
Metadherin Regulates Inflammatory Breast Cancer Invasion and Metastasis

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
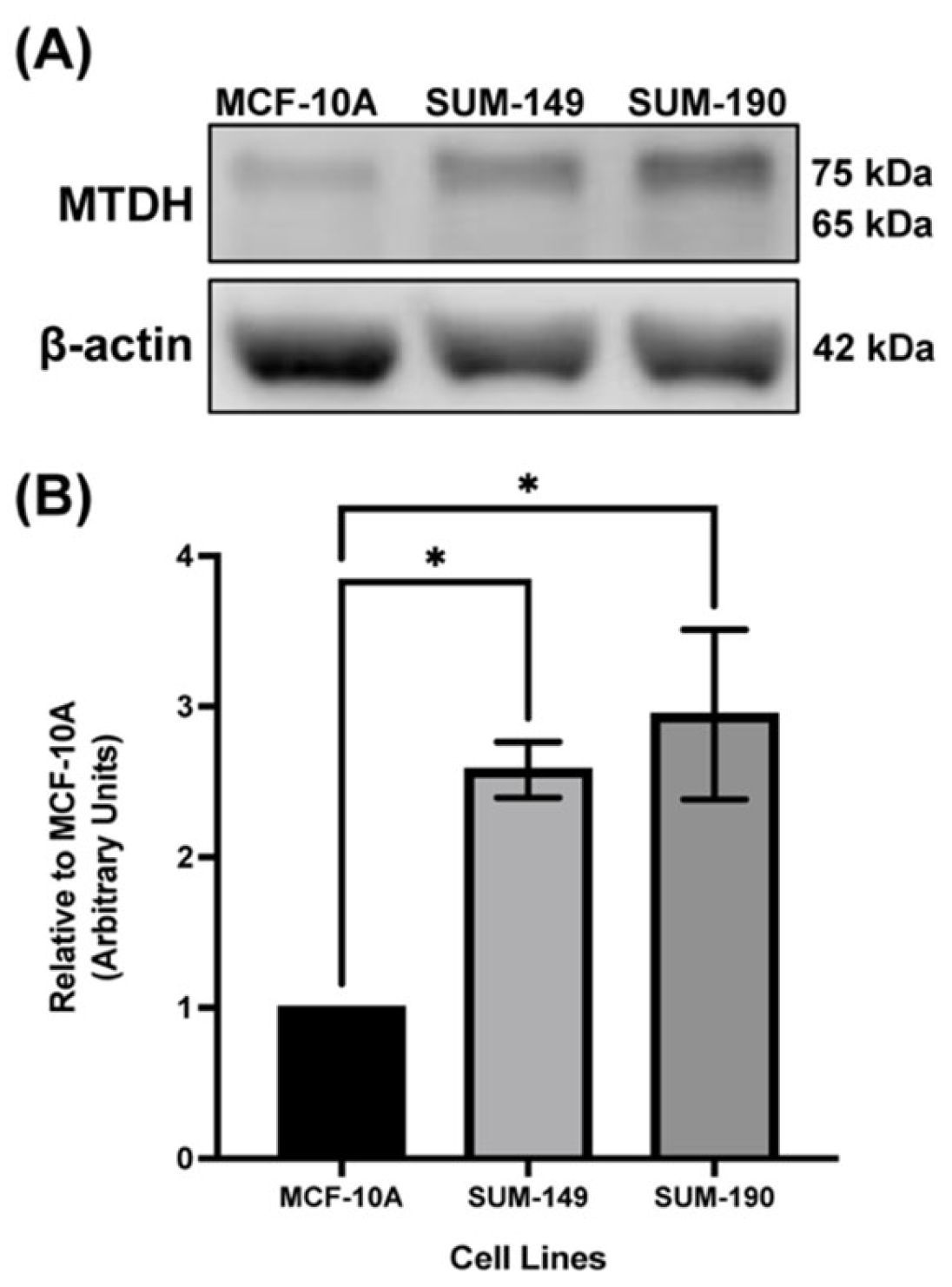
2.1. MTDH Is Overexpressed in IBC Cell Line Models Compared to the Non-Cancerous Mammary Epithelial Cell Line, MCF-10A
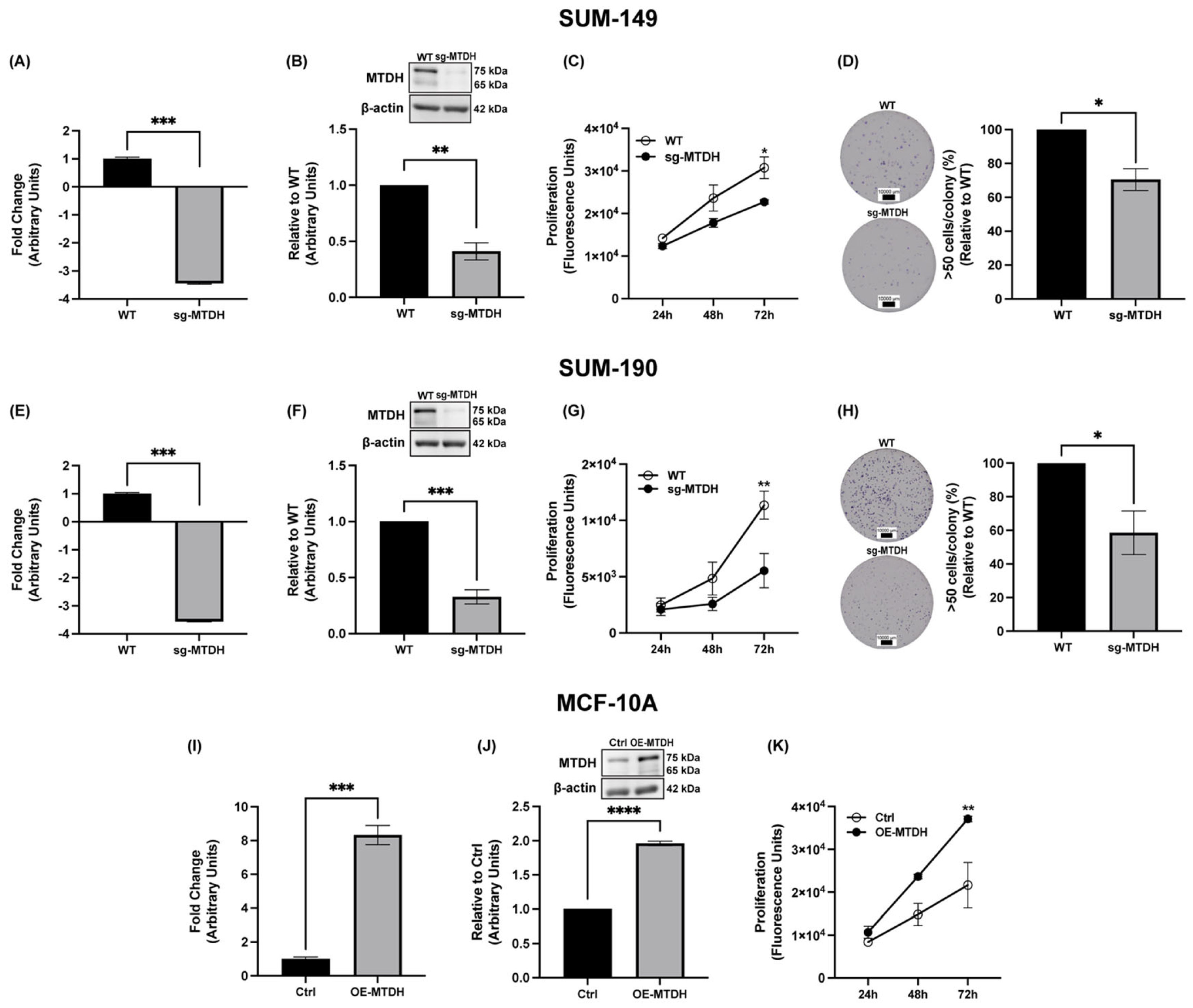
2.2. MTDH Knockout Results in Reduced IBC Cell Proliferation and Colony Formation
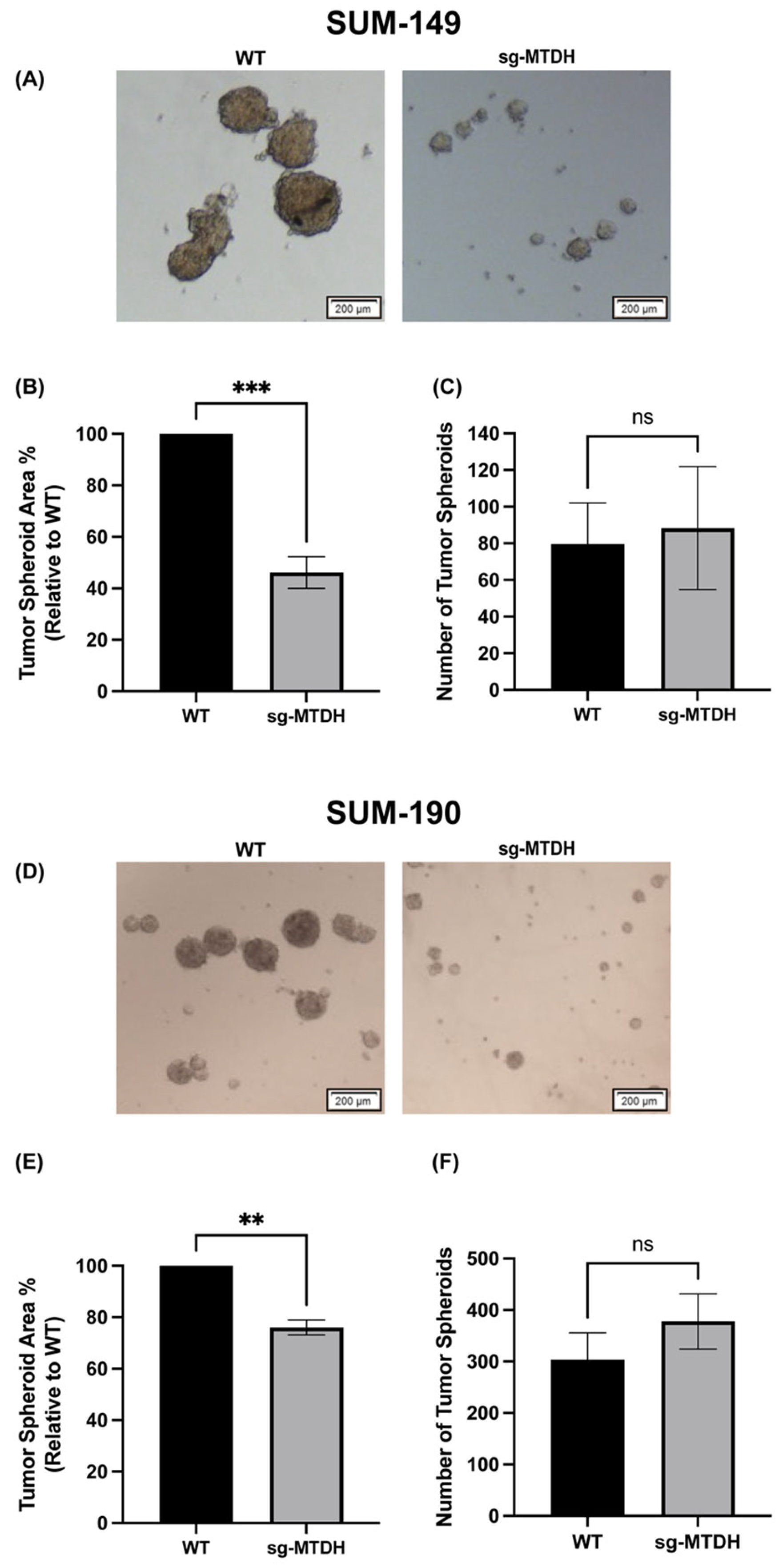
2.3. MTDH Knockout Reduces the Size of IBC-Derived Tumor Spheroids
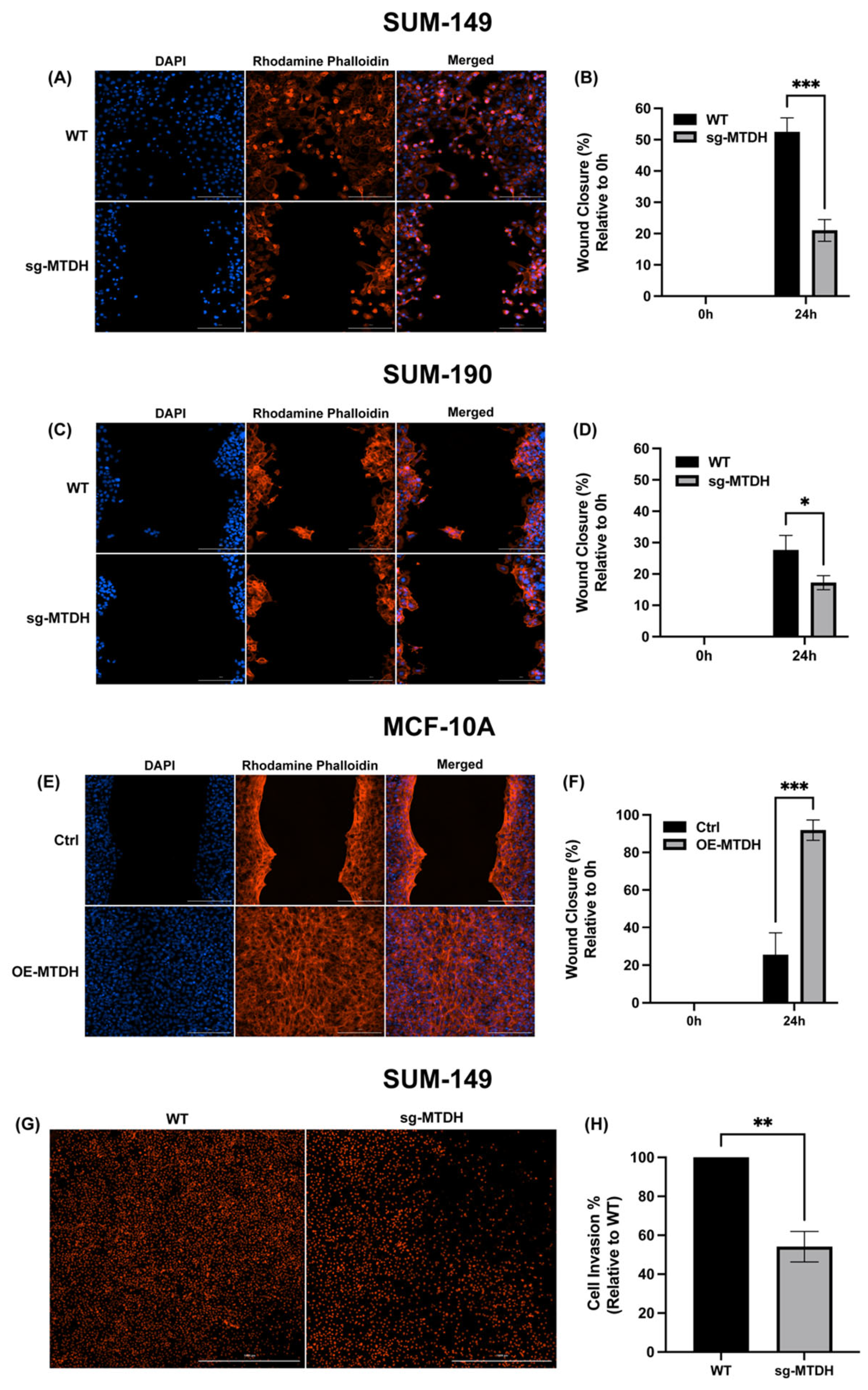
2.4. MTDH Modulates the Migration and Invasive Capacity of IBC Cells
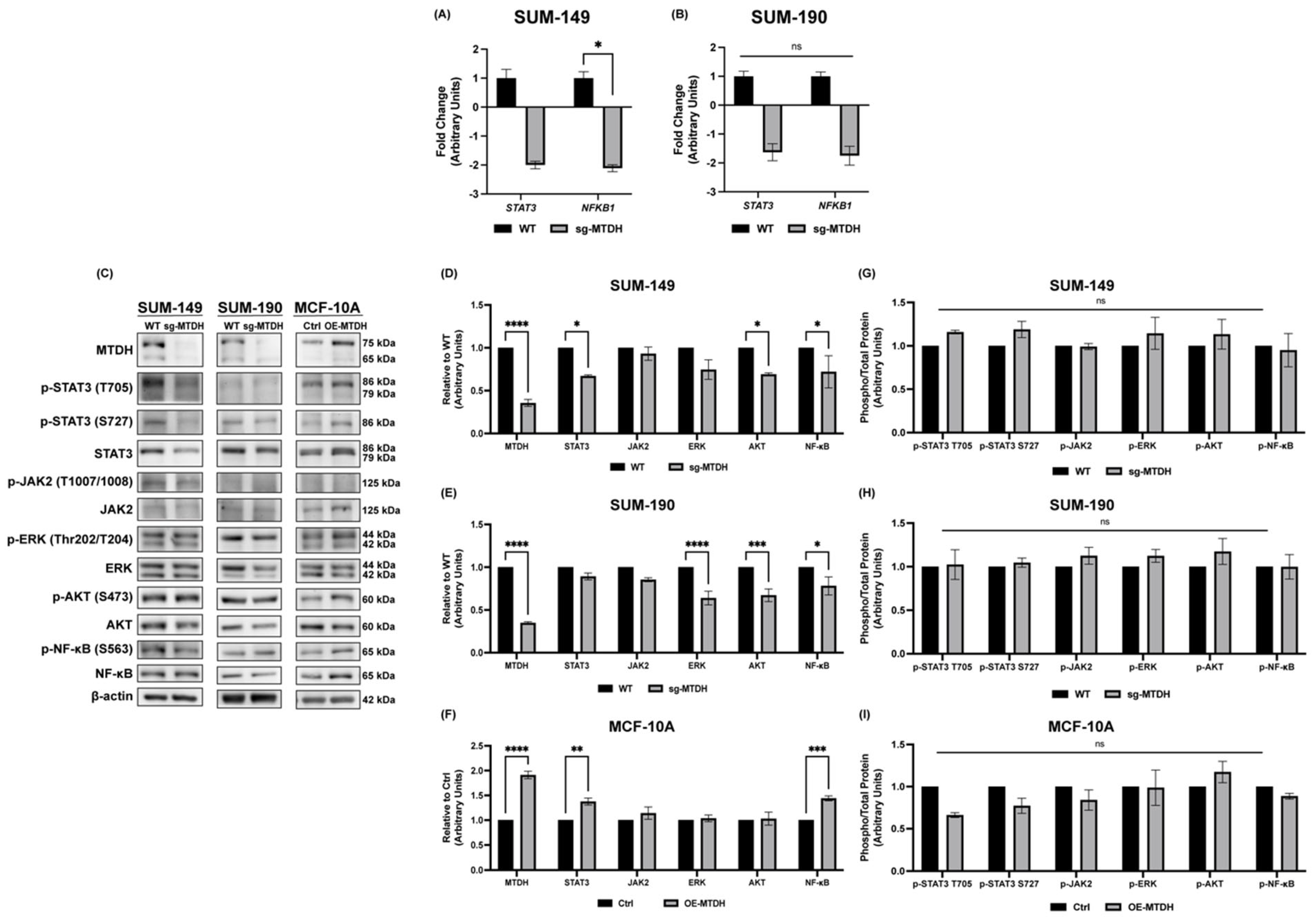
2.5. Silencing of MTDH Modulates the Expression of STAT3, NF-κB, and AKT Expression
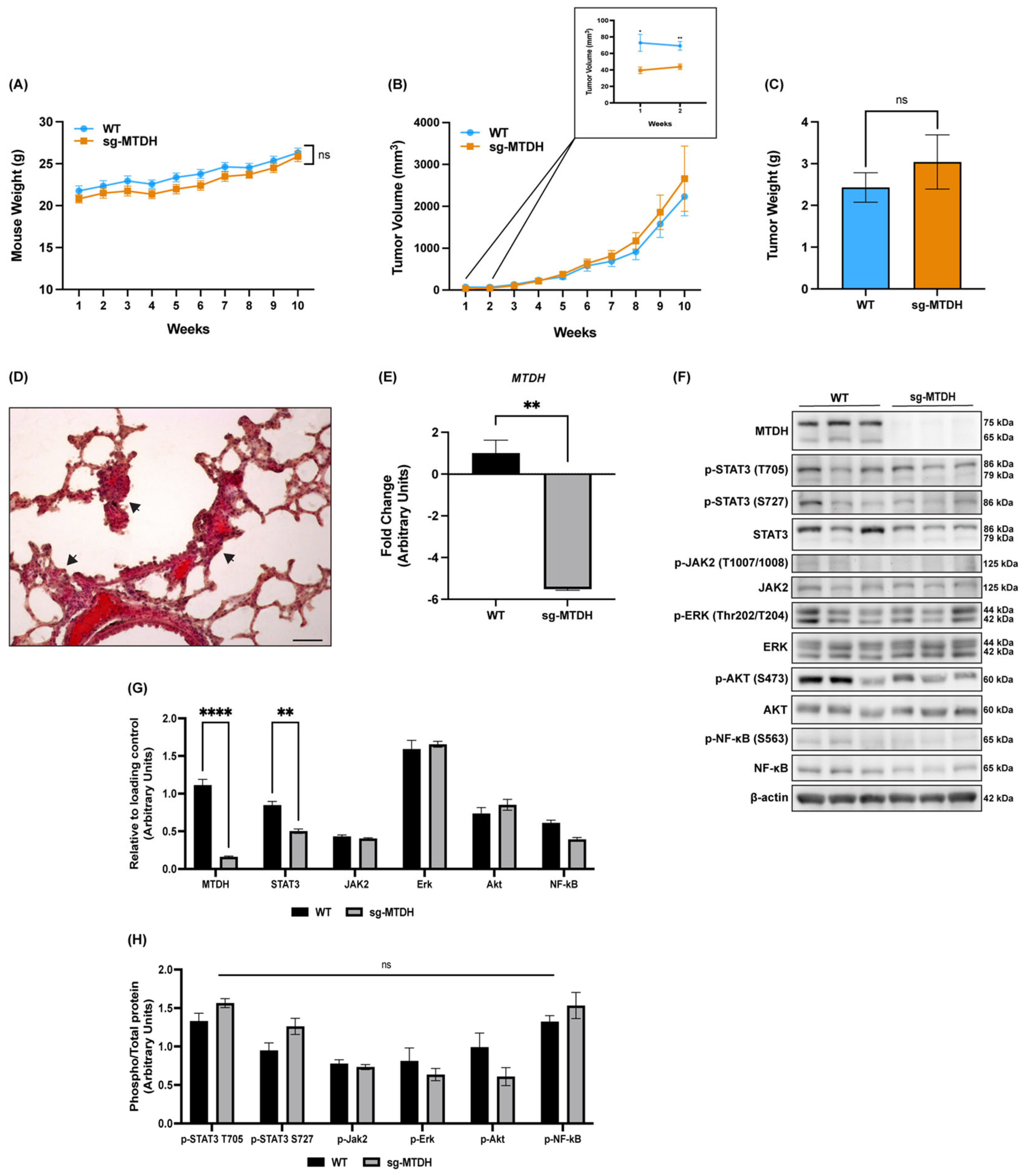
2.6. MTDH Editing in IBC Xenograft Models Delays Tumor Development and Decreases STAT3 Expression and Metastasis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Plasmids, Lentiviral Particle Generation, and Mammalian Cells Transduction
4.3. RNA Isolation and Quantitative RT-qPCR Assays
4.4. Immunoblotting for Cells and Tumors
4.5. Proliferation Assay
4.6. Colony Formation Assay
4.7. Wound Healing Assay
4.8. Invasion Assay
4.9. IBC-Dderived Tumor Spheroids Assay
4.10. In Vivo Study
4.11. Lung Sectioning and Hematoxylin & Eosin (H & E) Staining
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robertson, F.M.; Bondy, M.; Yang, W.; Yamauchi, H.; Wiggins, S.; Kamrudin, S.; Krishnamurthy, S.; Le-Petross, H.; Bidaut, L.; Player, A.N.; et al. Inflammatory breast cancer: The disease, the biology, the treatment. CA Cancer J. Clin. 2010, 60, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Levine, P.H.; Veneroso, C. The epidemiology of inflammatory breast cancer. In Seminars in Oncology; WB Saunders: Philadelphia, PA, USA, 2008; Volume 35, pp. 11–16. [Google Scholar]
- Manai, M.; Finetti, P.; Mejri, N.; Athimni, S.; Birnbaum, D.; Bertucci, F.; Rahal, K.; Gamoudi, A.; Chaffanet, M.; Manai, M.; et al. Inflammatory breast cancer in 210 patients: A retrospective study on epidemiological, anatomo-clinical features and therapeutic results. Mol. Clin. Oncol. 2018, 10, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Quirindongo-Rivera, C.; Rullán-Varela, V.; Underill, Z.; Rivera, M.; Ortiz-Ortiz, K.J.; Martínez-Montemayor, M.M. Characterization of Inflammatory Breast Cancer in Hispanic Women from Puerto Rico. J. Cancer 2022, 13, 3495–3502. [Google Scholar] [CrossRef] [PubMed]
