
Analysis of Dysferlin Direct Interactions with Putative Repair Proteins Links Apoptotic Signaling to Ca2+ Elevation via PDCD6 and FKBP8

Abstract
1. Introduction
2. Results
2.1. Surface Plasmon Resonance (SPR) Analysis of Dysferlin Direct Protein–Protein Interactions Implicated in Dysferlin-Mediated Membrane Repair
2.2. Dysferlin Interacts with Proteins Related to Apoptosis/Anti-Apoptosis, Linking PDCD6 and FKBP8 Pathways

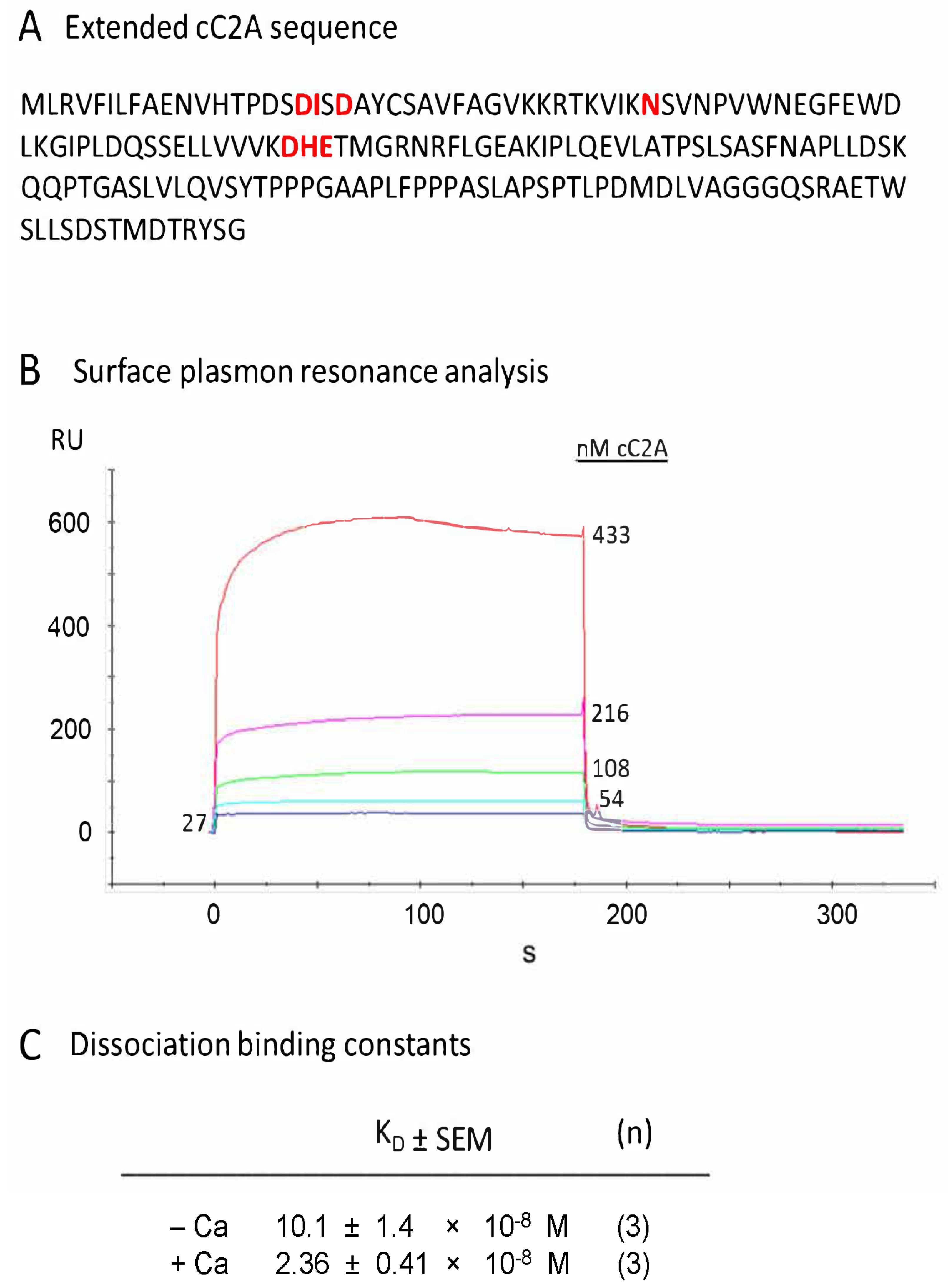
2.3. Calcium-Binding Properties of Extended cC2A Construct
2.4. Localization of Putative Dysferlin Repair-Linked Proteins in Myofibers
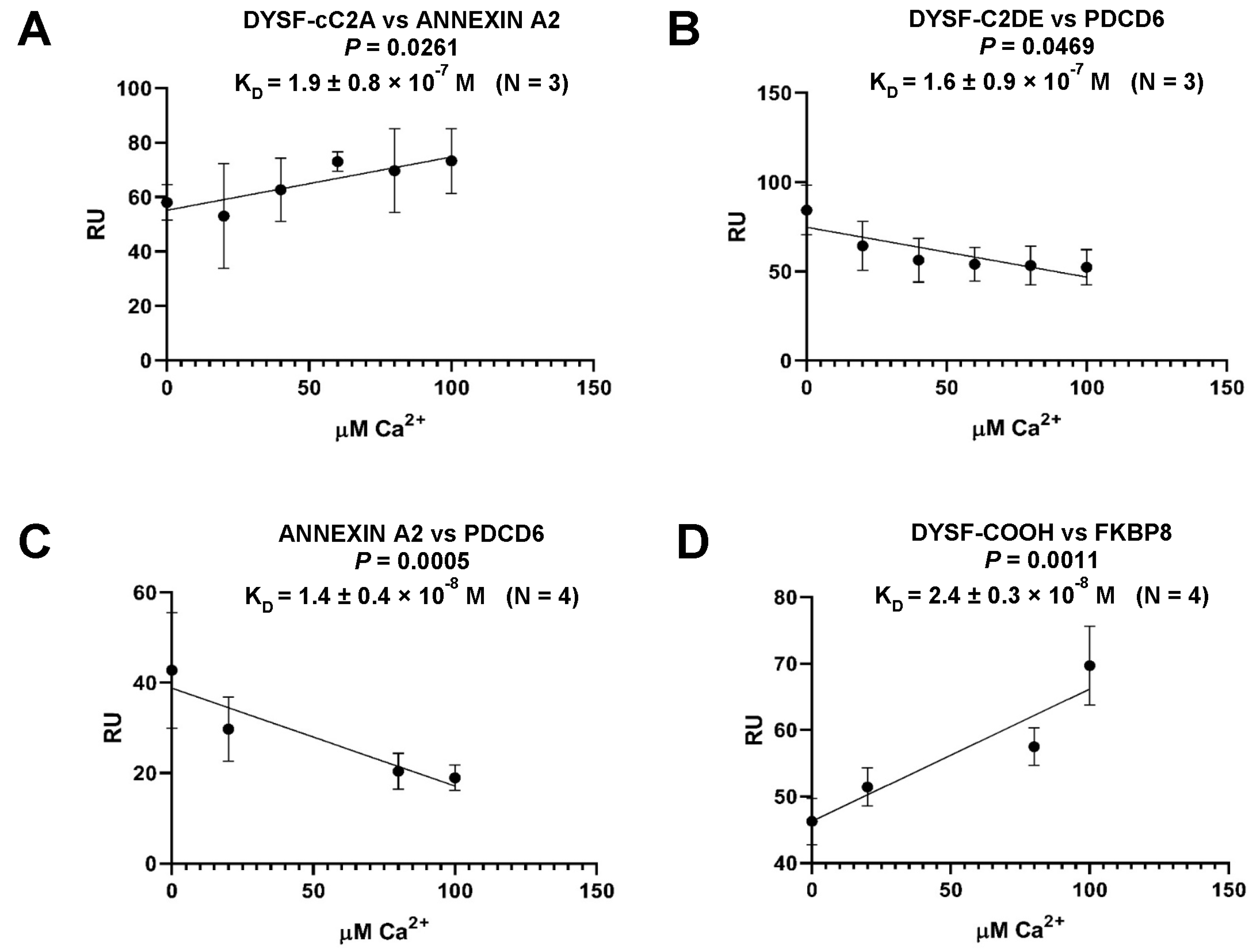
2.5. Dysferlin C2 Domain Interactions and Dependence on [Ca2+]
3. Discussion
3.1. Binding of Dysferlin C2 Domains to Repair Complex Proteins
3.2. Dysferlin-Binding Proteins Whose Ca2+-Dependent Binding Would Influence Cell Membrane Repair
3.3. Dysferlin-Interacting Proteins Related to Apoptosis/Anti-Apoptosis: PDCD6 (ALG-2), FKBP8, Annexin A2, and Links to Mitochondria
4. Materials and Methods
4.1. Synthesis of Proteins for Binding Studies
4.2. Western Blot Analysis
4.3. Surface Plasmon Resonance (SPR) Binding Analysis
4.4. Immunohistochemistry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Straub, V.; Murphy, A.; Udd, B. LGMD workshop study group. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H., Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [PubMed]
- Rescher, U.; Gerke, V. Annexins—unique membrane binding proteins with diverse functions. J. Cell Sci. 2004, 117, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, C.; Kameyama, K.; Tagawa, K.; Ogawa, M.; Suzuki, A.; Yamaji, S.; Okamoto, H.; Nishino, I.; Hayashi, Y.K. Dysferlin interacts with affixin (beta-parvin) at the sarcolemma. J. Neuropathol. Exp. Neurol. 2005, 64, 334–340. [Google Scholar] [CrossRef]
- Glover, L.; Brown, R.H., Jr. Dysferlin in membrane trafficking and patch repair. Traffic 2007, 8, 785–794. [Google Scholar] [CrossRef]
- Kobayashi, K.; Izawa, T.; Kuwamura, M.; Yamate, J. Dysferlin and animal models for dysferlinopathy. J. Toxicol. Pathol. 2012, 25, 135–147. [Google Scholar] [CrossRef]
- Cooper, S.T.; McNeil, P.L. Membrane repair: Mechanisms and pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef]
