
Electrical Remodeling in Right Ventricular Failure Due to Pulmonary Hypertension: Unraveling Novel Therapeutic Targets

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
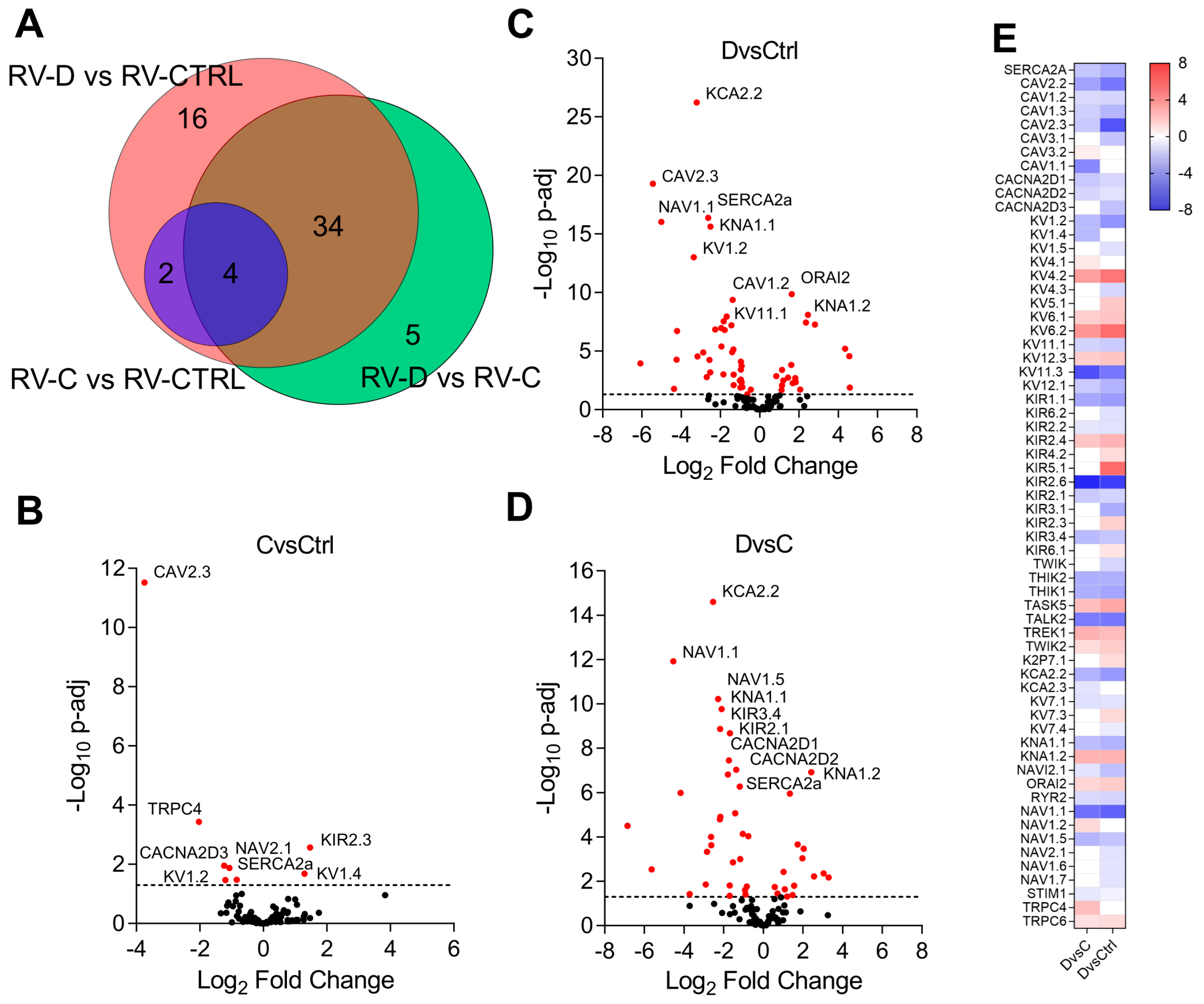
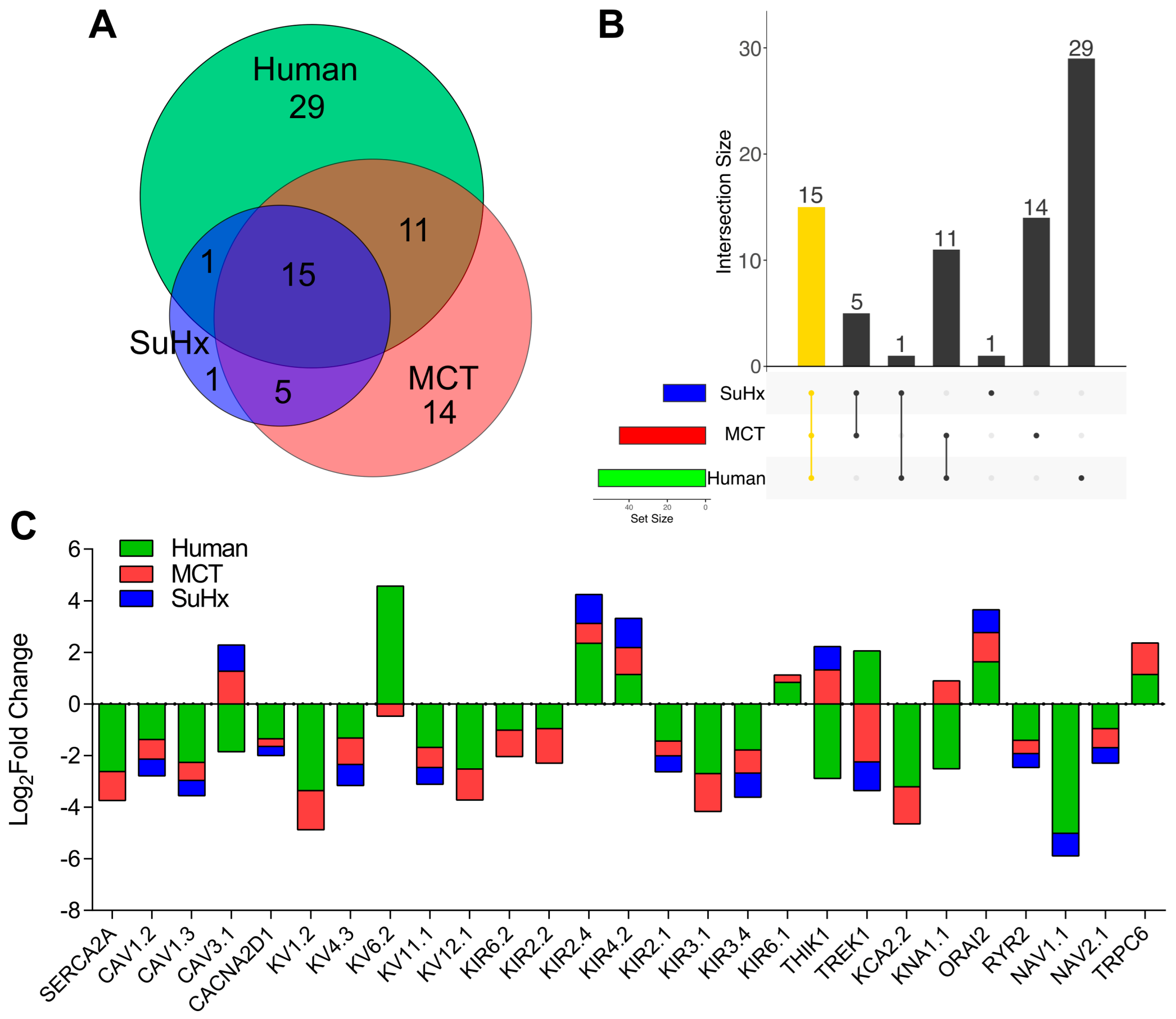
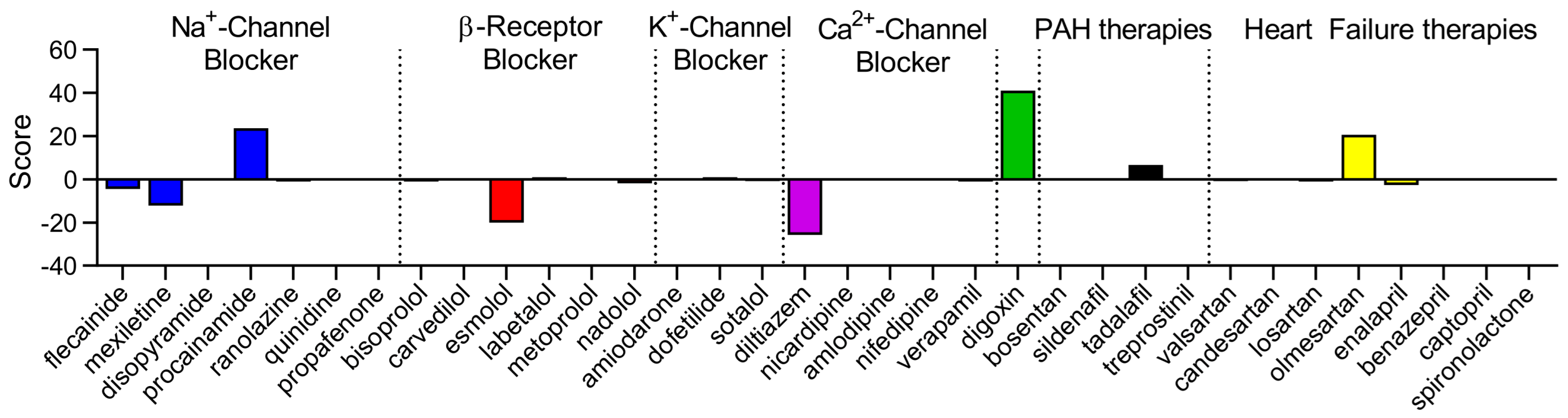
2. Results
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ren, X.; Johns, R.A.; Gao, W.D. Right Heart in Pulmonary Hypertension: From Adaptation to Failure. Pulm. Circ. 2019, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zelt, J.G.E.; Chaudhary, K.R.; Cadete, V.J.; Mielniczuk, L.M.; Stewart, D.J. Medical Therapy for Heart Failure Associated With Pulmonary Hypertension. Circ. Res. 2019, 124, 1551–1567. [Google Scholar] [CrossRef] [PubMed]
- Rajdev, A.; Garan, H.; Biviano, A. Arrhythmias in Pulmonary Arterial Hypertension. Prog. Cardiovasc. Dis. 2012, 55, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.T.; Maulik, A.; Lewis, R.; Kiely, D.G.; Toshner, M.; Charalampopoulos, A.; Kyriacou, A.; Rothman, A. Arrhythmic Burden and Outcomes in Pulmonary Arterial Hypertension. Front. Med. 2019, 6, 169. [Google Scholar] [CrossRef]
- Temple, I.P. Arrhythmias in Pulmonary Arterial Hypertension. J. Congenit. Cardiol. 2017, 1, 2. [Google Scholar] [CrossRef]
- Umar, S.; Lee, J.-H.; de Lange, E.; Iorga, A.; Partow-Navid, R.; Bapat, A.; van der Laarse, A.; Saggar, R.; Saggar, R.; Ypey, D.L.; et al. Spontaneous Ventricular Fibrillation in Right Ventricular Failure Secondary to Chronic Pulmonary Hypertension. Circ. Arrhythm. Electrophysiol. 2012, 5, 181–190. [Google Scholar] [CrossRef]