- Faldoni, F.L.C.; Rainho, C.A.; Rogatto, S.R. Epigenetics in Inflammatory Breast Cancer: Biological Features and Therapeutic Perspectives. Cells 2020, 9, 1164. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S.; Lei, X.; Dent, R.; Gupta, S.; Sirohi, B.; Cortes, J.; Cristofanilli, M.; Buchholz, T.; Gonzalez-Angulo, A.M. Survival of women with inflammatory breast cancer: A large population-based study. Ann. Oncol. 2014, 25, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Arroyo, I.J.; Feliz-Mosquea, Y.R.; Perez-Laspiur, J.; Arju, R.; Giashuddin, S.; Maldonado-Martinez, G.; Cubano, L.A.; Schneider, R.J.; Martinez-Montemayor, M.M. The proteome signature of the inflammatory breast cancer plasma membrane identifies novel molecular markers of disease. Am. J. Cancer Res. 2016, 6, 1720–1740. [Google Scholar] [PubMed]
- Dhiman, G.; Srivastava, N.; Goyal, M.; Rakha, E.; Lothion-Roy, J.; Mongan, N.P.; Miftakhova, R.R.; Khaiboullina, S.F.; Rizvanov, A.A.; Baranwal, M. Metadherin: A Therapeutic Target in Multiple Cancers. Front. Oncol. 2019, 9, 349. [Google Scholar] [CrossRef]
- Yoo, B.K.; Emdad, L.; Lee, S.G.; Su, Z.Z.; Santhekadur, P.; Chen, D.; Gredler, R.; Fisher, P.B.; Sarkar, D. Astrocyte elevated gene-1 (AEG-1): A multifunctional regulator of normal and abnormal physiology. Pharmacol. Ther. 2011, 130, 1–8. [Google Scholar] [CrossRef]
- Lee, S.G.; Kang, D.C.; DeSalle, R.; Sarkar, D.; Fisher, P.B. AEG-1/MTDH/LYRIC, the beginning: Initial cloning, structure, expression profile, and regulation of expression. Adv. Cancer Res. 2013, 120, 1–38. [Google Scholar]
- Sutherland, H.G.; Lam, Y.W.; Briers, S.; Lamond, A.I.; Bickmore, W.A. 3D3/lyric: A novel transmembrane protein of the endoplasmic reticulum and nuclear envelope, which is also present in the nucleolus. Exp. Cell Res. 2004, 294, 94–105. [Google Scholar] [CrossRef]
- Britt, D.E.; Yang, D.F.; Yang, D.Q.; Flanagan, D.; Callanan, H.; Lim, Y.P.; Lin, S.H.; Hixson, D.C. Identification of a novel protein, LYRIC, localized to tight junctions of polarized epithelial cells. Exp. Cell Res. 2004, 300, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.C.; Su, Z.Z.; Sarkar, D.; Emdad, L.; Volsky, D.J.; Fisher, P.B. Cloning and characterization of HIV-1-inducible astrocyte elevated gene-1, AEG-1. Gene 2005, 353, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Sarkar, D. The Scope of Astrocyte Elevated Gene-1/Metadherin (AEG-1/MTDH) in Cancer Clinicopathology: A Review. Genes 2021, 12, 308. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Ruoslahti, E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell 2004, 5, 365–374. [Google Scholar] [CrossRef]
- Su, Z.Z.; Kang, D.C.; Chen, Y.; Pekarskaya, O.; Chao, W.; Volsky, D.J.; Fisher, P.B. Identification and cloning of human astrocyte genes displaying elevated expression after infection with HIV-1 or exposure to HIV-1 envelope glycoprotein by rapid subtraction hybridization, RaSH. Oncogene 2002, 21, 3592–3602. [Google Scholar] [CrossRef]
