
Transcriptomic Analyses of Brains of RBM8A Conditional Knockout Mice at Different Developmental Stages Reveal Conserved Signaling Pathways Contributing to Neurodevelopmental Diseases
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
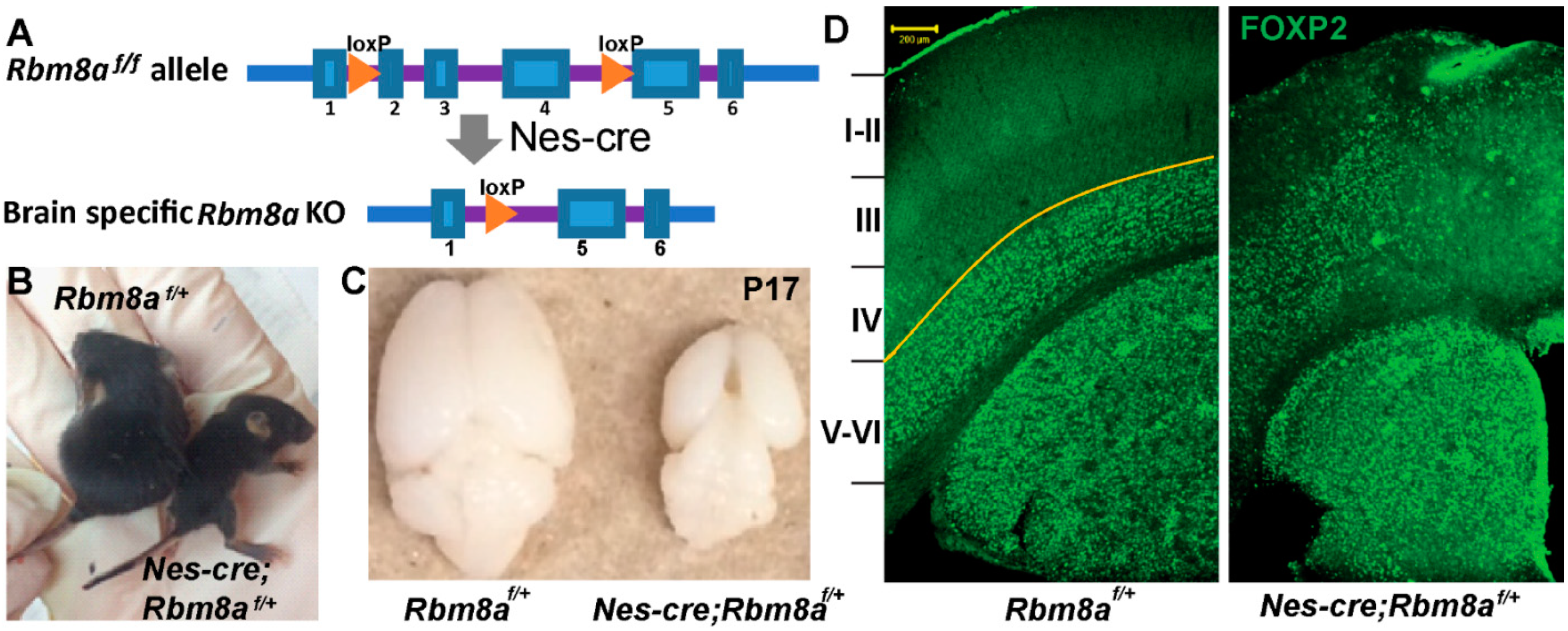
2.1. Rbm8a cKO Mouse Model
