Hepatic Energy Metabolism under the Local Control of the Thyroid Hormone System

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Diet-Induced Alterations of the TH System
2.1. Fasting and Energy Restriction
2.1.1. Systemic Alterations
2.1.2. Local Alterations in Liver
2.2. Dietary Interventions
3. Effects of THs on Metabolic Pathways in the Liver
3.1. Carbohydrate Metabolism
3.1.1. T3 Is the Pacemaker for Global Energy Demand
3.1.2. Local T3 Signal Modulation in Oxidative Tissue Affects Glucose Demand by the Liver
3.1.3. Gluconeogenesis Responds to Various Tissue-Specific Effects of T3 Signaling and via Direct T3 Target Genes
3.2. Lipid Metabolism
3.2.1. T3 Orchestrates the Fatty Acid Uptake in a Tissue-Specific Manner
3.2.2. TH Tightly Regulate Various Stages of de novo Lipogenesis via Canonical and Non-Canonical Action
3.2.3. TH Signaling Is the Master Regulator of Transcription Factors Controlling Hepatic Fatty Acid Metabolism
3.2.4. TH Regulate the Release of Fatty Acids from Intracellular Stores during Lipophagy
3.2.5. TH Regulate the Conversion of Fatty Acids into Building Blocks for Energy Production
3.2.6. TH Regulate Targets in Fatty Acid Catabolism either Directly or through Prolonged Mitochondrial Signaling Axis
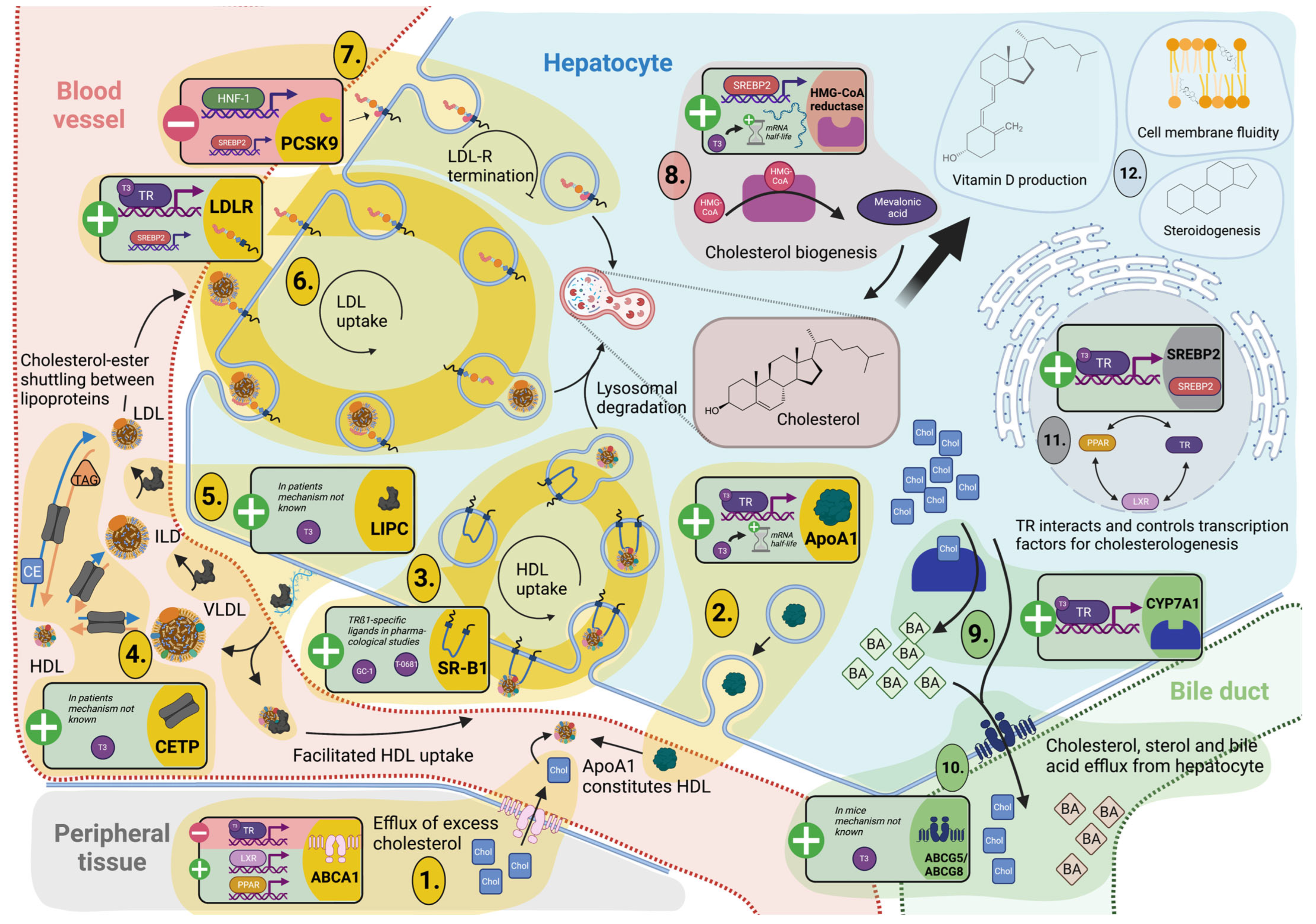
3.3. Cholesterol Metabolism and Turnover
3.3.1. TH Tightly Regulate Hepatic Cholesterol Formation and Peripheral Secretion
3.3.2. TH-Mediated Processing of Circulating Cholesterol Protects against Hypercholesterolemia
3.4. Reverse Cholesterol Transport
3.4.1. Cholesterol Shuttling from Peripheral Cells Is TH Dependent
3.4.2. TH Control Hepatic Lipoprotein Secretion and Uptake for Reverse Cholesterol Transport and Modulation of Lipoprotein Fractions
4. Future Challenges
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hönes, G.S.; Geist, D.; Moeller, L.C. Noncanonical Action of Thyroid Hormone Receptors α and β. Exp. Clin. Endocrinol. Diabetes 2020, 128, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.H.; Ganguly, S.; Abdalla, A.; Manning Fox, J.E.; Halestrap, A.P.; Visser, T.J. Identification of Monocarboxylate Transporter 8 as a Specific Thyroid Hormone Transporter. J. Biol. Chem. 2003, 278, 40128–40135. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, A.M.; Liao, X.-H.; Best, T.B.; Brockmann, K.; Refetoff, S. A Novel Syndrome Combining Thyroid and Neurological Abnormalities Is Associated with Mutations in a Monocarboxylate Transporter Gene. Am. J. Hum. Genet. 2004, 74, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J.; Frädrich, C. Deiodinases control local cellular and systemic thyroid hormone availability. Free Radic. Biol. Med. 2022, 193, 59–79. [Google Scholar] [CrossRef]
- Fonseca, T.L.; Fernandes, G.W.; McAninch, E.A.; Bocco, B.M.L.C.; Abdalla, S.M.; Ribeiro, M.O.; Mohácsik, P.; Fekete, C.; Li, D.; Xing, X.; et al. Perinatal deiodinase 2 expression in hepatocytes defines epigenetic susceptibility to liver steatosis and obesity. Proc. Natl. Acad. Sci. USA 2015, 112, 14018–14023. [Google Scholar] [CrossRef]
- Visser, T.J.; van Buuren, J.C.; Rutgers, M.; Rooda, S.J.E.; de Herder, W.W. The role of sulfation in thyroid hormone metabolism. Trends Endocrinol. Metab. 1990, 1, 211–218. [Google Scholar] [CrossRef]
- Dentice, M.; Ambrosio, R.; Damiano, V.; Sibilio, A.; Luongo, C.; Guardiola, O.; Yennek, S.; Zordan, P.; Minchiotti, G.; Colao, A.; et al. Intracellular Inactivation of Thyroid Hormone Is a Survival Mechanism for Muscle Stem Cell Proliferation and Lineage Progression. Cell Metab. 2014, 20, 1038–1048. [Google Scholar] [CrossRef]
- Ogawa-Wong, A.; Carmody, C.; Le, K.; Marschner, R.A.; Larsen, P.R.; Zavacki, A.M.; Wajner, S.M. Modulation of Deiodinase Types 2 and 3 during Skeletal Muscle Regeneration. Metabolites 2022, 12, 612. [Google Scholar] [CrossRef]
- Kaplan, M.M.; Utiger, R.D. Iodothyronine Metabolism in Rat Liver Homogenates. J. Clin. Investig. 1978, 61, 459–471. [Google Scholar] [CrossRef]
- Balsam, A.; Ingbar, S.H.; Sexton, F. The Influence of Fasting, Diabetes, and Several Pharmacological Agents on the Pathways of Thyroxine Metabolism in Rat Liver. J. Clin. Investig. 1978, 62, 415–424. [Google Scholar] [CrossRef]
- Russo, S.C.; Salas-Lucia, F.; Bianco, A.C. Deiodinases and the Metabolic Code for Thyroid Hormone Action. Endocrinology 2021, 162, bqab059. [Google Scholar] [CrossRef]
- Galton, V.A.; Hernandez, A.; Germain, D.L.S. The 5′-Deiodinases Are Not Essential for the Fasting-Induced Decrease in Circulating Thyroid Hormone Levels in Male Mice: Possible Roles for the Type 3 Deiodinase and Tissue Sequestration of Hormone. Endocrinology 2014, 155, 3172–3181. [Google Scholar] [CrossRef]
- Cordeiro, A.; Souza, L.; Oliveira, L.S.; Faustino, L.C.; Santiago, L.A.; Bloise, F.F.; Ortiga-Carvalho, T.M.; Almeida, N.A.D.S.; Pazos-Moura, C.C. Thyroid hormone regulation of Sirtuin 1 expression and implications to integrated responses in fasted mice. J. Endocrinol. 2012, 216, 181–193. [Google Scholar] [CrossRef]
- De Vries, E.M.; van Beeren, H.C.; van Wijk, A.C.W.A.; Kalsbeek, A.; Romijn, J.A.; Fliers, E.; Boelen, A. Regulation of type 3 deiodinase in rodent liver and adipose tissue during fasting. Endocr. Connect. 2020, 9, 552–562. [Google Scholar] [CrossRef]
- Van der Wal, A.M.; Bakker, O.; Wiersinga, W.M. The decrease of liver LDL receptor mRNA during fasting is related to the decrease in serum T3. Int. J. Biochem. Cell Biol. 1998, 30, 209–215. [Google Scholar] [CrossRef]
- De Vries, E.M.; Eggels, L.; Van Beeren, H.C.; Ackermans, M.T.; Kalsbeek, A.; Fliers, E.; Boelen, A. Fasting-Induced Changes in Hepatic Thyroid Hormone Metabolism in Male Rats Are Independent of Autonomic Nervous Input to the Liver. Endocrinology 2014, 155, 5033–5041. [Google Scholar] [CrossRef][Green Version]
- De Vries, E.M.; Van Beeren, H.C.; Ackermans, M.T.; Kalsbeek, A.; Fliers, E.; Boelen, A. Differential effects of fasting vs food restriction on liver thyroid hormone metabolism in male rats. J. Endocrinol. 2014, 224, 25–35. [Google Scholar] [CrossRef]