- Emdad, L.; Sarkar, D.; Su, Z.Z.; Randolph, A.; Boukerche, H.; Valerie, K.; Fisher, P.B. Activation of the nuclear factor kappaB pathway by astrocyte elevated gene-1: Implications for tumor progression and metastasis. Cancer Res. 2006, 66, 1509–1516. [Google Scholar] [CrossRef]
- Lee, S.G.; Su, Z.Z.; Emdad, L.; Sarkar, D.; Franke, T.F.; Fisher, P.B. Astrocyte elevated gene-1 activates cell survival pathways through PI3K-Akt signaling. Oncogene 2008, 27, 1114. [Google Scholar] [CrossRef]
- Yoo, B.K.; Emdad, L.; Su, Z.Z.; Villanueva, A.; Chiang, D.Y.; Mukhopadhyay, N.D.; Mills, A.S.; Waxman, S.; Fisher, R.A.; Llovet, J.M.; et al. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J. Clin. Investig. 2009, 119, 465–477. [Google Scholar] [CrossRef]
- Lee, S.-G.; Su, Z.-Z.; Emdad, L.; Sarkar, D.; Fisher, P.B. Astrocyte elevated gene-1 (AEG-1) is a target gene of oncogenic Ha-ras requiring phosphatidylinositol 3-kinase and c-Myc. Proc. Natl. Acad. Sci. USA 2006, 103, 17390–17395. [Google Scholar] [CrossRef]
- Emdad, L.; Das, S.K.; Dasgupta, S.; Hu, B.; Sarkar, D.; Fisher, P.B. AEG-1/MTDH/LYRIC: Signaling pathways, downstream genes, interacting proteins, and regulation of tumor angiogenesis. Adv. Cancer Res. 2013, 120, 75–111. [Google Scholar]
- Kong, X.; Moran, M.S.; Zhao, Y.; Yang, Q. Inhibition of metadherin sensitizes breast cancer cells to AZD6244. Cancer Biol. Ther. 2012, 13, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Li, Z.; Chen, W.; Ma, C.; Zhan, F.; Wu, W.; Peng, Y. AEG-1 participates in TGF-beta1-induced EMT through p38 MAPK activation. Cell Biol. Int. 2013, 37, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Solomon, J.; Samson, A.O.; Gil-Henn, H. Invasion and Metastasis as a Central Hallmark of Breast Cancer. J. Clin. Med. 2021, 10, 3498. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Valero, V.; Buzdar, A.U.; Kau, S.-W.; Broglio, K.R.; Gonzalez-Angulo, A.M.; Sneige, N.; Islam, R.; Ueno, N.T.; Buchholz, T.A.; et al. Inflammatory breast cancer (IBC) and patterns of recurrence: Understainding the biology of a unique disease. Cancer 2007, 110, 1436–1444. [Google Scholar] [CrossRef]
- Walshe, J.M.; Swain, S.M. Clinical aspects of inflammatory breast cancer. Breast Dis. 2005, 22, 35–44. [Google Scholar] [CrossRef]
- Van Uden, D.J.P.; Van Maaren, M.C.; Strobbe, L.J.A.; Bult, P.; Van Der Hoeven, J.J.; Siesling, S.; De Wilt, J.H.W.; Blanken-Peeters, C.F.J.M. Metastatic behavior and overall survival according to breast cancer subtypes in stage IV inflammatory breast cancer. Breast Cancer Res. 2019, 21, 113. [Google Scholar] [CrossRef]
- Hu, G.; Chong, R.A.; Yang, Q.; Wei, Y.; Blanco, M.A.; Li, F.; Reiss, M.; Au, J.L.; Haffty, B.G.; Kang, Y. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell 2009, 15, 9–20. [Google Scholar] [CrossRef]