- Benoist, D.; Stones, R.; Drinkhill, M.; Bernus, O.; White, E. Arrhythmogenic Substrate in Hearts of Rats with Monocrotaline-Induced Pulmonary Hypertension and Right Ventricular Hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2230–H2237. [Google Scholar] [CrossRef]
- Andersen, A.; van der Feen, D.E.; Andersen, S.; Schultz, J.G.; Hansmann, G.; Bogaard, H.J. Animal Models of Right Heart Failure. Cardiovasc. Diagn. 2020, 10, 1561–1579. [Google Scholar] [CrossRef]
- Park, J.F.; Clark, V.R.; Banerjee, S.; Hong, J.; Razee, A.; Williams, T.; Fishbein, G.; Saddic, L.; Umar, S. Transcriptomic Analysis of Right Ventricular Remodeling in Two Rat Models of Pulmonary Hypertension: Identification and Validation of EMT in Human Right Ventricular Failure. Circ. Heart Fail. 2021, 14, e007058. [Google Scholar] [CrossRef]
- Boucherat, O.; Yokokawa, T.; Krishna, V.; Kalyana-Sundaram, S.; Martineau, S.; Breuils-Bonnet, S.; Azhar, N.; Bonilla, F.; Gutstein, D.; Potus, F.; et al. Identification of LTBP-2 as a Plasma Biomarker for Right Ventricular Dysfunction in Human Pulmonary Arterial Hypertension. Nat. Cardiovasc. Res. 2022, 1, 748–760. [Google Scholar] [CrossRef]