- Naito, K.; Inada, M.; Mashio, Y.; Tanaka, K.; Ishii, H.; Ishikawa, M.; Imura, H. Modulation of T4 5’-monodeiodination in rat anterior pituitary and liver homogenates by thyroid states and fasting. Endocrinol. Jpn. 1981, 28, 793–798. [Google Scholar] [CrossRef]
- Visser, T.J. Pathways of thyroid hormone metabolism. Acta Med. Austriaca 1996, 23, 10–16. [Google Scholar]
- Giacco, A.; Paoli, G.D.; Simiele, R.; Caterino, M.; Ruoppolo, M.; Bloch, W.; Kraaij, R.; Uitterlinden, A.G.; Santillo, A.; Senese, R.; et al. Exercise with food withdrawal at thermoneutrality impacts fuel use, the microbiome, AMPK phosphorylation, muscle fibers, and thyroid hormone levels in rats. Physiol. Rep. 2020, 8, e14354. [Google Scholar] [CrossRef]
- Visser, T.J.; van Haasteren, G.A.; Linkels, E.; Kaptein, E.; van Toor, H.; de Greef, W.J. Gender-specific changes in thyroid hormone-glucuronidating enzymes in rat liver during short-term fasting and long-term food restriction. Eur. J. Endocrinol. 1996, 135, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Young, R.A.; Rajatanavin, R.; Moring, A.F.; Braverman, L.E. Fasting Induces the Generation of Serum Thyronine-Binding Globulin in Zucker Rats*. Endocrinology 1985, 116, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Wade, S.; Bleiberg-Daniel, F.; Le Moullac, B. Rat Transthyretin: Effects of Acute Short-Term Food Deprivation and Refeeding on Serum and Cerebrospinal Fluid Concentration and on Hepatic mRNA Level. J. Nutr. 1988, 118, 199–205. [Google Scholar] [CrossRef]
- Gereben, B.; Zavacki, A.M.; Ribich, S.; Kim, B.W.; Huang, S.A.; Simonides, W.S.; Zeöld, A.; Bianco, A.C. Cellular and Molecular Basis of Deiodinase-Regulated Thyroid Hormone Signaling1. Endocr. Rev. 2008, 29, 898–938. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.C.; Kim, B.W. Deiodinases: Implications of the local control of thyroid hormone action. J. Clin. Investig. 2006, 116, 2571–2579. [Google Scholar] [CrossRef]
- Köhrle, J. Local activation and inactivation of thyroid hormones: The deiodinase family. Mol. Cell. Endocrinol. 1999, 151, 103–119. [Google Scholar] [CrossRef]
- Kester, M.H.A.; Van Dijk, C.H.; Tibboel, D.; Hood, A.M.; Rose, N.J.M.; Meinl, W.; Pabel, U.; Glatt, H.; Falany, C.N.; Coughtrie, M.; et al. Sulfation of Thyroid Hormone by Estrogen Sulfotransferase. J. Clin. Endocrinol. Metab. 1999, 84, 2577–2580. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Green, W.L.; Huang, W.-S.; Hays, M.T.; Chopra, I.J. Alternate Pathways of Thyroid Hormone Metabolism. Thyroid 2005, 15, 943–958. [Google Scholar] [CrossRef]
- Visser, T.J. Role of sulfation in thyroid hormone metabolism. Chem. Interact. 1994, 92, 293–303. [Google Scholar] [CrossRef]
- Distefano, J.J.; Sternlicht, M.; Harris, D.R. Rat Enterohepatic Circulation and Intestinal Distribution of Enterally Infused Thyroid Hormones*. Endocrinology 1988, 123, 2526–2539. [Google Scholar] [CrossRef]
- De Herder, W.W.; Hazenberg, M.P.; Pennock-Schröder, A.M.; Hennemann, G.; Visser, T.J. Rapid and bacteria-dependent in vitro hydrolysis of iodothyronine-conjugates by intestinal contents of humans and rats. Med. Biol. 1986, 64, 31–35. [Google Scholar]
- Virili, C.; Centanni, M. “With a little help from my friends”—The role of microbiota in thyroid hormone metabolism and enterohepatic recycling. Mol. Cell. Endocrinol. 2017, 458, 39–43. [Google Scholar] [CrossRef]
- De Jong, M.; Docter, R.; Van Der Hoek, H.J.; Vos, R.A.; Krenning, E.P.; Hennemann, G. Transport of 3,5,3’-triiodothyronine into the perfused rat liver and subsequent metabolism are inhibited by fasting. Endocrinology 1992, 131, 463–470. [Google Scholar] [CrossRef]
- Kester, M.H.A.; Kaptein, E.; Roest, T.J.; van Dijk, C.H.; Tibboel, D.; Meinl, W.; Glatt, H.; Coughtrie, M.W.H.; Visser, T.J. Characterization of rat iodothyronine sulfotransferases. Am. J. Physiol. Metab. 2003, 285, E592–E598. [Google Scholar] [CrossRef]
- Darras, V.M.; Hume, R.; Visser, T.J. Regulation of thyroid hormone metabolism during fetal development. Mol. Cell. Endocrinol. 1999, 151, 37–47. [Google Scholar] [CrossRef]
- Visser, T.J.; Kaptein, E.; Gijzel, A.L.; De Herder, W.W.; Ebner, T.; Burchell, B. Glucuronidation of thyroid hormone by human bilirubin and phenol UDP-glucuronyltransferase isoenzymes. FEBS Lett. 1993, 324, 358–360. [Google Scholar] [CrossRef]
- Fujita, K.-I.; Nagata, K.; Ozawa, S.; Sasano, H.; Yamazoe, Y. Molecular Cloning and Characterization of Rat ST1B1 and Human ST1B2 cDNAs, Encoding Thyroid Hormone Sulfotransferases. J. Biochem. 1997, 122, 1052–1061. [Google Scholar] [CrossRef]
- Blanchard, R.L.; Freimuth, R.; Buck, J.; Weinshilboum, R.M.; Coughtrie, M. A proposed nomenclature system for the cytosolic sulfotransferase (SULT) superfamily. Pharmacogenetics 2004, 14, 199–211. [Google Scholar] [CrossRef]
- Mackenzie, P.I.; Bock, K.W.; Burchell, B.; Guillemette, C.; Ikushiro, S.-I.; Iyanagi, T.; Miners, J.O.; Owens, I.S.; Nebert, D.W. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet. Genom. 2005, 15, 677–685. [Google Scholar] [CrossRef]
- Maglich, J.M.; Watson, J.; McMillen, P.J.; Goodwin, B.; Willson, T.M.; Moore, J.T. The Nuclear Receptor CAR Is a Regulator of Thyroid Hormone Metabolism during Caloric Restriction. J. Biol. Chem. 2004, 279, 19832–19838. [Google Scholar] [CrossRef]
- Vella, K.R.; Ramadoss, P.; Lam, F.S.; Harris, J.C.; Ye, F.D.; Same, P.D.; O’Neill, N.F.; Maratos-Flier, E.; Hollenberg, A.N. NPY and MC4R Signaling Regulate Thyroid Hormone Levels during Fasting through Both Central and Peripheral Pathways. Cell Metab. 2011, 14, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Küblbeck, J.; Niskanen, J.; Honkakoski, P. Metabolism-Disrupting Chemicals and the Constitutive Androstane Receptor CAR. Cells 2020, 9, 2306. [Google Scholar] [CrossRef] [PubMed]
- Everts, M.E.; de Jong, M.; Lim, C.-F.; Docter, R.; Krenning, E.P.; Visser, T.J.; Hennemann, G. Different Regulation of Thyroid Hormone Transport in Liver and Pituitary: Its Possible Role in the Maintenance of Low T3 Production during Nonthyroidal Illness and Fasting in Man. Thyroid 1996, 6, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.G.; Martin, I.V.; Porn, A.C.; Voigt, S.; Gartung, C.; Trautwein, C.; Geier, A. Fasting induces basolateral uptake transporters of the SLC family in the liver via HNF4α and PGC1α. Am. J. Physiol. Liver Physiol. 2007, 293, G585–G590. [Google Scholar] [CrossRef]