- Hu, G.; Wei, Y.; Kang, Y. The multifaceted role of MTDH/AEG-1 in cancer progression. Clin. Cancer Res. 2009, 15, 5615–5620. [Google Scholar] [CrossRef]
- Zhang, E.Y.; Cristofanilli, M.; Robertson, F.; Reuben, J.M.; Mu, Z.; Beavis, R.C.; Im, H.; Snyder, M.; Hofree, M.; Ideker, T.; et al. Genome wide proteomics of ERBB2 and EGFR and other oncogenic pathways in inflammatory breast cancer. J. Proteome Res. 2013, 12, 2805–2817. [Google Scholar] [CrossRef]
- Sarkar, D.; Fisher, P.B. AEG-1/MTDH/LYRIC: Clinical significance. Adv. Cancer Res. 2013, 120, 39–74. [Google Scholar]
- Chen, Y.; Huang, S.; Guo, R.; Chen, D. Metadherin-mediated mechanisms in human malignancies. Biomark. Med. 2021, 15, 1769–1783. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeyer, M.R.; Wall, K.M.; Dharmawardhane, S.F. In vitro analysis of the invasive phenotype of SUM 149, an inflammatory breast cancer cell line. Cancer Cell Int. 2005, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Lehman, H.L.; Dashner, E.J.; Lucey, M.; Vermeulen, P.; Dirix, L.; Laere, S.V.; Van Golen, K.L. Modeling and characterization of inflammatory breast cancer emboli grown in vitro. Int. J. Cancer 2013, 132, 2283–2294. [Google Scholar] [CrossRef] [PubMed]
- Allensworth, J.L.; Evans, M.K.; Bertucci, F.; Aldrich, A.J.; Festa, R.A.; Finetti, P.; Ueno, N.T.; Safi, R.; McDonnell, D.P.; Thiele, D.J.; et al. Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer. Mol. Oncol. 2015, 9, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, X. The role of MTDH/AEG-1 in the progression of cancer. Int. J. Clin. Exp. Med. 2015, 8, 4795. [Google Scholar]
- Van Laere, S.J.; Van der Auwera, I.; Van den Eynden, G.G.; Elst, H.J.; Weyler, J.; Harris, A.L.; van Dam, P.; Van Marck, E.A.; Vermeulen, P.B.; Dirix, L.Y. Nuclear factor-kappaB signature of inflammatory breast cancer by cDNA microarray validated by quantitative real-time reverse transcription-PCR, immunohistochemistry, and nuclear factor-kappaB DNA-binding. Clin. Cancer Res. 2006, 12, 3249–3256. [Google Scholar] [CrossRef]
- Van Laere, S.; Limame, R.; Van Marck, E.A.; Vermeulen, P.B.; Dirix, L.Y. Is there a role for mammary stem cells in inflammatory breast carcinoma?: A review of evidence from cell line, animal model, and human tissue sample experiments. Cancer 2010, 116, 2794–2805. [Google Scholar] [CrossRef]
- Stevens, L.E.; Peluffo, G.; Qiu, X.; Temko, D.; Fassl, A.; Li, Z.; Trinh, A.; Seehawer, M.; Jovanović, B.; Alečković, M.; et al. JAK-STAT Signaling in Inflammatory Breast Cancer Enables Chemotherapy-Resistant Cell States. Cancer Res. 2023, 83, 264–284. [Google Scholar] [CrossRef]
- Rios-Fuller, T.J.; Ortiz-Soto, G.; Lacourt-Ventura, M.; Maldonado-Martinez, G.; Cubano, L.A.; Schneider, R.J.; Martinez-Montemayor, M.M. Ganoderma lucidum extract (GLE) impairs breast cancer stem cells by targeting the STAT3 pathway. Oncotarget 2018, 9, 35907–35921. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef]
