellular traps-induced invasion via cyclic GMP-AMP synthase and the non-canonical NF-κB pathway in diabetic hepatocellular carcinoma. Clin. Transl. Immunol. 2022, 11, e1386. [Google Scholar] [CrossRef]
- Wang, S.; Ma, H.; Li, X.; Mo, X.; Zhang, H.; Yang, L.; Deng, Y.; Yan, Y.; Yang, G.; Liu, X.; et al. DNASE1L3 as an indicator of favorable survival in hepatocellular carcinoma patients following resection. Aging 2020, 12, 1171–1185. [Google Scholar] [CrossRef]
- Angeletti, A.; Volpi, S.; Bruschi, M.; Lugani, F.; Vaglio, A.; Prunotto, M.; Gattorno, M.; Schena, F.; Verrina, E.; Ravelli, A.; et al. Neutrophil Extracellular Traps-DNase Balance and Autoimmunity. Cells 2021, 10, 2667. [Google Scholar] [CrossRef]
- Papalian, M.; Lafer, E.; Wong, R.; Stollar, B.D. Reaction of systemic lupus erythematosus antinative DNA antibodies with native DNA fragments from 20 to 1,200 base pairs. J. Clin. Investig. 1980, 65, 469–477. [Google Scholar] [CrossRef]
- Seredkina, N.; Zykova, S.N.; Rekvig, O.P. Progression of Murine Lupus Nephritis Is Linked to Acquired Renal Dnase1 Deficiency and Not to Up-Regulated Apoptosis. Am. J. Pathol. 2009, 175, 97–106. [Google Scholar] [CrossRef]
- Sisirak, V.; Sally, B.; D’Agati, V.; Martinez-Ortiz, W.; Özçakar, Z.B.; David, J.; Rashidfarrokhi, A.; Yeste, A.; Panea, C.; Chida, A.S.; et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 2016, 166, 88–101. [Google Scholar] [CrossRef]
- Yasutomo, K.; Horiuchi, T.; Kagami, S.; Tsukamoto, H.; Hashimura, C.; Urushihara, M.; Kuroda, Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 2001, 28, 313–314. [Google Scholar] [CrossRef]
- Napirei, M.; Ludwig, S.; Mezrhab, J.; Klöckl, T.; Mannherz, H.G. Murine serum nucleases—Contrasting effects of plasmin and heparin on the activities of DNase1 and DNase1-like 3 (DNase1l3). FEBS J. 2009, 276, 1059–1073. [Google Scholar] [CrossRef]
- Skiljevic, D.; Jeremic, I.; Nikolic, M.; Andrejevic, S.; Sefik-Bukilica, M.; Stojimirovic, B.; Bonaci-Nikolic, B. Serum DNase I activity in systemic lupus erythematosus: Correlation with immunoserological markers, the disease activity and organ involvement. Clin. Chem. Lab. Med. 2013, 51, 1083–1091. [Google Scholar] [CrossRef]
- Sogkas, G.; Atschekzei, F.; Adriawan, I.R.; Dubrowinskaja, N.; Witte, T.; Schmidt, R.E. Cellular and molecular mechanisms breaking immune tolerance in inborn errors of immunity. Cell. Mol. Immunol. 2021, 18, 1122–1140. [Google Scholar] [CrossRef]
- Bruschi, M.; Moroni, G.; Sinico, R.A.; Franceschini, F.; Fredi, M.; Vaglio, A.; Cavagna, L.; Petretto, A.; Pratesi, F.; Migliorini, P.; et al. Neutrophil Extracellular Traps in the Autoimmunity Context. Front. Med. 2021, 8, 614829. [Google Scholar] [CrossRef]
- Hartl, J.; Serpas, L.; Wang, Y.; Rashidfarrokhi, A.; Perez, O.A.; Sally, B.; Sisirak, V.; Soni, C.; Khodadadi-Jamayran, A.; Tsirigos, A.; et al. Autoantibody-mediated impairment of DNASE1L3 activity in sporadic systemic lupus erythematosus. J. Exp. Med. 2021, 218, e20201138. [Google Scholar] [CrossRef]
- Zhou, W.; Richmond-Buccola, D.; Wang, Q.; Kranzusch, P.J. Structural basis of human TREX1 DNA degradation and autoimmune disease. Nat. Commun. 2022, 13, 4277. [Google Scholar] [CrossRef]