- Tian, L.; Song, Y.; Xing, M.; Zhang, W.; Ning, G.; Li, X.; Yu, C.; Qin, C.; Liu, J.; Tian, X.; et al. A novel role for thyroid-stimulating hormone: Up-regulation of hepatic 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase expression through the cyclic adenosine monophosphate/protein kinase A/cyclic adenosine monophosphate-responsive element binding protei. Hepatology 2010, 52, 1401–1409. [Google Scholar] [CrossRef]
- Zhang, W.; Tian, L.-M.; Han, Y.; Ma, H.-Y.; Wang, L.-C.; Guo, J.; Gao, L.; Zhao, J.-J. Presence of thyrotropin receptor in hepatocytes: Not a case of illegitimate transcription. J. Cell. Mol. Med. 2009, 13, 4636–4642. [Google Scholar] [CrossRef]
- Wang, T.; Xu, J.; Bo, T.; Zhou, X.; Jiang, X.; Gao, L.; Zhao, J. Decreased fasting blood glucose is associated with impaired hepatic glucose production in thyroid-stimulating hormone receptor knockout mice. Endocr. J. 2013, 60, 941–950. [Google Scholar] [CrossRef]
- Elgadi, A.; Zemack, H.; Marcus, C.; Norgren, S. Tissue-specific knockout of TSHr in white adipose tissue increases adipocyte size and decreases TSH-induced lipolysis. Biochem. Biophys. Res. Commun. 2010, 393, 526–530. [Google Scholar] [CrossRef]
- Zhou, L.; Wu, K.; Zhang, L.; Gao, L.; Chen, S. Liver-specific deletion of TSHR inhibits hepatic lipid accumulation in mice. Biochem. Biophys. Res. Commun. 2018, 497, 39–45. [Google Scholar] [CrossRef]
- González-Ramos, S.; Paz-García, M.; Fernández-García, V.; Portune, K.J.; Acosta-Medina, E.F.; Sanz, Y.; Castrillo, A.; Martín-Sanz, P.; Obregon, M.J.; Boscá, L. NOD1 deficiency promotes an imbalance of thyroid hormones and microbiota homeostasis in mice fed high fat diet. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef]
- Han, H.; Xin, P.; Zhao, L.; Xu, J.; Xia, Y.; Yang, X.; Sun, X.; Hao, L. Excess Iodine and High-Fat Diet Combination Modulates Lipid Profile, Thyroid Hormone, and Hepatic LDLr Expression Values in Mice. Biol. Trace Element Res. 2012, 147, 233–239. [Google Scholar] [CrossRef]
- Lopez, A.N.; Geissler, C.; Naujack, A.-M.; Chen, Y.; Taege, N.; Britsemmer, J.H.; Vinicius, M.D.A.L.; Oster, H.; Spranger, J.; Katrin, W.E.; et al. Deiodinase type I (DIO1) regulation in non-alcoholic fatty liver disease (NAFLD). Endocr. Abstr. 2022, 84, PS2-09-80. [Google Scholar] [CrossRef]
- Bruinstroop, E.; Zhou, J.; Tripathi, M.; Yau, W.W.; Boelen, A.; Singh, B.K.; Yen, P.M. Early induction of hepatic deiodinase type 1 inhibits hepatosteatosis during NAFLD progression. Mol. Metab. 2021, 53, 101266. [Google Scholar] [CrossRef]
- Lietzow, J.; Golchert, J.; Homuth, G.; Völker, U.; Jonas, W.; Köhrle, J. 3,5-T2 alters murine genes relevant for xenobiotic, steroid, and thyroid hormone metabolism. J. Mol. Endocrinol. 2016, 56, 311–323. [Google Scholar] [CrossRef]
- Wu, J.; Wang, C.; Li, S.; Li, S.; Wang, W.; Li, J.; Chi, Y.; Yang, H.; Kong, X.; Zhou, Y.; et al. Thyroid hormone-responsive SPOT 14 homolog promotes hepatic lipogenesis, and its expression is regulated by Liver X receptor α through a sterol regulatory element-binding protein 1c-dependent mechanism in mice. Hepatology 2013, 58, 617–628. [Google Scholar] [CrossRef]
- LaFave, L.T.; Augustin, L.B.; Mariash, C.N. S14: Insights from Knockout Mice. Endocrinology 2006, 147, 4044–4047. [Google Scholar] [CrossRef]
- Jornayvaz, F.; Lee, H.-Y.; Jurczak, M.; Alves, T.C.; Guebre-Egziabher, F.; Guigni, B.; Zhang, D.; Samuel, V.T.; Silva, J.E.; Shulman, G.I. Thyroid Hormone Receptor-α Gene Knockout Mice Are Protected from Diet-Induced Hepatic Insulin Resistance. Endocrinology 2012, 153, 583–591. [Google Scholar] [CrossRef]
- Goldfarb, C.N.; Karri, K.; Pyatkov, M.; Waxman, D.J. Interplay Between GH-regulated, Sex-biased Liver Transcriptome and Hepatic Zonation Revealed by Single-Nucleus RNA Sequencing. Endocrinology 2022, 163, bqac059. [Google Scholar] [CrossRef]
- Smati, S.; Polizzi, A.; Fougerat, A.; Ellero-Simatos, S.; Blum, Y.; Lippi, Y.; Régnier, M.; Laroyenne, A.; Huillet, M.; Arif, M.; et al. Integrative study of diet-induced mouse models of NAFLD identifies PPARα as a sexually dimorphic drug target. Gut 2021, 71, 807–821. [Google Scholar] [CrossRef]
- Riese, C.; Michaelis, M.; Mentrup, B.; Götz, F.; Köhrle, J.; Schweizer, U.; Schomburg, L. Selenium-Dependent Pre- and Posttranscriptional Mechanisms Are Responsible for Sexual Dimorphic Expression of Selenoproteins in Murine Tissues. Endocrinology 2006, 147, 5883–5892. [Google Scholar] [CrossRef]
- Desai-Yajnik, V.; Zeng, J.; Omori, K.; Sherman, J.; Morimoto, T. The effect of thyroid hormone treatment on the gene expression and enzyme activity of rat liver sodium-potassium dependent adenosine triphosphatase. Endocrinology 1995, 136, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.; Jakab, G.; Kranias, E.G.; Edes, I. Thyroid hormone-induced alterations in phospholamban protein expression. Regulatory effects on sarcoplasmic reticulum Ca2+ transport and myocardial relaxation. Circ. Res. 1994, 75, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.P.; O’Boyle, E.; Fisher, M.; Haber, R.S. Regulation of GLUT2 glucose transporter expression in liver by thyroid hormone: Evidence for hormonal regulation of the hepatic glucose transport system. Endocrinology 1994, 135, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Stalmans, W. The Role of the Liver in the Homeostasis of Blood Glucose. Curr. Top. Cell. Regul. 1976, 11, 51–97. [Google Scholar] [CrossRef]
- Kim, B.W.; Zavacki, A.M.; Curcio-Morelli, C.; Dentice, M.; Harney, J.W.; Larsen, P.R.; Bianco, A.C. Endoplasmic Reticulum-Associated Degradation of the Human Type 2 Iodothyronine Deiodinase (D2) is Mediated via an Association between Mammalian UBC7 and the Carboxyl Region of D2. Mol. Endocrinol. 2003, 17, 2603–2612. [Google Scholar] [CrossRef]