- Barthélémy, F.; Defour, A.; Lévy, N.; Krahn, M.; Bartoli, M. Muscle cells fix breaches by orchestrating a membrane repair ballet. J. Neuromuscul. Dis. 2018, 5, 21–28. [Google Scholar] [CrossRef]
- Nalefski, E.A.; Falke, J.J. The C2 domain calcium-binding motif: Structural and functional diversity. Protein Sci. 1996, 5, 2375–2390. [Google Scholar] [CrossRef]
- Ramakrishnan, N.A.; Drescher, M.J.; Morley, B.J.; Kelley, P.M.; Drescher, D.G. Calcium regulates molecular interactions of otoferlin with SNARE proteins required for hair cell exocytosis. J. Biol. Chem. 2014, 289, 8750–8766. [Google Scholar] [CrossRef]
- Krebs, J.I.; Saremaslani, P.; Caduff, R. ALG-2: A Ca2+-binding modulator protein involved in cell proliferation and in cell death. Biochim. Biophys. Acta 2002, 1600, 68–73. [Google Scholar] [CrossRef]
- Shirane, M.; Nakayama, K.I. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 2003, 5, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P. Use of surface plasmon resonance to probe the equilibrium and dynamic aspects of interactions between biological macromolecules. Ann. Rev. Biophys. Struct. 1997, 26, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Drescher, D.G.; Ramakrishnan, N.A.; Drescher, M.J. Surface plasmon resonance (SPR) analysis of binding interactions of proteins in inner-ear sensory epithelia. Methods Mol. Biol. 2009, 493, 323–343. [Google Scholar] [CrossRef] [PubMed]
- Drescher, D.G.; Dakshnamurthy, S.; Drescher, M.J.; Ramakrishnan, N.A. Surface plasmon resonance (SPR) analysis of binding interactions of inner-ear proteins. Methods Mol. Biol. 2016, 1427, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Drescher, D.G.; Drescher, M.J. Protein interaction analysis by surface plasmon resonance. In Advanced Methods in Structural Biology; Springer Nature: Berlin/Heidelberg, Germany, 2023; In press. [Google Scholar]
- Lek, A.; Evesson, F.J.; Sutton, R.B.; North, K.N.; Cooper, S.T. Ferlins: Regulators of vesicle fusion for auditory neurotransmission, receptor trafficking and membrane repair. Traffic 2012, 13, 185–194. [Google Scholar] [CrossRef]
- Azakir, B.A.; Di Fulvio, S.; Salomon, S.; Brockhoff, M.; Therrien, C.; Sinnreich, M. Modular dispensability of dysferlin C2 domains reveals rational design for mini-dysferlin molecules. J. Biol. Chem. 2012, 287, 27629–27636. [Google Scholar] [CrossRef]