- Li, J.; Zhang, N.; Song, L.B.; Liao, W.T.; Jiang, L.L.; Gong, L.Y.; Wu, J.; Yuan, J.; Zhang, H.Z.; Zeng, M.S.; et al. Astrocyte elevated gene-1 is a novel prognostic marker for breast cancer progression and overall patient survival. Clin. Cancer Res. 2008, 14, 3319–3326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lafortune, T.A.; Krishnamurthy, S.; Esteva, F.J.; Cristofanilli, M.; Liu, P.; Lucci, A.; Singh, B.; Hung, M.-C.; Hortobagyi, G.N.; et al. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Reverses Mesenchymal to Epithelial Phenotype and Inhibits Metastasis in Inflammatory Breast Cancer. Clin. Cancer Res. 2009, 15, 6639–6648. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Yu, L.; Mi, Y.; Zhang, J.; Wang, K.; Hu, L. Association of MTDH immunohistochemical expression with metastasis and prognosis in female reproduction malignancies: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 38365. [Google Scholar] [CrossRef]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, L.; Song, L.; Xiong, H.; Wang, L.; Yan, X.; Yuan, J.; Wu, J.; Li, M. Astrocyte elevated gene-1 is a proliferation promoter in breast cancer via suppressing transcriptional factor FOXO1. Oncogene 2009, 28, 3188–3196. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Zhang, Q.; Yang, Q. Immunohistochemical analysis of Metadherin in proliferative and cancerous breast tissue. Diagn. Pathol. 2010, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Offiah, G.; McSherry, E.A.; Hopkins, A.M. Tight Junctions: A Barrier to the Initiation and Progression of Breast Cancer? J. Biomed. Biotechnol. 2010, 2010, 460607. [Google Scholar] [CrossRef]
- Wang, F.; Ke, Z.-F.; Sun, S.-J.; Chen, W.-F.; Yang, S.-C.; Li, S.-H.; Mao, X.-P.; Wang, L.-T. Oncogenic roles of astrocyte elevated gene-1 (AEG-1) in osteosarcoma progression and prognosis. Cancer Biol. Ther. 2011, 12, 539–548. [Google Scholar] [CrossRef]
- Vartak-Sharma, N.; Ghorpade, A. Astrocyte elevated gene-1 regulates astrocyte responses to neural injury: Implications for reactive astrogliosis and neurodegeneration. J. Neuroinflammation 2012, 9, 195. [Google Scholar] [CrossRef]
- Kleer, C.G.; van Golen, K.L.; Braun, T.; Merajver, S.D. Persistent E-cadherin expression in inflammatory breast cancer. Mod. Pathol. 2001, 14, 458–464. [Google Scholar] [CrossRef]