- Salvatore, D.; Bartha, T.; Harney, J.W.; Larsen, P.R. Molecular biological and biochemical characterization of the human type 2 selenodeiodinase. Endocrinology 1996, 137, 3308–3315. [Google Scholar] [CrossRef]
- Torrance, C.J.; Devente, J.E.; Jones, J.P.; Dohm, G.L. Effects of Thyroid Hormone on GLUT4 Glucose Transporter Gene Expression and NIDDM in Rats. Endocrinology 1997, 138, 1204–1214. [Google Scholar] [CrossRef]
- Torrance, C.J.; Usala, S.J.; Pessin, J.E.; Dohm, G.L. Characterization of a Low Affinity Thyroid Hormone Receptor Binding Site within the Rat GLUT4 Gene Promoter. Endocrinology 1997, 138, 1215–1223. [Google Scholar] [CrossRef][Green Version]
- Weinstein, S.P.; O’Boyle, E.; Haber, R.S. Thyroid Hormone Increases Basal and Insulin-Stimulated Glucose Transport in Skeletal Muscle: The Role of GLUT4 Glucose Transporter Expression. Diabetes 1994, 43, 1185–1189. [Google Scholar] [CrossRef]
- Leonard, J.L. Dibutyryl cAMP induction of type II 5′deiodinase activity in rat brain astrocytes in culture. Biochem. Biophys. Res. Commun. 1988, 151, 1164–1172. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef]
- Klieverik, L.P.; Janssen, S.F.; van Riel, A.; Foppen, E.; Bisschop, P.H.; Serlie, M.J.; Boelen, A.; Ackermans, M.T.; Sauerwein, H.P.; Fliers, E.; et al. Thyroid hormone modulates glucose production via a sympathetic pathway from the hypothalamic paraventricular nucleus to the liver. Proc. Natl. Acad. Sci. USA 2009, 106, 5966–5971. [Google Scholar] [CrossRef]
- Carter, W.J.; Benjamin, W.S.V.D.W.; Faas, F.H. Effect of experimental hyperthyroidism on skeletal-muscle proteolysis. Biochem. J. 1981, 194, 685–690. [Google Scholar] [CrossRef]
- Felig, P. Amino Acid Metabolism in Man. Annu. Rev. Biochem. 1975, 44, 933–955. [Google Scholar] [CrossRef]
- Suh, J.H.; Sieglaff, U.H.; Zhang, A.; Xia, X.; Cvoro, A.; Winnier, G.E.; Webb, P. SIRT1 is a Direct Coactivator of Thyroid Hormone Receptor β1 with Gene-Specific Actions. PLoS ONE 2013, 8, e70097. [Google Scholar] [CrossRef]
- Attia, R.R.; Connnaughton, S.; Boone, L.R.; Wang, F.; Elam, M.B.; Ness, G.C.; Cook, G.A.; Park, E.A. Regulation of Pyruvate Dehydrogenase Kinase 4 (PDK4) by Thyroid Hormone: Role of the peroxisome proliferator-activated receptor gamma coactivator (PGC-1 alpha). J. Biol. Chem. 2010, 285, 2375–2385. [Google Scholar] [CrossRef]
- Thakran, S.; Sharma, P.; Attia, R.R.; Hori, R.T.; Deng, X.; Elam, M.B.; Park, E.A. Role of Sirtuin 1 in the Regulation of Hepatic Gene Expression by Thyroid Hormone. J. Biol. Chem. 2013, 288, 807–818. [Google Scholar] [CrossRef]
- Erion, D.M.; Yonemitsu, S.; Nie, Y.; Nagai, Y.; Gillum, M.P.; Hsiao, J.J.; Iwasaki, T.; Stark, R.; Weismann, D.; Yu, X.X.; et al. SirT1 knockdown in liver decreases basal hepatic glucose production and increases hepatic insulin responsiveness in diabetic rats. Proc. Natl. Acad. Sci. USA 2009, 106, 11288–11293. [Google Scholar] [CrossRef]
- Bratusch-Marrain, P.R.; Komjati, M.; Waldhäusl, W.K. Glucose Metabolism in Noninsulin-Dependent Diabetic Patients with Experimental Hyperthyroidism*. J. Clin. Endocrinol. Metab. 1985, 60, 1063–1068. [Google Scholar] [CrossRef]
- Moon, S.W.; Hahm, J.R.; Lee, G.-W.; Kang, M.Y.; Jung, J.H.; Jung, T.S.; Lee, K.W.; Jung, K.A.; Ahn, Y.J.; Kim, S.; et al. A Case of Hyperglycemic Hyperosmolar State Associated with Graves’ Hyperthyroidism: A Case Report. J. Korean Med. Sci. 2006, 21, 765–767. [Google Scholar] [CrossRef]
- Wintergerst, K.A.; Rogers, E.S.; Foster, M.B. Hyperthyroidism presenting with hyperglycemia in an adolescent female. J. Pediatr. Endocrinol. Metab. 2011, 24, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.D.; Raptis, S.A. Thyroid hormone excess and glucose intolerance. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. S2), S225–S239. [Google Scholar] [CrossRef] [PubMed]
- Battarbee, H.D. The Effects of Thyroid State on Rat Liver Glucose-6-Phosphatase Activity and Glycogen Content. Exp. Biol. Med. 1974, 147, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Burton, S.D.; Robbins, E.; Byers, S.O. Effect of Hyperthyroidism on Glycogen Content of the Isolated Rat Liver. Am. J. Physiol. Content 1957, 188, 509–513. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Lu, C.; Cheng, S.-Y. Thyroid hormone receptors regulate adipogenesis and carcinogenesis via crosstalk signaling with peroxisome proliferator-activated receptors. J. Mol. Endocrinol. 2009, 44, 143–154. [Google Scholar] [CrossRef]
- Wierzbicki, M.; Chabowski, A.; Zendzian-Piotrowska, M.; Górski, J. Differential effects of in vivo PPAR alpha and gamma activation on fatty acid transport proteins expression and lipid content in rat liver. J. Physiol. Pharmacol. 2009, 60, 99–106. [Google Scholar]
- Doege, H.; Grimm, D.; Falcon, A.; Tsang, B.; Storm, T.A.; Xu, H.; Ortegon, A.M.; Kazantzis, M.; Kay, M.A.; Stahl, A. Silencing of Hepatic Fatty Acid Transporter Protein 5 in Vivo Reverses Diet-induced Non-alcoholic Fatty Liver Disease and Improves Hyperglycemia. J. Biol. Chem. 2008, 283, 22186–22192. [Google Scholar] [CrossRef]
- Klieverik, L.P.; Coomans, C.P.; Endert, E.; Sauerwein, H.P.; Havekes, L.M.; Voshol, P.J.; Rensen, P.C.N.; Romijn, J.A.; Kalsbeek, A.; Fliers, E. Thyroid Hormone Effects on Whole-Body Energy Homeostasis and Tissue-Specific Fatty Acid Uptake in Vivo. Endocrinology 2009, 150, 5639–5648. [Google Scholar] [CrossRef]
- Nakagawa, S.; Kawashima, Y.; Hirose, A.; Kozuka, H. Regulation of hepatic level of fatty-acid-binding protein by hormones and clofibric acid in the rat. Biochem. J. 1994, 297, 581–584. [Google Scholar] [CrossRef]
- Santana-Farré, R.; Mirecki-Garrido, M.; Bocos, C.; Henríquez-Hernández, L.A.; Kahlon, N.; Herrera, E.; Norstedt, G.; Parini, P.; Flores-Morales, A.; Fernández-Pérez, L. Influence of Neonatal Hypothyroidism on Hepatic Gene Expression and Lipid Metabolism in Adulthood. PLoS ONE 2012, 7, e37386. [Google Scholar] [CrossRef]
- Petty, K.J.; Desvergne, B.; Mitsuhashi, T.; Nikodem, V.M. Identification of a thyroid hormone response element in the malic enzyme gene. J. Biol. Chem. 1990, 265, 7395–7400. [Google Scholar] [CrossRef]