- Mariano, A.; Henning, A.; Han, R. Dysferlin-deficient muscular dystrophy and innate immune activation. FEBS J. 2013, 280, 4165–4176. [Google Scholar] [CrossRef]
- Kerr, J.P.; Ward, C.W.; Bloch, R.J. Dysferlin at transverse tubules regulates Ca2+ homeostasis in skeletal muscle. Front. Physiol. 2014, 5, 89. [Google Scholar] [CrossRef]
- Fuson, K.; Rice, A.; Mahling, R.; Snow, A.; Nayak, K.; Shanbhogue, P.; Meyer, A.G.; Redpath, G.M.I.; Hinderliter, A.; Cooper, S.T.; et al. Alternate splicing of dysferlin C2A confers Ca2⁺-dependent and Ca2⁺-independent binding for membrane repair. Structure 2014, 22, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Harsini, F.M.; Bui, A.A.; Rice, A.M.; Chebrolu, S.; Fuson, K.L.; Turtoi, A.; Bradberry, M.; Chapman, E.R.; Sutton, R.B. Structural basis for the distinct membrane binding activity of the homologous C2A domains of myoferlin and dysferlin. J. Mol. Biol. 2019, 431, 2112–2126. [Google Scholar] [CrossRef] [PubMed]
- Heidrych, P. Novel Interaction Partners for Otoferlin, a Functional Member of Acoustic Transmission in the Inner Ear. Ph.D. Thesis, University of Tubingen, Tubingen, Germany, 2008. [Google Scholar]
- Satoh, H.; Nakano, Y.; Shibata, H.; Maki, M. The penta-EF-hand domain of ALG-2 interacts with amino-terminal domains of annexin VII and annexin XI in a Ca2+-dependent manner. Biochim. Biophys. Acta 2002, 1600, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.B.; Little, A.J.; Williams, R.B.; Milner, J.R. Interpretation of serum calcium in patients with abnormal serum proteins. Brit. Med. J. 1973, 4, 643–646. [Google Scholar] [CrossRef]
- Roche, J.A.; Ru, L.W.; O’Neill, A.M.; Resneck, W.G.; Lovering, R.M.; Bloch, R.J. Unmasking potential intracellular roles for dysferlin through improved immunolabeling methods. J. Histochem. Cytochem. 2011, 59, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; White, K.; Turnbull, D.M. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: A quantitative three-dimensional electron microscopy study. J. App. Physiol. 2013, 114, 161–171. [Google Scholar] [CrossRef]
- Labay, V.; Weichert, R.M.; Makishima, T.; Griffith, A.J. Topology of transmembrane channel-like gene 1 protein. Biochemistry 2010, 49, 8592–8598. [Google Scholar] [CrossRef]