- Lee, J.-K.; Nishiyama, A.; Kambe, F.; Seo, H.; Takeuchi, S.; Kamiya, K.; Kodama, I.; Toyama, J. Downregulation of Voltage-Gated K+ Channels in Rat Heart with Right Ventricular Hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 1999, 277, H1725–H1731. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Mercier, O.; Humbert, M.; Sabourin, J. Excitation-Contraction Coupling and Relaxation Alteration in Right Ventricular Remodelling Caused by Pulmonary Arterial Hypertension. Arch. Cardiovasc. Dis. 2020, 113, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, K.C.; Hong, Y.M. Change of Voltage-Gated Potassium Channel 1.7 Expressions in Monocrotaline-Induced Pulmonary Arterial Hypertension Rat Model. Korean J. Pediatr. 2018, 61, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Reilly, L.; Eckhardt, L.L. Cardiac Potassium Inward Rectifier Kir2: Review of Structure, Regulation, Pharmacology, and Arrhythmogenesis. Heart Rhythm 2021, 18, 1423–1434. [Google Scholar] [CrossRef]
- Shah, K.; Seeley, S.; Schulz, C.; Fisher, J.; Gururaja Rao, S. Calcium Channels in the Heart: Disease States and Drugs. Cells 2022, 11, 943. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Soong, T.W. CaV1.2 Channelopathies: From Arrhythmias to Autism, Bipolar Disorder, and Immunodeficiency. Pflug. Arch. 2010, 460, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Le Quang, K.; Benito, B.; Naud, P.; Qi, X.Y.; Shi, Y.F.; Tardif, J.-C.; Gillis, M.-A.; Dobrev, D.; Charpentier, F.; Nattel, S. T-Type Calcium Current Contributes to Escape Automaticity and Governs the Occurrence of Lethal Arrhythmias After Atrioventricular Block in Mice. Circ. Arrhythm. Electrophysiol. 2013, 6, 799–808. [Google Scholar] [CrossRef]