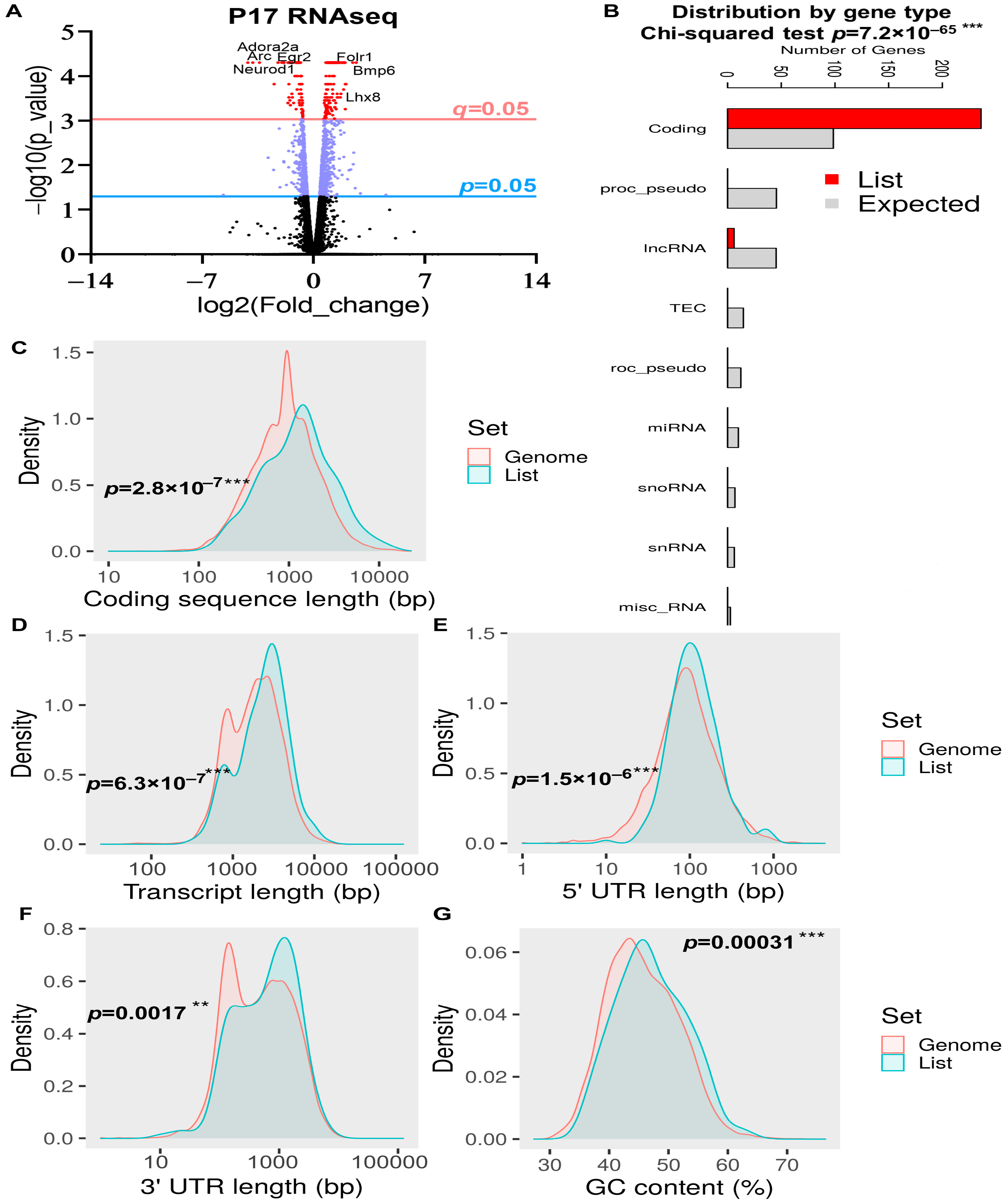
2.2. General DEG Analysis of the Whole Brain at P17
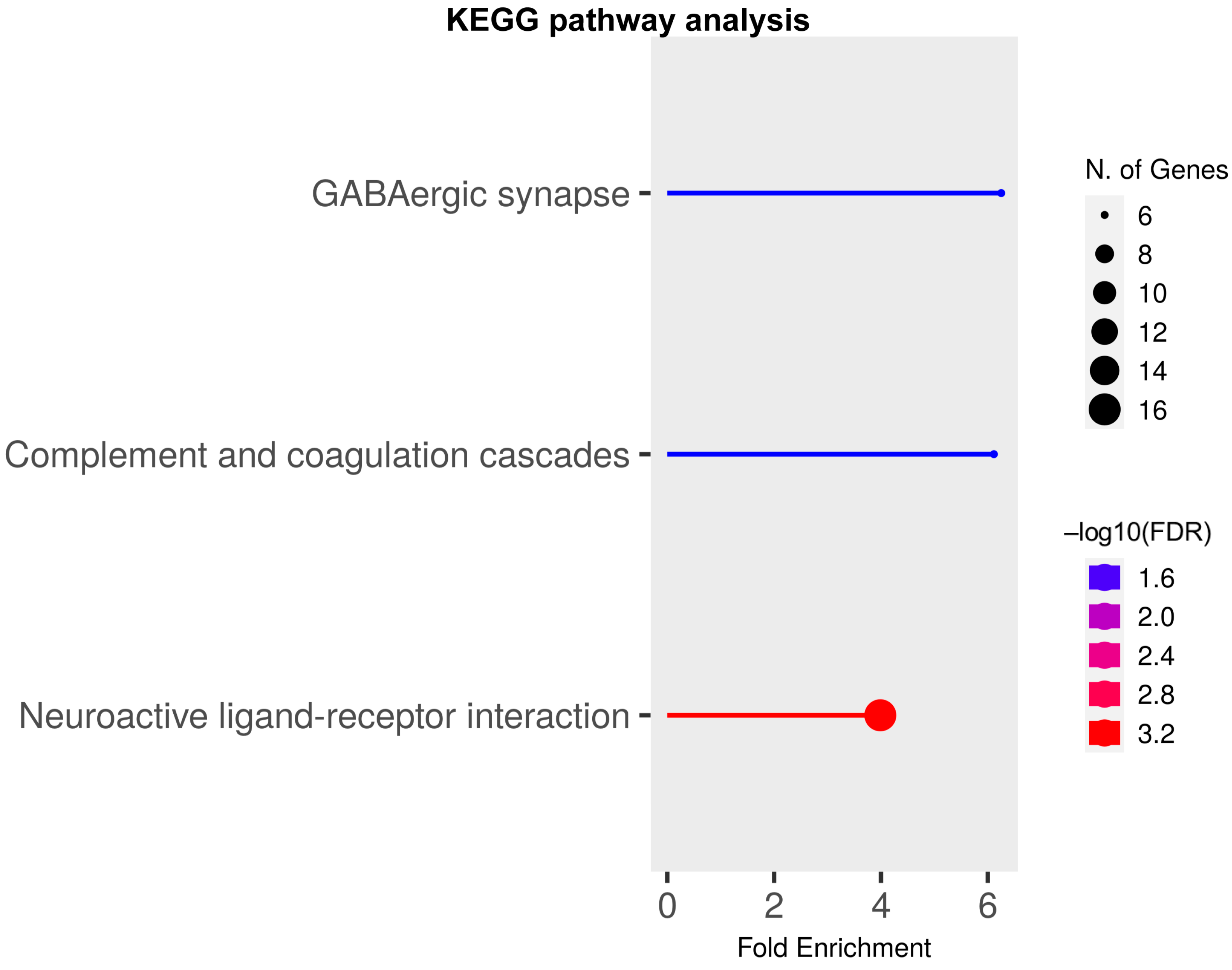