- Selvakumar, D.; Drescher, M.J.; Deckard, N.; Ramakrishnan, N.A.; Drescher, D.G. Dopamine D1A directly interacts with otoferlin synaptic pathway proteins: Phosphorylation underlies a molecular switch from NSF to AP2mu1 interactions. Biochem. J. 2017, 474, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Kourie, J.I.; Wood, H.B. Biophysical and molecular properties of annexin-formed channels. Prog. Biophys. Mol. Biol. 2000, 73, 91–134. [Google Scholar] [CrossRef]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef]
- Stewart, S.E.; Ashkenazi, A.; Williamson, A.; Rubinsztein, D.C.; Moreau, K. Transbilayer phospholipid movement facilitates the translocation of annexin across membranes. J. Cell Sci. 2018, 131, jcs217034. [Google Scholar] [CrossRef] [PubMed]
- Bizzarro, V.; Belvedere, R.; Dal Piaz, F.; Parente, L.; Petrel, A. Annexin A1 induces skeletal muscle cell migration acting through formyl peptide receptors. PLoS ONE 2012, 7, e48246. [Google Scholar] [CrossRef] [PubMed]
- Croissant, C.; Carmeille, R.; Brévart, C.; Bouter, A. Annexins and membrane repair dysfunctions in muscular dystrophies. Int. J. Mol. Sci. 2021, 22, 5276. [Google Scholar] [CrossRef]
- McNeil, A.K.; Rescher, U.; Gerke, V.; McNeil, P.L. Requirement for annexin A1 in plasma membrane repair. J. Biol. Chem. 2006, 281, 35202–35207. [Google Scholar] [CrossRef]
- Potez, S.; Luginbühl, M.; Monastyrskaya, K.; Hostettler, A.; Draeger, A.; Babiychuk, E.B. Tailored protection against plasmalemmal injury by annexins with different Ca2+ sensitivities. J. Biol. Chem. 2011, 286, 17982–17991. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Atanassoff, A.P.; Monastyrskaya, K.; Brandenberger, C.; Studer, D.; Allemann, C.; Draeger, A. The targeting of plasmalemmal ceramide to mitochondria during apoptosis. PLoS ONE 2011, 6, e23706. [Google Scholar] [CrossRef]
- Kalinec, F.; Webster, P.; Maricle, A.; Guerrero, D.; Chakravarti, D.N.; Chakravarti, B.; Gellibolian, R.; Kalinec, G. Glucocorticoid-stimulated, transcription-independent release of annexin A1 by cochlear Hensen cells. Br. J. Pharmacol. 2009, 158, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; de Morrée, A.; van Remoortere, A.; Bushby, K.; Frants, R.R.; den Dunnen, J.T.; van der Maarel, S.M. Calpain 3 is a modulator of the dysferlin protein complex in skeletal muscle. Hum. Mol. Genet. 2008, 17, 1855–1866. [Google Scholar] [CrossRef]