- Daimi, H.; Lozano-Velasco, E.; Aranega, A.; Franco, D. Genomic and Non-Genomic Regulatory Mechanisms of the Cardiac Sodium Channel in Cardiac Arrhythmias. Int. J. Mol. Sci. 2022, 23, 1381. [Google Scholar] [CrossRef]
- Li, W.; Yin, L.; Shen, C.; Hu, K.; Ge, J.; Sun, A. SCN5A Variants: Association With Cardiac Disorders. Front. Physiol. 2018, 9, 1372. [Google Scholar] [CrossRef]
- Perros, F.; de Man, F.S.; Bogaard, H.J.; Antigny, F.; Simonneau, G.; Bonnet, S.; Provencher, S.; Galiè, N.; Humbert, M. Use of β-Blockers in Pulmonary Hypertension. Circ. Heart Fail. 2017, 10, e003703. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Bajaj, N.S.; Zein, J.; Minai, O.A.; Dweik, R.A. Outcomes of β-Blocker Use in Pulmonary Arterial Hypertension: A Propensity-Matched Analysis. Eur. Respir. J. 2015, 46, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.; Badagliacca, R.; Berger, R.; Brida, M.; Carlsen, J.; Coats, A.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension: Developed by the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on rare respiratory diseases (ERN-LUNG). Eur. Heart J. Oxf. Acad. 2022, 43, 3618–3731. Available online: https://academic.oup.com/eurheartj/article/43/38/3618/6673929?login=false#384349316 (accessed on 13 January 2023).