- López, M.; Lelliott, C.J.; Vidal-Puig, A. Hypothalamic fatty acid metabolism: A housekeeping pathway that regulates food intake. Bioessays 2007, 29, 248–261. [Google Scholar] [CrossRef]
- Pender, C.; Trentadue, A.R.; Pories, W.J.; Dohm, G.L.; Houmard, J.A.; Youngren, J.F. Expression of genes regulating Malonyl-CoA in human skeletal muscle. J. Cell. Biochem. 2006, 99, 860–867. [Google Scholar] [CrossRef]
- Kreuz, S.; Schoelch, C.; Thomas, L.; Rist, W.; Rippmann, J.F.; Neubauer, H. Acetyl-CoA carboxylases 1 and 2 show distinct expression patterns in rats and humans and alterations in obesity and diabetes. Diabetes/Metab. Res. Rev. 2009, 25, 577–586. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, L.; Hillgartner, F.B. Thyroid Hormone Stimulates Acetyl-CoA Carboxylase-α Transcription in Hepatocytes by Modulating the Composition of Nuclear Receptor Complexes Bound to a Thyroid Hormone Response Element. J. Biol. Chem. 2001, 276, 974–983. [Google Scholar] [CrossRef]
- Blennemann, B.; Leahy, P.; Kim, T.-S.; Freake, H.C. Tissue-specific regulation of lipogenic mRNAs by thyroid hormone. Mol. Cell. Endocrinol. 1995, 110, 1–8. [Google Scholar] [CrossRef]
- López, M.; Varela, L.; Vázquez, M.J.; Rodríguez-Cuenca, S.; González, C.R.; Velagapudi, V.R.; Morgan, D.A.; Schoenmakers, E.; Agassandian, K.; Lage, R.; et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat. Med. 2010, 16, 1001–1008. [Google Scholar] [CrossRef]
- Zhu, Q.; Mariash, A.; Margosian, M.R.; Gopinath, S.; Fareed, M.T.; Anderson, G.W.; Mariash, C.N. Spot 14 Gene Deletion Increases Hepatic de Novo Lipogenesis. Endocrinology 2001, 142, 4363–4370. [Google Scholar] [CrossRef]
- Campbell, M.C.; Anderson, G.W.; Mariash, C.N. Human Spot 14 Glucose and Thyroid Hormone Response: Characterization and Thyroid Hormone Response Element Identification. Endocrinology 2003, 144, 5242–5248. [Google Scholar] [CrossRef]
- Paton, C.M.; Ntambi, J.M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol. Metab. 2009, 297, E28–E37. [Google Scholar] [CrossRef] [PubMed]
- Radenne, A.; Akpa, M.; Martel, C.; Sawadogo, S.; Mauvoisin, D.; Mounier, C. Hepatic regulation of fatty acid synthase by insulin and T3: Evidence for T3 genomic and nongenomic actions. Am. J. Physiol. Metab. 2008, 295, E884–E894. [Google Scholar] [CrossRef] [PubMed]
- Hönes, G.S.; Rakov, H.; Logan, J.; Liao, X.-H.; Werbenko, E.; Pollard, A.S.; Præstholm, S.M.; Siersbæk, M.S.; Rijntjes, E.; Gassen, J.; et al. Noncanonical thyroid hormone signaling mediates cardiometabolic effects in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E11323–E11332. [Google Scholar] [CrossRef] [PubMed]
- Shabtai, Y.; Nagaraj, N.K.; Batmanov, K.; Cho, Y.-W.; Guan, Y.; Jiang, C.; Remsberg, J.; Forrest, D.; Lazar, M.A. A coregulator shift, rather than the canonical switch, underlies thyroid hormone action in the liver. Genes Dev. 2021, 35, 367–378. [Google Scholar] [CrossRef]
- Ritter, M.J.; Amano, I.; Imai, N.; De Oliveira, L.S.; Vella, K.R.; Hollenberg, A.N. Nuclear Receptor CoRepressors, NCOR1 and SMRT, are required for maintaining systemic metabolic homeostasis. Mol. Metab. 2021, 53, 101315. [Google Scholar] [CrossRef]
- Mendoza, A.; Tang, C.; Choi, J.; Acuña, M.; Logan, M.; Martin, A.G.; Al-Sowaimel, L.; Desai, B.N.; Tenen, D.E.; Jacobs, C.; et al. Thyroid hormone signaling promotes hepatic lipogenesis through the transcription factor ChREBP. Sci. Signal. 2021, 14, eabh3839. [Google Scholar] [CrossRef]
- Song, S.; Attia, R.R.; Connaughton, S.; Niesen, M.I.; Ness, G.C.; Elam, M.B.; Hori, R.T.; Cook, G.A.; Park, E.A. Peroxisome proliferator activated receptor α (PPARα) and PPAR gamma coactivator (PGC-1α) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol. Cell. Endocrinol. 2010, 325, 54–63. [Google Scholar] [CrossRef]
- Adams, A.C.; Astapova, I.; Fisher, F.M.; Badman, M.K.; Kurgansky, K.E.; Flier, J.S.; Hollenberg, A.N.; Maratos-Flier, E. Thyroid Hormone Regulates Hepatic Expression of Fibroblast Growth Factor 21 in a PPARα-dependent Manner. J. Biol. Chem. 2010, 285, 14078–14082. [Google Scholar] [CrossRef]
- Chau, M.D.L.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK–SIRT1–PGC-1α pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558. [Google Scholar] [CrossRef]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.-S.; Lindberg, R.A.; et al. Fibroblast Growth Factor 21 Reverses Hepatic Steatosis, Increases Energy Expenditure, and Improves Insulin Sensitivity in Diet-Induced Obese Mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef]
- Chi, H.-C.; Tsai, C.-Y.; Tsai, M.-M.; Yeh, C.-T.; Lin, K.-H. Molecular functions and clinical impact of thyroid hormone-triggered autophagy in liver-related diseases. J. Biomed. Sci. 2019, 26, 24. [Google Scholar] [CrossRef]
- Sinha, R.A.; You, S.-H.; Zhou, J.; Siddique, M.M.; Bay, B.-H.; Zhu, X.; Privalsky, M.L.; Cheng, S.-Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Investig. 2012, 122, 2428–2438. [Google Scholar] [CrossRef]
- Coates, P.M.; Brown, S.A.; Lau, H.; Krulich, L.; Koldovský, O. Effect of thyroxine on acid lipase activity of adult rat liver. FEBS Lett. 1978, 86, 45–48. [Google Scholar] [CrossRef]
- Singh, B.K.; Sinha, R.A.; Zhou, J.; Tripathi, M.; Ohba, K.; Wang, M.-E.; Astapova, I.; Ghosh, S.; Hollenberg, A.N.; Gauthier, K.; et al. Hepatic FOXO1 Target Genes Are Co-regulated by Thyroid Hormone via RICTOR Protein Deacetylation and MTORC2-AKT Protein Inhibition. J. Biol. Chem. 2016, 291, 198–214. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Zhou, J.; Wu, Y.; Farah, B.L.; Ohba, K.; Lesmana, R.; Gooding, J.; Bay, B.-H.; Yen, P.M. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 2015, 11, 1341–1357. [Google Scholar] [CrossRef]
- Chi, H.-C.; Chen, S.-L.; Lin, S.-L.; Tsai, C.-Y.; Chuang, W.-Y.; Lin, Y.-H.; Huang, Y.-H.; Tsai, M.-M.; Yeh, C.-T.; Lin, K.-H. Thyroid hormone protects hepatocytes from HBx-induced carcinogenesis by enhancing mitochondrial turnover. Oncogene 2017, 36, 5274–5284. [Google Scholar] [CrossRef]