- Ahn, M.K.; Lee, K.J.; Cai, C.; Huang, M.; Cho, C.-H.; Ma, J.; Lee, E.H. Mitsugumin 53 regulates extracellular Ca2+ entry and intracellular Ca2+ release via Orai1 and RyR1 in skeletal muscle. Sci. Rep. 2016, 6, 36909. [Google Scholar] [CrossRef]
- Hernández-Deviez, D.J.; Howes, M.T.; Laval, S.H.; Bushby, K.; Hancock, J.F.; Parton, R.G. Caveolin regulates endocytosis of the muscle repair protein, dysferlin. J. Biol. Chem. 2008, 283, 6476–6488. [Google Scholar] [CrossRef]
- Sumitani, S.; Ramlal, T.; Liu, Z.; Klip, A. Expression of syntaxin 4 in rat skeletal muscle and rat skeletal muscle cells in culture. Biochem. Biophys. Res. Comm. 1995, 213, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, N.A.; Drescher, M.J.; Drescher, D.G. Direct interaction of otoferlin with syntaxin 1A, SNAP-25, and the L-type voltage-gated calcium channel CaV1.3. J. Biol. Chem. 2009, 284, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Evesson, F.J.; Peat, R.A.; Lek, A.; Brilot, F.; Lo, H.P.; Dale, R.C.; Parton, R.G.; North, K.N.; Cooper, S.T. Reduced plasma membrane expression of dysferlin mutants is attributed to accelerated endocytosis via a syntaxin-4-associated pathway. J. Biol. Chem. 2010, 285, 28529–28539. [Google Scholar] [CrossRef] [PubMed]
- Codding, S.J.; Marty, N.; Abdullah, N.; Johnson, C.P. Dysferlin binds SNARES (soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptors) and stimulates membrane fusion in a calcium-sensitive manner. J. Biol. Chem. 2016, 291, 14575–14584. [Google Scholar] [CrossRef]
- Huang, Y.; Laval, S.H.; van Remoortere, A.; Baudier, J.; Benaud, C.; Anderson, L.V.; Straub, V.; Deelder, A.; Frants, R.R.; den Dunnen, J.T.; et al. AHNAK, a novel component of the dysferlin protein complex, redistributes to the cytoplasm with dysferlin during skeletal muscle regeneration. FASEB J. 2007, 21, 732–742. [Google Scholar] [CrossRef]
- Han, W.-Q.; Xia, M.; Xu, M.; Boini, K.M.; Ritter, J.K.; Li, N.-J.; Li, P.-L. Lysosome fusion to the cell membrane is mediated by the dysferlin C2A domain in coronary arterial endothelial cells. J. Cell Sci. 2012, 125, 1225–1234. [Google Scholar] [CrossRef]