- Colpaert, C.G.; Vermeulen, P.B.; Benoy, I.; Soubry, A.; van Roy, F.; van Beest, P.; Goovaerts, G.; Dirix, L.Y.; van Dam, P.; Fox, S.B.; et al. Inflammatory breast cancer shows angiogenesis with high endothelial proliferation rate and strong E-cadherin expression. Br. J. Cancer 2003, 88, 718–725. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Lewis-Tuffin, L.J.; Anastasiadis, P.Z. E-cadherin’s dark side: Possible role in tumor progression. Biochim. Biophys. Acta 2012, 1826, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Debeb, B.G.; Aceto, N.; Farach-Carson, M.C.; Woodward, W.A.; Levine, H. Inflammatory breast cancer: A model for investigating cluster-based dissemination. NPJ Breast Cancer 2017, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Merkhofer, E.C.; Cogswell, P.; Baldwin, A.S. Her2 activates NF-κB and induces invasion through the canonical pathway involving IKKα. Oncogene 2010, 29, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Yang, X.-Y.; Glass, O.; Lei, G.; Osada, T.; Dave, S.S.; Morse, M.A.; Clay, T.M.; Lyerly, H.K. HER2 Overexpression Elicits a Proinflammatory IL-6 Autocrine Signaling Loop That Is Critical for Tumorigenesis. Cancer Res. 2011, 71, 4380–4391. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Park, E.S.; Emdad, L.; Lee, S.G.; Su, Z.Z.; Fisher, P.B. Molecular basis of nuclear factor-kappaB activation by astrocyte elevated gene-1. Cancer Res. 2008, 68, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Bièche, I.; Lerebours, F.; Tozlu, S.; Espie, M.; Marty, M.; Lidereau, R. Molecular profiling of inflammatory breast cancer: Identification of a poor-prognosis gene expression signature. Clin. Cancer Res. 2004, 10, 6789–6795. [Google Scholar] [CrossRef]
- Lee, H.; Herrmann, A.; Deng, J.-H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently Activated Stat3 Maintains Constitutive NF-κB Activity in Tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, H.; Wang, X.; Wang, C.; Gan, L.; Zhu, J.; Tong, J.; Li, Z. Huaier Granule extract inhibit the proliferation and metastasis of lung cancer cells through down-regulation of MTDH, JAK2/STAT3 and MAPK signaling pathways. Biomed. Pharmacother. 2018, 101, 311–321. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, W.; Kone, B.C. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor κB. Biochem. J. 2002, 367, 97–105. [Google Scholar] [CrossRef]
- Wang, W.; Nag, S.; Zhang, R. Targeting the NFkB Signaling Pathways for Breast Cancer Prevention and Therapy. Curr. Med. Chem. 2014, 22, 264–289. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Yi, X.; Liu, W.; Han, T.; Liu, Z.; Ding, Z.; Zheng, Z.; Piao, Y.; Yuan, J.; Han, Y.; et al. MTDH mediates trastuzumab resistance in HER2 positive breast cancer by decreasing PTEN expression through an NFkappaB-dependent pathway. BMC Cancer 2014, 14, 869. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D. Animal Models and the Tumor Microenvironment: Studies of Tumor–Host Symbiosis. In Seminars in Oncology; WB Saunders: Philadelphia, PA, USA, 2014; Volume 41, pp. 146–155. [Google Scholar]