- Chi, H.-C.; Chen, S.-L.; Tsai, C.-Y.; Chuang, W.-Y.; Huang, Y.-H.; Tsai, M.-M.; Wu, S.-M.; Sun, C.-P.; Yeh, C.-T.; Lin, K.-H. Thyroid hormone suppresses hepatocarcinogenesis via DAPK2 and SQSTM1-dependent selective autophagy. Autophagy 2016, 12, 2271–2285. [Google Scholar] [CrossRef]
- Tseng, Y.-H.; Ke, P.-Y.; Liao, C.-J.; Wu, S.-M.; Chi, H.-C.; Tsai, C.-Y.; Chen, C.-Y.; Lin, Y.-H.; Lin, K.-H. Chromosome 19 open reading frame 80 is upregulated by thyroid hormone and modulates autophagy and lipid metabolism. Autophagy 2013, 10, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ellis, J.M.; Wolfgang, M.J. Adipose Fatty Acid Oxidation Is Required for Thermogenesis and Potentiates Oxidative Stress-Induced Inflammation. Cell Rep. 2015, 10, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A.; Ellis, J.M. Peroxisomal acyl-CoA synthetases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Jackson-Hayes, L.; Song, S.; Lavrentyev, E.N.; Jansen, M.S.; Hillgartner, F.B.; Tian, L.; Wood, P.A.; Cook, G.A.; Park, E.A. A Thyroid Hormone Response Unit Formed between the Promoter and First Intron of the Carnitine Palmitoyltransferase-Iα Gene Mediates the Liver-specific Induction by Thyroid Hormone. J. Biol. Chem. 2003, 278, 7964–7972. [Google Scholar] [CrossRef]
- Weitzel, J.M.; Iwen, K.A. Coordination of mitochondrial biogenesis by thyroid hormone. Mol. Cell. Endocrinol. 2011, 342, 1–7. [Google Scholar] [CrossRef]
- Djouadi, F.; Riveau, B.; Merlet-Benichou, C.; Bastin, J. Tissue-specific regulation of medium-chain acyl-CoA dehydrogenase gene by thyroid hormones in the developing rat. Biochem. J. 1997, 324, 289–294. [Google Scholar] [CrossRef]
- Jekabsons, M.B.; Gregoire, F.M.; Schonfeld-Warden, N.A.; Warden, C.H.; Horwitz, B.A. T3 stimulates resting metabolism and UCP-2 and UCP-3 mRNA but not nonphosphorylating mitochondrial respiration in mice. Am. J. Physiol. Metab. 1999, 277, E380–E389. [Google Scholar] [CrossRef]
- Hoffstedt, J.; Folkesson, R.; Wahrenberg, H.; Wennlund, A.; van Harmelen, V.; Arner, P. A Marked Upregulation of Uncoupling Protein 2 Gene Expression in Adipose Tissue of Hyperthyroid Subjects. Horm. Metab. Res. 2000, 32, 475–479. [Google Scholar] [CrossRef]
- Fozzatti, L.; Lu, C.; Kim, D.-W.; Cheng, S.-Y. Differential Recruitment of Nuclear Coregulators Directs the Isoform-Dependent Action of Mutant Thyroid Hormone Receptors. Mol. Endocrinol. 2011, 25, 908–921. [Google Scholar] [CrossRef]
- Shin, D.-J.; Osborne, T.F. Thyroid Hormone Regulation and Cholesterol Metabolism Are Connected through Sterol Regulatory Element-binding Protein-2 (SREBP-2). J. Biol. Chem. 2003, 278, 34114–34118. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Ness, G.C.; Chambers, C.M. Feedback and Hormonal Regulation of Hepatic 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase: The Concept of Cholesterol Buffering Capacity. Proc. Soc. Exp. Boil. Med. 2008, 224, 8–19. [Google Scholar] [CrossRef]
- Ness, G.C.; Dugan, R.E.; Lakshmanan, M.R.; Nepokroeff, C.M.; Porter, J.W. Stimulation of Hepatic β-Hydroxy-β-methylglutaryl Coenzyme A Reductase Activity in Hypophysectomized Rats by L-Triiodothyronine. Proc. Natl. Acad. Sci. USA 1973, 70, 3839–3842. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Choi, H.S. The Regulatory Effects of Thyroid Hormone on The Activity Of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase. Endocr. Res. 2000, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ness, G.C.; Lopez, D.; Chambers, C.M.; Newsome, W.P.; Cornelius, P.; Long, C.A.; Harwood, H. Effects of l-Triiodothyronine and the Thyromimetic L-94901 on Serum Lipoprotein Levels and Hepatic Low-Density Lipoprotein Receptor, 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase, and Apo A-I Gene Expression. Biochem. Pharmacol. 1998, 56, 121–129. [Google Scholar] [CrossRef]
- Lopez, D.; Socarrás, J.F.A.; Bedi, M.; Ness, G.C. Activation of the hepatic LDL receptor promoter by thyroid hormone. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2007, 1771, 1216–1225. [Google Scholar] [CrossRef]
- Smith, J.R.; Osborne, T.F.; Goldstein, J.L.; Brown, M.S. Identification of nucleotides responsible for enhancer activity of sterol regulatory element in low density lipoprotein receptor gene. J. Biol. Chem. 1990, 265, 2306–2310. [Google Scholar] [CrossRef]
- Ogura, M. PCSK9 inhibition in the management of familial hypercholesterolemia. J. Cardiol. 2018, 71, 1–7. [Google Scholar] [CrossRef]
- Bonde, Y.; Breuer, O.; Lütjohann, D.; Sjöberg, S.; Angelin, B.; Rudling, M. Thyroid hormone reduces PCSK9 and stimulates bile acid synthesis in humans. J. Lipid Res. 2014, 55, 2408–2415. [Google Scholar] [CrossRef]
- Yildirim, A.M.; Koca, A.O.; Beyan, E.; Dogan, O.; Karakaya, S.; Aksoz, Z.; Ertugrul, D.T. Association of serum proprotein convertase Subtilisin/Kexin Type 9 (PCSK9) level with thyroid function disorders. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 5511–5517. [Google Scholar] [CrossRef]
- Sadik, N.A.; Rashed, L.A.; El-Sawy, S.S. The Relationship of Circulating Proprotein Convertase Subtilisin/Kexin Type 9 With TSH and Lipid Profile in Newly Diagnosed Patients with Subclinical and Overt Hypothyroidism. Clin. Med. Insights Endocrinol. Diabetes 2022, 15, 1177271918765137. [Google Scholar] [CrossRef]
- Li, H.; Dong, B.; Park, S.W.; Lee, H.-S.; Chen, W.; Liu, J. Hepatocyte Nuclear Factor 1α Plays a Critical Role in PCSK9 Gene Transcription and Regulation by the Natural Hypocholesterolemic Compound Berberine. J. Biol. Chem. 2009, 284, 28885–28895. [Google Scholar] [CrossRef]
- Gong, Y.; Ma, Y.; Ye, Z.; Fu, Z.; Yang, P.; Gao, B.; Guo, W.; Hu, D.; Ye, J.; Ma, S.; et al. Thyroid stimulating hormone exhibits the impact on LDLR/LDL-c via up-regulating hepatic PCSK9 expression. Metabolism 2017, 76, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Matozel, M.; Boehme, S.; Kong, B.; Nilsson, L.-M.; Guo, G.; Ellis, E.; Chiang, J.Y.L. Overexpression of cholesterol 7α-hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology 2011, 53, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhao, M.; Zhang, H.; Zhang, X.; Zhao, J.; Xu, J.; Gao, L. Thyroid-stimulating Hormone Levels Are Inversely Associated With Serum Total Bile Acid Levels: A Cross-Sectional Study. Endocr. Pract. 2016, 22, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.L.; Hunt, H.M.; Hurxthal, L. Blood Cholesterol Values in Hyperthyroidism and Hypothyroidism—Their Significance. N. Engl. J. Med. 1930, 203, 1273–1278. [Google Scholar] [CrossRef]