- Maki, M.; Suzuki, S.; Shibata, H. Structure and function of ALG-2, a penta-EF-hand calcium-dependent adaptor protein. Sci. China 2011, 54, 770–779. [Google Scholar] [CrossRef]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca2+-triggeredESCRT assembly and regulation of cell membrane repair. Nat. Commun. 2014, 5, 5646. [Google Scholar] [CrossRef]
- Maestre-Martínez, M.; Haupt, K.; Edlich, F.; Neumann, P.; Parthier, C.; Stubbs, M.T.; Fischer, G.; Lücke, C. A charge-sensitive loop in the FKBP38 catalytic domain modulates Bcl-2 binding. J. Mol. Recognit. 2011, 24, 23–34. [Google Scholar] [CrossRef]
- Haupt, K.; Jahreis, G.; Linnert, M.; Maestre-Martínez, M.; Malesevic, M.; Pechstein, A.; Edlich, F.; Lücke, C. The FKBP38 catalytic domain binds to Bcl-2 via a charge-sensitive loop. J. Biol. Chem. 2012, 287, 19665–19673. [Google Scholar] [CrossRef]
- Jayaraman, T.; Brillantes, A.M.; Timerman, A.P.; Fleischer, S.; Erdjument-Bromage, H.; Tempst, P.; Marks, A.R. FK506 binding protein associated with the calcium release channel (ryanodine receptor). J. Biol. Chem. 1992, 267, 9474–9477. [Google Scholar] [CrossRef]
- Vervliet, T.; Parys, J.B.; Bultynck, G. Bcl-2 and FKBP12 bind to IP3 and ryanodine receptors at overlapping sites: The complexity of protein–protein interactions for channel regulation. Biochem. Soc. Trans. 2015, 43, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Saita, S.; Shirane, M.; Nakayama, N.I. Selective escape of proteins from the mitochondria during mitophagy. Nat. Commun. 2012, 4, 1410. [Google Scholar] [CrossRef] [PubMed]
- Therrien, C.; Di Fulvio, S.; Pickles, S.; Sinnreich, M. Characterization of lipid binding specificities of dysferlin C2 domains reveals novel interactions with phosphoinositides. Biochemistry 2009, 48, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Golbek, T.W.; Otto, S.C.; Roeters, S.J.; Weidner, T.; Johnson, C.P.; Baio, J.E. Direct evidence that mutations within dysferlin’s C2A domain inhibit lipid clustering. J. Phys. Chem. B 2021, 125, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 mediates dysferlin accumulation and muscle cell membrane repair. Cells 2020, 9, 1919. [Google Scholar] [CrossRef]
- Báez-Matus, X.; Figueroa-Cares, C.; Gónzalez-Jamett, A.M.; Almarza-Salazar, H.; Arriagada, C.; Maldifassi, M.C.; Guerra, M.J.; Mouly, V.; Bigot, A.; Caviedes, P.; et al. Defects in G-actin incorporation into filaments in myoblasts derived from dysferlinopathy patients are restored by dysferlin C2 domains. Int. J. Mol. Sci. 2020, 21, 37. [Google Scholar] [CrossRef]