- Sestier, M.; Hillis, C.; Fraser, G.; Leong, D. Bruton’s Tyrosine Kinase Inhibitors and Cardiotoxicity: More Than Just Atrial Fibrillation. Curr. Oncol. Rep. 2021, 23, 113. [Google Scholar] [CrossRef]
- Giudice, V.; Vecchione, C.; Selleri, C. Cardiotoxicity of Novel Targeted Hematological Therapies. Life 2020, 10, 344. [Google Scholar] [CrossRef] [PubMed]
- Zhabyeyev, P.; Chen, X.; Vanhaesebroeck, B.; Oudit, G.Y. PI3Kα in Cardioprotection: Cytoskeleton, Late Na+ Current, and Mechanism of Arrhythmias. Channels 2019, 13, 520–532. [Google Scholar] [CrossRef]
- Berghausen, E.M.; Janssen, W.; Vantler, M.; Gnatzy-Feik, L.L.; Krause, M.; Behringer, A.; Joseph, C.; Zierden, M.; ten Freyhaus, H.; Klinke, A.; et al. Disrupted PI3K Subunit P110α Signaling Protects against Pulmonary Hypertension and Reverses Established Disease in Rodents. J. Clin. Investig. 2021, 131, e136939. [Google Scholar] [CrossRef]
- Hof, T.; Chaigne, S.; Récalde, A.; Sallé, L.; Brette, F.; Guinamard, R. Transient Receptor Potential Channels in Cardiac Health and Disease. Nat. Rev. Cardiol. 2019, 16, 344–360. [Google Scholar] [CrossRef]
- Yue, Z.; Xie, J.; Yu, A.S.; Stock, J.; Du, J.; Yue, L. Role of TRP Channels in the Cardiovascular System. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H157–H182. [Google Scholar] [CrossRef]
- Kuo, I.Y.; Chapman, A.B. Polycystins, ADPKD, and Cardiovascular Disease. Kidney Int. Rep. 2020, 5, 396–406. [Google Scholar] [CrossRef]
- Altamirano, F.; Schiattarella, G.G.; French, K.M.; Kim, S.Y.; Engelberger, F.; Kyrychenko, S.; Villalobos, E.; Tong, D.; Schneider, J.W.; Ramirez-Sarmiento, C.A.; et al. Polycystin-1 Assembles With Kv Channels to Govern Cardiomyocyte Repolarization and Contractility. Circulation 2019, 140, 921–936. [Google Scholar] [CrossRef]
- Zhou, S.; Tan, A.Y.; Paz, O.; Ogawa, M.; Chou, C.-C.; Hayashi, H.; Nihei, M.; Fishbein, M.C.; Chen, L.S.; Lin, S.-F.; et al. Antiarrhythmic Effects of Beta3-Adrenergic Receptor Stimulation in a Canine Model of Ventricular Tachycardia. Heart Rhythm 2008, 5, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.Y.M.; Farah, C.; Balligand, J.-L. The Beta3 Adrenergic Receptor in Healthy and Pathological Cardiovascular Tissues. Cells 2020, 9, 2584. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.; Nielsen-Kudsk, J.E.; Vonk Noordegraaf, A.; de Man, F.S. Right Ventricular Fibrosis. Circulation 2019, 139, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, J.; Trenor, B.; Saiz, J.; Dössel, O.; Loewe, A. Fibrotic Remodeling during Persistent Atrial Fibrillation: In Silico Investigation of the Role of Calcium for Human Atrial Myofibroblast Electrophysiology. Cells 2021, 10, 2852. [Google Scholar] [CrossRef] [PubMed]
- Rydman, R.; Gatzoulis, M.A.; Ho, S.Y.; Ernst, S.; Swan, L.; Li, W.; Wong, T.; Sheppard, M.; McCarthy, K.P.; Roughton, M.; et al. Systemic Right Ventricular Fibrosis Detected by Cardiovascular Magnetic Resonance Is Associated with Clinical Outcome, Mainly New-Onset Atrial Arrhythmia, in Patients after Atrial Redirection Surgery for Transposition of the Great Arteries. Circ. Cardiovasc. Imaging 2015, 8, e002628. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.F.; Liang, J.; Umar, S. Electrical Remodeling in Right Ventricular Failure Due to Pulmonary Hypertension: Unraveling Novel Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 4633. https://doi.org/10.3390/ijms24054633
Park JF, Liang J, Umar S. Electrical Remodeling in Right Ventricular Failure Due to Pulmonary Hypertension: Unraveling Novel Therapeutic Targets. International Journal of Molecular Sciences. 2023; 24(5):4633. https://doi.org/10.3390/ijms24054633
Chicago/Turabian StylePark, John F., Justine Liang, and Soban Umar. 2023. "Electrical Remodeling in Right Ventricular Failure Due to Pulmonary Hypertension: Unraveling Novel Therapeutic Targets" International Journal of Molecular Sciences 24, no. 5: 4633. https://doi.org/10.3390/ijms24054633
APA StylePark, J. F., Liang, J., & Umar, S. (2023). Electrical Remodeling in Right Ventricular Failure Due to Pulmonary Hypertension: Unraveling Novel Therapeutic Targets. International Journal of Molecular Sciences, 24(5), 4633. https://doi.org/10.3390/ijms24054633