- Honda, A.; Yamashita, K.; Numazawa, M.; Ikegami, T.; Doy, M.; Matsuzaki, Y.; Miyazaki, H. Highly sensitive quantification of 7α-hydroxy-4-cholesten-3-one in human serum by LC-ESI-MS/MS. J. Lipid Res. 2007, 48, 458–464. [Google Scholar] [CrossRef]
- Lin, J.Z.; Martagón, A.J.; Hsueh, W.A.; Baxter, J.D.; Gustafsson, J.; Webb, P.; Phillips, K.J. Thyroid Hormone Receptor Agonists Reduce Serum Cholesterol Independent of the LDL Receptor. Endocrinology 2012, 153, 6136–6144. [Google Scholar] [CrossRef]
- Lindemann, J.A.L.; Angajala, A.; Engler, D.A.; Webb, P.; Ayers, S.D. Thyroid hormone induction of human cholesterol 7 alpha-hydroxylase (Cyp7a1) in vitro. Mol. Cell. Endocrinol. 2014, 388, 32–40. [Google Scholar] [CrossRef]
- Berge, K.E.; Tian, H.; Graf, G.A.; Yu, L.; Grishin, N.V.; Schultz, J.; Kwiterovich, P.; Shan, B.; Barnes, R.; Hobbs, H.H. Accumulation of Dietary Cholesterol in Sitosterolemia Caused by Mutations in Adjacent ABC Transporters. Science 2000, 290, 1771–1775. [Google Scholar] [CrossRef]
- Grefhorst, A.; Verkade, H.J.; Groen, A.K. The TICE Pathway: Mechanisms and Lipid-Lowering Therapies. Methodist DeBakey Cardiovasc. J. 2019, 15, 70–76. [Google Scholar] [CrossRef]
- Gälman, C.; Bonde, Y.; Matasconi, M.; Angelin, B.; Rudling, M. Dramatically Increased Intestinal Absorption of Cholesterol Following Hypophysectomy Is Normalized by Thyroid Hormone. Gastroenterology 2008, 134, 1127–1136. [Google Scholar] [CrossRef]
- Wang, D.; Tosevska, A.; Heiß, E.H.; Ladurner, A.; Mölzer, C.; Wallner, M.; Bulmer, A.; Wagner, K.; Dirsch, V.M.; Atanasov, A.G. Bilirubin Decreases Macrophage Cholesterol Efflux and ATP-Binding Cassette Transporter A1 Protein Expression. J. Am. Heart Assoc. 2017, 6, e005520. [Google Scholar] [CrossRef]
- Hafiane, A.; Gasbarrino, K.; Daskalopoulou, S.S. The role of adiponectin in cholesterol efflux and HDL biogenesis and metabolism. Metabolism 2019, 100, 153953. [Google Scholar] [CrossRef]
- Schmitz, G.; Langmann, T. Transcriptional regulatory networks in lipid metabolism control ABCA1 expression. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2005, 1735, 1–19. [Google Scholar] [CrossRef]
- Huuskonen, J.; Vishnu, M.; Pullinger, C.R.; Fielding, P.E.; Fielding, C.J. Regulation of ATP-Binding Cassette Transporter A1 Transcription by Thyroid Hormone Receptor. Biochemistry 2004, 43, 1626–1632. [Google Scholar] [CrossRef]
- Mauerer, R.; Ebert, S.; Langmann, T. High glucose, unsaturated and saturated fatty acids differentially regulate expression of ATP-binding cassette transporters ABCA1 and ABCG1 in human macrophages. Exp. Mol. Med. 2009, 41, 126–132. [Google Scholar] [CrossRef]
- Strobl, W.; Gorder, N.L.; Lin-Lee, Y.C.; Gotto, A.M.; Patsch, W. Role of thyroid hormones in apolipoprotein A-I gene expression in rat liver. J. Clin. Investig. 1990, 85, 659–667. [Google Scholar] [CrossRef]
- Romney, J.S.; Chan, J.; Carr, F.E.; Mooradian, A.D.; Wong, N.C. Identification of the thyroid hormone-responsive messenger RNA spot 11 as apolipoprotein-A1 messenger RNA and effects of the hormone on the promoter. Mol. Endocrinol. 1992, 6, 943–950. [Google Scholar] [CrossRef]
- Vandenbrouck, Y.; Janvier, B.; Loriette, C.; Bereziat, G.; Mangeney-Andreani, M. Thyroid Hormone Modulates Apolipoprotein-AI Gene Expression at the Post-Transcriptional Level in Hep G2 Cells. JBIC J. Biol. Inorg. Chem. 1995, 231, 126–132. [Google Scholar] [CrossRef]
- Post, A.; Garcia, E.; Gruppen, E.G.; Kremer, D.; Connelly, M.A.; Bakker, S.J.L.; Dullaart, R.P.F. Higher Free Triiodothyronine Is Associated With Higher HDL Particle Concentration and Smaller HDL Particle Size. J. Clin. Endocrinol. Metab. 2022, 107, e1807–e1815. [Google Scholar] [CrossRef]
- Peppa, M.; Betsi, G.; Dimitriadis, G. Lipid Abnormalities and Cardiometabolic Risk in Patients with Overt and Subclinical Thyroid Disease. J. Lipids 2011, 2011, 1–9. [Google Scholar] [CrossRef]
- Duntas, L.H. Thyroid Disease and Lipids. Thyroid 2002, 12, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Duntas, L.H.; Mantzou, E.; Koutras, D.A. Circulating Levels of Oxidized Low-Density Lipoprotein in Overt and Mild Hypothyroidism. Thyroid 2002, 12, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Halley, P.; Kadakkuzha, B.M.; Faghihi, M.A.; Magistri, M.; Zeier, Z.; Khorkova, O.; Coito, C.; Hsiao, J.; Lawrence, M.; Wahlestedt, C. Regulation of the Apolipoprotein Gene Cluster by a Long Noncoding RNA. Cell Rep. 2014, 6, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, Y.; Jadhav, K.; Zhu, Y.; Yin, L.; Zhang, Y. Hepatic Forkhead Box Protein A3 Regulates ApoA-I (Apolipoprotein A-I) Expression, Cholesterol Efflux, and Atherogenesis. Arter. Thromb. Vasc. Biol. 2019, 39, 1574–1587. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Fernández-Hernando, C. Regulation of cholesterol homeostasis. Cell. Mol. Life Sci. 2011, 69, 915–930. [Google Scholar] [CrossRef]
- Tan, K.C.B.; Shiu, S.W.M.; Kung, A.W.C. Plasma Cholesteryl Ester Transfer Protein Activity in Hyper- and Hypothyroidism 1. J. Clin. Endocrinol. Metab. 1998, 83, 140–143. [Google Scholar] [CrossRef]
- Ito, M.; Arishima, T.; Kudo, T.; Nishihara, E.; Ohye, H.; Kubota, S.; Fukata, S.; Amino, N.; Kuma, K.; Sasaki, I.; et al. Effect of Levo-Thyroxine Replacement on Non-High-Density Lipoprotein Cholesterol in Hypothyroid Patients. J. Clin. Endocrinol. Metab. 2006, 92, 608–611. [Google Scholar] [CrossRef]
- Brenta, G.; Berg, G.; Arias, P.; Zago, V.; Schnitman, M.; Muzzio, M.L.; Sinay, I.; Schreier, L. Lipoprotein Alterations, Hepatic Lipase Activity, and Insulin Sensitivity in Subclinical Hypothyroidism: Response to L-T4 Treatment. Thyroid 2007, 17, 453–460. [Google Scholar] [CrossRef]