- Grieves, J.L.; Fye, J.M.; Harvey, S.; Grayson, J.M.; Hollis, T.; Perrino, F.W. Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proc. Natl. Acad. Sci. USA 2015, 112, 5117–5122. [Google Scholar] [CrossRef]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tydén, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil Extracellular Traps That Are Not Degraded in Systemic Lupus Erythematosus Activate Complement Exacerbating the Disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef]
- Leffler, J.; Gullstrand, B.; Jönsen, A.; Nilsson, J.; Martin, M.; Blom, A.M.; Bengtsson, A.A. Degradation of neutrophil extracellular traps co-varies with disease activity in patients with systemic lupus erythematosus. Thromb. Haemost. 2013, 15, R84. [Google Scholar] [CrossRef]
- Martin, J.E.; Assassi, S.; Diaz-Gallo, L.-M.; Broen, J.C.; Simeon, C.P.; Castellvi, I.; Vicente-Rabaneda, E.; Fonollosa, V.; Ortego-Centeno, N.; González-Gay, M.A.; et al. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum. Mol. Genet. 2013, 22, 4021–4029. [Google Scholar] [CrossRef]
- Ueki, M.; Takeshita, H.; Fujihara, J.; Iida, R.; Yuasa, I.; Kato, H.; Panduro, A.; Nakajima, T.; Kominato, Y.; Yasuda, T. Caucasian-specific allele in non-synonymous single nucleotide polymorphisms of the gene encoding deoxyribonuclease I-like 3, potentially relevant to autoimmunity, produces an inactive enzyme. Clin. Chim. Acta 2009, 407, 20–24. [Google Scholar] [CrossRef]
- Nakazawa, D.; Shida, H.; Tomaru, U.; Yoshida, M.; Nishio, S.; Atsumi, T.; Ishizu, A. Enhanced Formation and Disordered Regulation of NETs in Myeloperoxidase-ANCA–Associated Microscopic Polyangiitis. J. Am. Soc. Nephrol. 2014, 25, 990–997. [Google Scholar] [CrossRef]
- Nielsen, C.T.; Østergaard, O.; Stener, L.; Iversen, L.V.; Truedsson, L.; Gullstrand, B.; Jacobsen, S.; Heegaard, N.H.H. Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum. 2012, 64, 1227–1236. [Google Scholar] [CrossRef]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2020, 61, 194–211. [Google Scholar] [CrossRef]
- He, Y.; Yang, F.-Y.; Sun, E.-W. Neutrophil Extracellular Traps in Autoimmune Diseases. Chin. Med. J. 2018, 131, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kaplan, M.J. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat. Rev. Nephrol. 2016, 12, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Pagnoux, C.; Korach, J.M.; Guillevin, L. Indications for plasma exchange in systemic lupus erythematosus in 2005. Lupus 2005, 14, 871–877. [Google Scholar] [CrossRef]
- De Buhr, N.; von Köckritz-Blickwede, M. The balance of neutrophil extracellular trap formation and nuclease degradation: An unknown role of bacterial coinfections in COVID-19 patients? MBio 2021, 12, e03304-20. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H.; Park, W.; Lee, Y.Y.; Kim, H.; Seo, H.S.; Choi, D.W.; Kwon, H.K.; Na, D.H.; Kim, T.H.; Choy, Y.B.; et al. Bioinspired DNase-1-coated melanin-like nanospheres for modulation of infection-associated NETosis dysregulation. Adv. Sci. 2020, 7, 2001940. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Y.; Park, H.H.; Park, W.; Kim, H.; Jang, J.G.; Hong, K.S.; Lee, J.Y.; Seo, H.S.; Na, D.H.; Kim, T.H.; et al. Long acting nanoparticle DN-ase-1 for effective suppression of SARS-CoV-2 mediated neutrophil activities and cytokine storm. Biomaterials 2021, 267, 120389. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Hippensteel, J.; Leavitt, A.; Maloney, J.P.; Beckham, D.; Garcia, C.; Li, Q.; Freed, B.M.; Ordway, D.; Sandhaus, R.A.; et al. Hypothesis: Alpha-1-antitrypsin is a promising treatment option for COVID-19. Med. Hypotheses 2020, 146, 110394. [Google Scholar] [CrossRef]