- Martinez-Montemayor, M.M.; Acevedo, R.R.; Otero-Franqui, E.; Cubano, L.A.; Dharmawardhane, S.F. Ganoderma lucidum (Reishi) inhibits cancer cell growth and expression of key molecules in inflammatory breast cancer. Nutr. Cancer 2011, 63, 1085–1094. [Google Scholar] [CrossRef]
- Acevedo-Díaz, A.; Morales-Cabán, B.M.; Zayas-Santiago, A.; Martínez-Montemayor, M.M.; Suárez-Arroyo, I.J. SCAMP3 Regulates EGFR and Promotes Proliferation and Migration of Triple-Negative Breast Cancer Cells through the Modulation of AKT, ERK, and STAT3 Signaling Pathways. Cancers 2022, 14, 2807. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Arroyo, I.J.; Rosario-Acevedo, R.; Aguilar-Perez, A.; Clemente, P.L.; Cubano, L.A.; Serrano, J.; Schneider, R.J.; Martinez-Montemayor, M.M. Anti-tumor effects of Ganoderma lucidum (reishi) in inflammatory breast cancer in in vivo and in vitro models. PLoS ONE 2013, 8, e57431. [Google Scholar] [CrossRef]
- Martínez-Montemayor, M.M.; Hill, G.M.; Raney, N.E.; Rilington, V.D.; Tempelman, R.J.; Link, J.E.; Wilkinson, C.P.; Ramos, A.M.; Ernst, C.W. Gene expression profiling in hepatic tissue of newly weaned pigs fed pharmacological zinc and phytase supplemented diets. BMC Genom. 2008, 9, 421. [Google Scholar] [CrossRef]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Kõressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef]
- Suarez-Arroyo, I.J.; Rios-Fuller, T.J.; Feliz-Mosquea, Y.R.; Lacourt-Ventura, M.; Leal-Alviarez, D.J.; Maldonado-Martinez, G.; Cubano, L.A.; Martinez-Montemayor, M.M. Ganoderma lucidum Combined with the EGFR Tyrosine Kinase Inhibitor, Erlotinib Synergize to Reduce Inflammatory Breast Cancer Progression. J. Cancer 2016, 7, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Díaz, A.; Ortiz-Soto, G.; Suárez-Arroyo, I.J.; Zayas-Santiago, A.; Martínez Montemayor, M.M. Ganoderma lucidum Extract Reduces the Motility of Breast Cancer Cells Mediated by the RAC–Lamellipodin Axis. Nutrients 2019, 11, 1116. [Google Scholar] [CrossRef] [PubMed]
- Allensworth, J.L.; Sauer, S.J.; Lyerly, H.K.; Morse, M.A.; Devi, G.R. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-α-independent mechanism. Breast Cancer Res. Treat. 2013, 137, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Arora, J.; Sauer, S.J.; Tarpley, M.; Vermeulen, P.; Rypens, C.; Van Laere, S.; Williams, K.P.; Devi, G.R.; Dewhirst, M.W. Inflammatory breast cancer tumor emboli express high levels of anti-apoptotic proteins: Use of a quantitative high content and high-throughput 3D IBC spheroid assay to identify targeting strategies. Oncotarget 2017, 8, 25848–25863. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Arroyo, I.J.; Acevedo-Díaz, A.; Ríos-Fuller, T.J.; Ortiz-Soto, G.; Vallejo-Calzada, R.; Reyes-Chea, J.; Maldonado-Martínez, G.; Schneider, R.J.; Martínez-Montemayor, M.M. Ganoderma lucidum enhances carboplatin chemotherapy effect by inhibiting the DNA damage response pathway and stemness. Am. J. Cancer Res. 2022, 12, 1282–1294. [Google Scholar] [PubMed]