- Zorov, B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef]
- Pathak, T.; Trebak, M. Mitochondrial Ca2+ signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Raavicharla, S.; Shah, S.; Cox, D.; Jaiswal, J.K. Mitochondrial fragmentation enables localized signaling required for cell repair. J. Cell Biol. 2020, 219, e201909154. [Google Scholar] [CrossRef]
- Yoo, S.-M.; Yamashita, S.; Kim, H.; Na, D.; Lee, H.; Kim, S.J.; Cho, D.-H.; Kanki, T.; Jung, Y.-K. FKBP8 LIRL-dependent mitochondrial fragmentation facilitates mitophagy under stress conditions. FASEB J. 2020, 34, 2944–2957. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.-T.; Xie, Z.-S.; Kuang, Y.-J.; Liu, S.-Y.; Zeng, C.; Li, P.; Liu, E.-H. Discovery of a potent FKBP38 agonist that ameliorates HFD-induced hyperlipidemia via mTOR/P70S6K/SREBPs pathway. Acta Pharm. Sin. B 2021, 11, 3542–3552. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, L.; Crabb, J.W.; Cox, J.; Durussel, I.; Walker, T.M.; van Ginkel, P.R.; Bhattacharya, S.; Dellaria, J.M.; Palczewski, K.; Polans, A.S. Ca2+ binding to EF hands 1 and 3 is essential for the interaction of apoptosis-linked gene-2 with Alix/AIP1 in ocular melanoma. Biochemistry 2004, 43, 11175–11186. [Google Scholar] [CrossRef]
- Sønder, S.L.; Boye, T.L.; Tölle, R.; Dengjel, J.; Maeda, K.; Jäättelä, M.; Simonsen, A.C.; Jaiswal, J.K.; Nylandsted, J. Annexin A7 is required for ESCRT III-mediated plasma membrane repair. Sci. Rep. 2019, 9, 6726. [Google Scholar] [CrossRef]
- Selvakumar, D.; Drescher, M.J.; Drescher, D.G. Cyclic nucleotide-gated channel α-3 (CNGA3) interacts with stereocilia tip-link cadherin 23 + exon 68 or alternatively with myosin VIIa, two proteins required for hair cell mechanotransduction. J. Biol. Chem. 2013, 288, 7215–7229. [Google Scholar] [CrossRef]
- Johnson, M. Detergents: Triton X-100, Tween-20, and more. Mater. Methods 2013, 3, 163–169. [Google Scholar] [CrossRef]
- Selvakumar, D.; Drescher, M.J.; Dowdall, J.R.; Khan, K.M.; Hatfield, J.S.; Ramakrishnan, N.A.; Drescher, D.G. CNGA3 is expressed in inner ear hair cells and binds to an intracellular carboxy terminus domain of EMILIN1. Biochem. J. 2012, 443, 463–476. [Google Scholar] [CrossRef]