- Brenta, G.; Berg, G.; Miksztowicz, V.; Lopez, G.; Lucero, D.; Faingold, C.; Murakami, M.; Machima, T.; Nakajima, K.; Schreier, L. Atherogenic Lipoproteins in Subclinical Hypothyroidism and Their Relationship with Hepatic Lipase Activity: Response to Replacement Treatment with Levothyroxine. Thyroid 2016, 26, 365–372. [Google Scholar] [CrossRef]
- Johansson, L.; Rudling, M.; Scanlan, T.S.; Lundåsen, T.; Webb, P.; Baxter, J.; Angelin, B.; Parini, P. Selective thyroid receptor modulation by GC-1 reduces serum lipids and stimulates steps of reverse cholesterol transport in euthyroid mice. Proc. Natl. Acad. Sci. USA 2005, 102, 10297–10302. [Google Scholar] [CrossRef]
- Tancevski, I.; Wehinger, A.; Demetz, E.; Hoefer, J.; Eller, P.; Huber, E.; Stanzl, U.; Duwensee, K.; Auer, K.; Schgoer, W.; et al. The thyromimetic T-0681 protects from atherosclerosis. J. Lipid Res. 2009, 50, 938–944. [Google Scholar] [CrossRef]
- Wirth, E.K.; Puengel, T.; Spranger, J.; Tacke, F. Thyroid hormones as a disease modifier and therapeutic target in nonalcoholic steatohepatitis. Expert Rev. Endocrinol. Metab. 2022, 17, 425–434. [Google Scholar] [CrossRef]
- Sane, R.; Wirth, E.K.; Köhrle, J. 3,5-T2-an Endogenous Thyroid Hormone Metabolite as Promising Lead Substance in Anti-Steatotic Drug Development? Metabolites 2022, 12, 582. [Google Scholar] [CrossRef]


{kind=link}
| Publication | Species | Sex | Age | Fasting | Serum TSH | Serum T4 | Serum T3 | Liver T4 | Liver T3 | Dio1 mRNA | Dio1 Activity | Dio3 mRNA | Dio3 Activity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Galton et al., 2014, [12] | mouse | male | 10–16 weeks | 30 h | ↔ | ↓ | ↓ | N/A | N/A | N/A | N/A | N/A | N/A |
| Galton et al., 2014, [12] | mouse | male | 10–16 weeks | 36 h | N/A | N/A | N/A | ↓ | ↓ | N/A | N/A | N/A | N/A |
| Cordeiro et al., 2013, [13] | mouse | male | 3 months | 48 h | N/A | not detectable | ↓ | N/A | N/A | N/A | N/A | N/A | N/A |
| de Vries et al., 2020, [14] | mouse | male | 12 weeks | 48 h | N/A | ↓ | ↓ | ↓ | ↓ | N/A | N/A | ↑ | ↔ |
| van der Wal et al., 1998, [15] | rat | N/A | N/A | 12 & 24 h | ↔ | ↔ | ↓ | N/A | N/A | ↓ | ↔ | N/A | N/A |
| de Vries et al., 2014, [16] | rat | male | N/A | 36 h | N/A | ↓ | ↓ | ↓ | ↔ | ↔ | ↔ | ↑ | ↑ |
| de Vries et al., 2015, [17] | rat | male | 8–12 weeks | 36 h | N/A | ↓ | ↓ | ↔ | ↓ | ↓ | ↔ | ↑ | ↑ |
| Naito et al., 1981, [18] | rat | male | N/A | 48 h | ↓ | ↓ | ↓ | lower T3 generation from T4 | N/A | N/A | N/A | N/A | |
| van der Wal et al., 1998, [15] | rat | N/A | N/A | 48 h | ↔ | ↓ | ↓ | N/A | N/A | ↓ | N/A | N/A | N/A |
| Visser et al., 1996, [19] | rat | male | N/A | 3 days | ↓ | ↓ | ↓ | N/A | N/A | N/A | ↓ | N/A | N/A |
| Visser et al., 1996, [19] | rat | female | N/A | 3 days | ↔ | ↓ | ↓ | N/A | N/A | N/A | ↓ | N/A | N/A |
| Giacco et al., 2020, [20] | rat | male | 3 months | 66 h | N/A | ↓ | N/A | N/A | N/A | ↓ | N/A | N/A | N/A |
| Publication | Species | Sex | Age | Food Restriction | Duration of Restriction | Serum TSH | Serum T4 | Serum T3 | Liver T4 | Liver T3 | Dio1 mRNA | Dio1 Activity | Dio3 mRNA | Dio3 Activity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Visser et al., 1996, [19] | rat | male and female | N/A | one-third of normal food intake | 3 weeks | ↓ | ↓ | ↓ | N/A | N/A | N/A | ↓ | N/A | N/A |
| de Vries et al., 2015, [17] | rat | male | 8–12 weeks | 50% of their individual baseline 24 h intake | 21 days | N/A | ↓ | ↓ | ↓ | ↓ | ↔ | ↔ | ↑ | ↑ |
| Publication | Species | Sex | Age | Genotype | Diet Composition | Duration of Dietary Intervention | Serum TSH | Serum T4 | Serum T3 | Dio1 mRNA | Dio1 Activity | Other TH-Related Genes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gonzalez-Ramos et al., 2020, [50] | mice | male | 12 weeks | Nod1-/- | HFD (10.2% hydrogenated coconut fat and 0.75% cholesterol) | 6 weeks | N/A | ↔ | ↔ | N/A | ↓ (independent of diet) | Glut4 ↑ |
| Gonzalez-Ramos et al., 2020, [50] | mice | male | 12 weeks | WT | HFD (10.2% hydrogenated coconut fat and 0.75% cholesterol) | 6 weeks | N/A | ↔ | ↔ | N/A | ↔ | |
| Han et al., 2012, [51] | mice | female | N/A (10–13 g) | WT | HFD (15% lard, 10% yolk powder, and 79% standard laboratory powder chow; with 1200 μg/L iodine in the form of potassium iodate (KIO3) in drinking water) | 6 months | ↓ | ↑ | ↑ | N/A | ↑ | |
| Lopez et al., 2022, [52] | mice | male | 5 weeks | WT | HFD (D12492; research diets) | 4–18 weeks | N/A | ↓ (12 weeks) | ↔ | ↑ | ↑ | |
| Bruinstroop et al., 2021, [53] | mice | male | 10 weeks | WT | WE supplemented with 15% weight/volume fructose in drinking water (D12079B; Research Diets) | 8 or 16 weeks | N/A | N/A | N/A | ↑ | ↑ | |
| Lietzow et al., 2016, [54] | mice | 20 weeks | WT | 2.5 µg/g bw; HFD: 60 kJ% fat; 9% soybean oil, 90% lard, D12492, Research Diets | 4 weeks | N/A | N/A | N/A | ↑ | N/A | Cyp1a2, Cyp39a1, Cyp46a1, Cyp51, Cyp2d9, Ces1(f,g) and 2a, Sult1b1, Slc13a3, Slc39a4, Gpx6, Cyp39a1 ↑,Cyp46a1 ↓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seifert, J.; Chen, Y.; Schöning, W.; Mai, K.; Tacke, F.; Spranger, J.; Köhrle, J.; Wirth, E.K. Hepatic Energy Metabolism under the Local Control of the Thyroid Hormone System. Int. J. Mol. Sci. 2023, 24, 4861. https://doi.org/10.3390/ijms24054861
Seifert J, Chen Y, Schöning W, Mai K, Tacke F, Spranger J, Köhrle J, Wirth EK. Hepatic Energy Metabolism under the Local Control of the Thyroid Hormone System. International Journal of Molecular Sciences. 2023; 24(5):4861. https://doi.org/10.3390/ijms24054861
Chicago/Turabian StyleSeifert, Joshua, Yingfu Chen, Wenzel Schöning, Knut Mai, Frank Tacke, Joachim Spranger, Josef Köhrle, and Eva Katrin Wirth. 2023. "Hepatic Energy Metabolism under the Local Control of the Thyroid Hormone System" International Journal of Molecular Sciences 24, no. 5: 4861. https://doi.org/10.3390/ijms24054861
APA StyleSeifert, J., Chen, Y., Schöning, W., Mai, K., Tacke, F., Spranger, J., Köhrle, J., & Wirth, E. K. (2023). Hepatic Energy Metabolism under the Local Control of the Thyroid Hormone System. International Journal of Molecular Sciences, 24(5), 4861. https://doi.org/10.3390/ijms24054861