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Preparation of slides and coverslips for microscopy. CSH Protoc. 2008, 2008, pdb.prot4988. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Primer Sequence |
|---|---|---|
| Beta-2-Microglobulin (B2M) | NM_004048.4 | FW 5′TGCTGTCTCCATGTTTGATGTATCT3′ REV 5′TCTCTGCTCCCCACCTCTAAGT3′ |
| Hypoxanthine Phosphoribosyltransferase (HPRT) | NM_000194.3 | FW 5′CCTGGCGTCGTGATTAGTGAT 3′ REV 5′AGACGTTCAGTCCTGTCCATAA 3′ |
| Metadherin (MTDH) | NM_178812.4 | FW 5′GAAACTGTCCGAGAAGCCCA3′ REV 5′GTCAATCTCTGGTGGCTGCT3′ |
| Signal Transducer and Activator of Transcription 3 (STAT3) | NM_139276.3 | FW 5′GCCAATTGTGATGCTTCC 3′ REV 5′TGGGTCTCTAGGTCAATCT 3′ |
| Ribosomal Protein L13a (RPL13A) | NM_012423.4 | FW 5′TGAAGCCTACAAGAAAGTTTGCCT 3′ REV 5′TAGCCTCATGAGCTGTTTCTTCTT 3′ |
| Nuclear factor kappa B subunit 1 (NFKB1) | NM_003998 | FW 5′ATAGCCTGCCATGTTTGCTGCT 3′ |
| REV 5′TGCCAATGAGATGTTGTCGTGC 3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Soto, G.; Babilonia-Díaz, N.S.; Lacourt-Ventura, M.Y.; Rivera-Rodríguez, D.M.; Quiñones-Rodríguez, J.I.; Colón-Vargas, M.; Almodóvar-Rivera, I.; Ferrer-Torres, L.E.; Suárez-Arroyo, I.J.; Martínez-Montemayor, M.M. Metadherin Regulates Inflammatory Breast Cancer Invasion and Metastasis. Int. J. Mol. Sci. 2023, 24, 4694. https://doi.org/10.3390/ijms24054694
Ortiz-Soto G, Babilonia-Díaz NS, Lacourt-Ventura MY, Rivera-Rodríguez DM, Quiñones-Rodríguez JI, Colón-Vargas M, Almodóvar-Rivera I, Ferrer-Torres LE, Suárez-Arroyo IJ, Martínez-Montemayor MM. Metadherin Regulates Inflammatory Breast Cancer Invasion and Metastasis. International Journal of Molecular Sciences. 2023; 24(5):4694. https://doi.org/10.3390/ijms24054694
Chicago/Turabian StyleOrtiz-Soto, Gabriela, Natalia S. Babilonia-Díaz, Mercedes Y. Lacourt-Ventura, Delmarie M. Rivera-Rodríguez, Jailenne I. Quiñones-Rodríguez, Mónica Colón-Vargas, Israel Almodóvar-Rivera, Luis E. Ferrer-Torres, Ivette J. Suárez-Arroyo, and Michelle M. Martínez-Montemayor. 2023. "Metadherin Regulates Inflammatory Breast Cancer Invasion and Metastasis" International Journal of Molecular Sciences 24, no. 5: 4694. https://doi.org/10.3390/ijms24054694
APA StyleOrtiz-Soto, G., Babilonia-Díaz, N. S., Lacourt-Ventura, M. Y., Rivera-Rodríguez, D. M., Quiñones-Rodríguez, J. I., Colón-Vargas, M., Almodóvar-Rivera, I., Ferrer-Torres, L. E., Suárez-Arroyo, I. J., & Martínez-Montemayor, M. M. (2023). Metadherin Regulates Inflammatory Breast Cancer Invasion and Metastasis. International Journal of Molecular Sciences, 24(5), 4694. https://doi.org/10.3390/ijms24054694