- Vianello, A.; Guarnieri, G.; Braccioni, F.; Lococo, S.; Molena, B.; Cecchetto, A.; Giraudo, C.; De Marchi, L.B.; Caminati, M.; Senna, G. The pathogenesis, epidemiology and biomarkers of susceptibility of pulmonary fibrosis in COVID-19 survivors. Clin. Chem. Lab. Med. 2021, 60, 307–316. [Google Scholar] [CrossRef]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine Deiminase Inhibition Reduces Vascular Damage and Modulates Innate Immune Responses in Murine Models of Atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef]
- Furumoto, Y.; Smith, C.K.; Blanco, L.; Zhao, W.; Brooks, S.R.; Thacker, S.G.; Zarzour, A.; Sciumè, G.; Tsai, W.L.; Trier, A.M.; et al. Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol. 2016, 69, 148–160. [Google Scholar] [CrossRef]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef]
- Handono, K.; Sidarta, Y.O.; Pradana, B.A.; Nugroho, R.A.; Hartono, I.A.; Kalim, H.; Endharti, A.T. Vitamin D prevents endothelial damage induced by increased neutrophil extracellular traps formation in patients with systemic lupus erythematosus. Acta medica Indones. 2014, 46, 189–198. [Google Scholar]
- Tolle, F.; Umansky, V.; Utikal, J.; Kreis, S.; Bréchard, S. Neutrophils in Tumorigenesis: Missing Targets for Successful Next Generation Cancer Therapies? Int. J. Mol. Sci. 2021, 22, 6744. [Google Scholar] [CrossRef]
- Ng, H.; Havervall, S.; Rosell, A.; Aguilera, K.; Parv, K.; Von Meijenfeldt, F.A.; Lisman, T.; Mackman, N.; Thålin, C.; Phillipson, M. Circulating Markers of Neutrophil Extracellular Traps Are of Prognostic Value in Patients With COVID-19. Arter. Thromb. Vasc. Biol. 2021, 41, 988–994. [Google Scholar] [CrossRef]
- Li, M.; Lin, C.; Deng, H.; Strnad, J.; Bernabei, L.; Vogl, D.T.; Burke, J.J.; Nefedova, Y. A Novel Peptidylarginine Deiminase 4 (PAD4) Inhibitor BMS-P5 Blocks Formation of Neutrophil Extracellular Traps and Delays Progression of Multiple Myeloma. Mol. Cancer Ther. 2020, 19, 1530–1538. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demkow, U. Molecular Mechanisms of Neutrophil Extracellular Trap (NETs) Degradation. Int. J. Mol. Sci. 2023, 24, 4896. https://doi.org/10.3390/ijms24054896
Demkow U. Molecular Mechanisms of Neutrophil Extracellular Trap (NETs) Degradation. International Journal of Molecular Sciences. 2023; 24(5):4896. https://doi.org/10.3390/ijms24054896
Chicago/Turabian StyleDemkow, Urszula. 2023. "Molecular Mechanisms of Neutrophil Extracellular Trap (NETs) Degradation" International Journal of Molecular Sciences 24, no. 5: 4896. https://doi.org/10.3390/ijms24054896
APA StyleDemkow, U. (2023). Molecular Mechanisms of Neutrophil Extracellular Trap (NETs) Degradation. International Journal of Molecular Sciences, 24(5), 4896. https://doi.org/10.3390/ijms24054896