- Drescher, M.J.; Cho, W.J.; Folbe, A.; Kewson, D.T.; Abu-Hamdan, M.; Oh, C.K.; Ramakrishnan, N.A.; Hatfield, J.S.; Khan, K.M.; Anne, S.; et al. An adenylyl cyclase signaling pathway predicts direct dopaminergic input to vestibular hair cells. Neuroscience 2010, 171, 1054–1074. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Partners | KD ± SEM | (n) | Ca2+ Dependence |
|---|---|---|---|
| Dysf C2A—Annexin A1 | 2.5 ± 0.8 × 10−8 | (3) | +++ |
| Dysf C2F—Annexin A1 | 3.3 ± 0.3 × 10−7 | (3) | + |
| Dysf C2A—Calpain-3 | 1.2 ± 0.4 × 10−6 | (3) | +++ |
| Dysf C2F—Calpain-3 | 2.2 ± 0.6 × 10−9 | (3) | ++ |
| Dysf C2A—Mitsugumin-53 | 4.7 ± 0.7 × 10−7 | (3) | +++ |
| Dysf C2F—Mitsugumin-53 | nd | 0 | |
| Dysf C2A—Affixin | 4.1 ± 1.1 × 10−5 | (5) | ++ |
| Dysf C2F—Affixin | 4.0 ± 1.0 × 10−7 | (4) | +++ |
| Dysf C2A—Caveolin-3 | 3.3 ± 0.6 × 10−8 | (3) | + |
| Dysf C2F—Caveolin-3 | 1.1 ± 0.5 × 10−7 | (3) | − |
| Dysf C2A—Syntaxin-4 | 4.4 ± 1.3 × 10−6 | (3) | + |
| Dysf C2F—Syntaxin-4 | 1.3 ± 0.4 × 10−7 | (3) | − |
| Dysf C2A—AHNAK1 | 3.8 ± 1.6 × 10−7 | (3) | + |
| Dysf C2F—AHNAK1 | nd | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drescher, D.G.; Drescher, M.J.; Selvakumar, D.; Annam, N.P. Analysis of Dysferlin Direct Interactions with Putative Repair Proteins Links Apoptotic Signaling to Ca2+ Elevation via PDCD6 and FKBP8. Int. J. Mol. Sci. 2023, 24, 4707. https://doi.org/10.3390/ijms24054707
Drescher DG, Drescher MJ, Selvakumar D, Annam NP. Analysis of Dysferlin Direct Interactions with Putative Repair Proteins Links Apoptotic Signaling to Ca2+ Elevation via PDCD6 and FKBP8. International Journal of Molecular Sciences. 2023; 24(5):4707. https://doi.org/10.3390/ijms24054707
Chicago/Turabian StyleDrescher, Dennis G., Marian J. Drescher, Dakshnamurthy Selvakumar, and Neeraja P. Annam. 2023. "Analysis of Dysferlin Direct Interactions with Putative Repair Proteins Links Apoptotic Signaling to Ca2+ Elevation via PDCD6 and FKBP8" International Journal of Molecular Sciences 24, no. 5: 4707. https://doi.org/10.3390/ijms24054707
APA StyleDrescher, D. G., Drescher, M. J., Selvakumar, D., & Annam, N. P. (2023). Analysis of Dysferlin Direct Interactions with Putative Repair Proteins Links Apoptotic Signaling to Ca2+ Elevation via PDCD6 and FKBP8. International Journal of Molecular Sciences, 24(5), 4707. https://doi.org/10.3390/ijms24054707