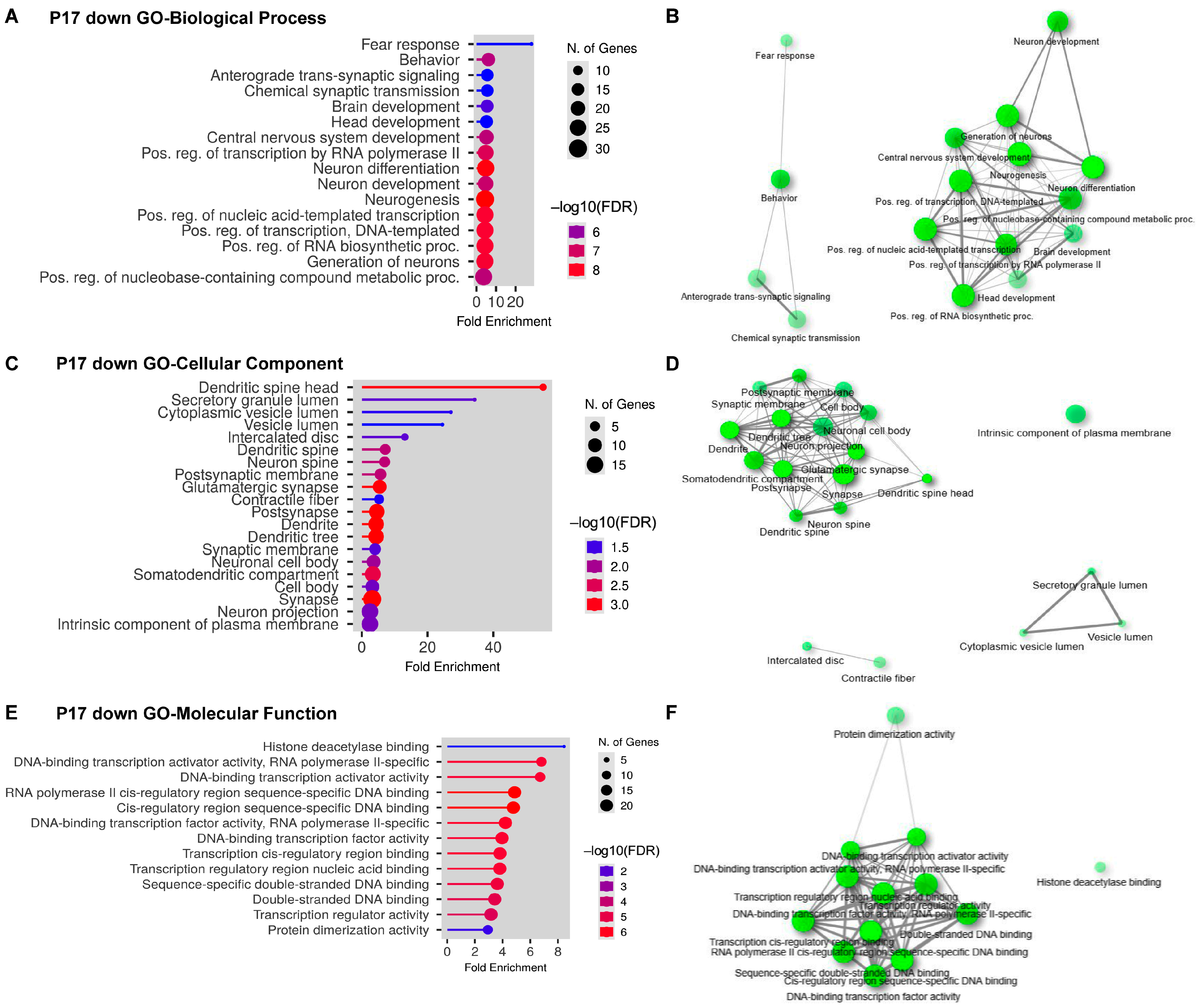
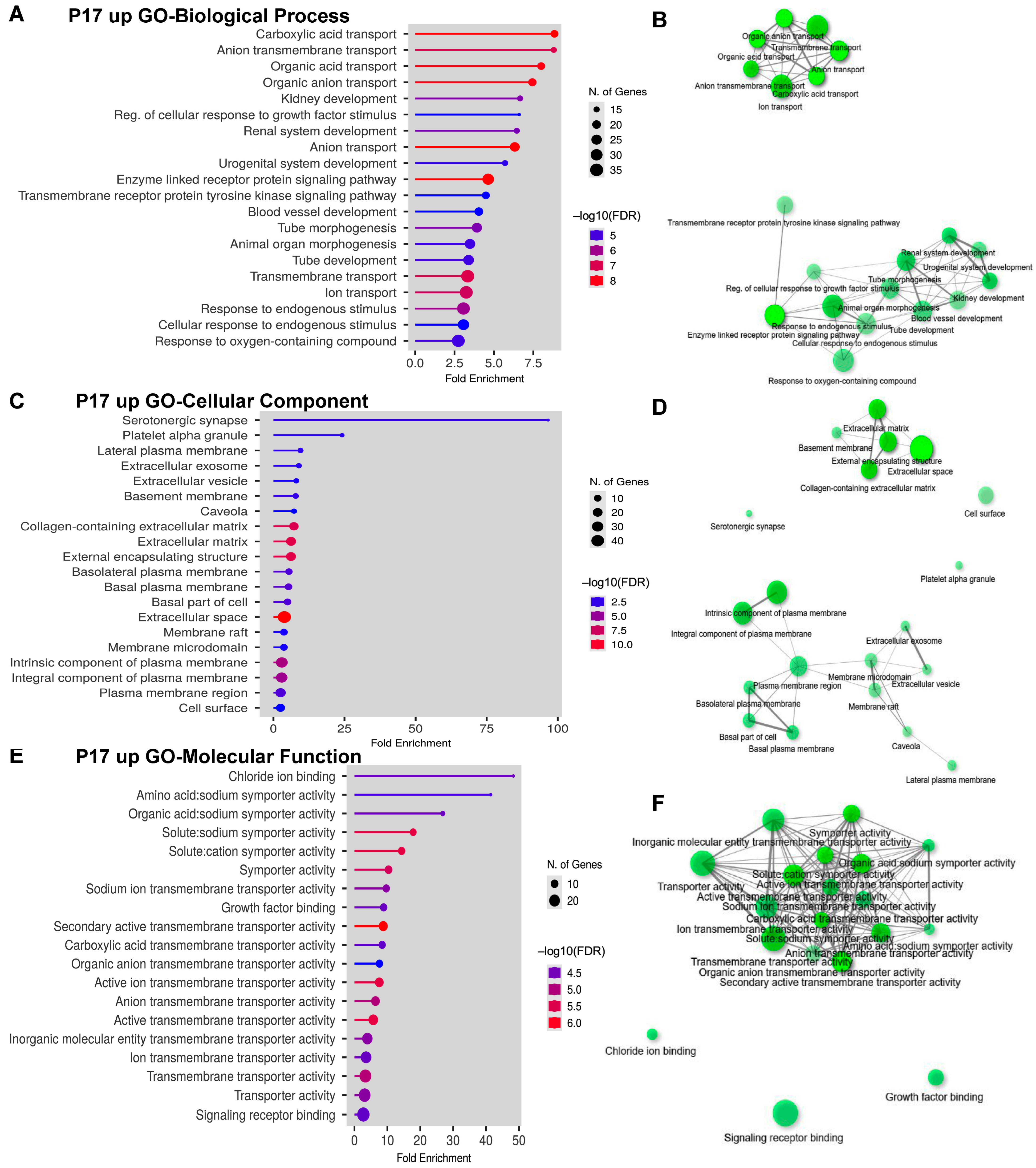
2.3. Gene Ontology (GO) Analyses on Upregulated and Downregulated DEGs at P17
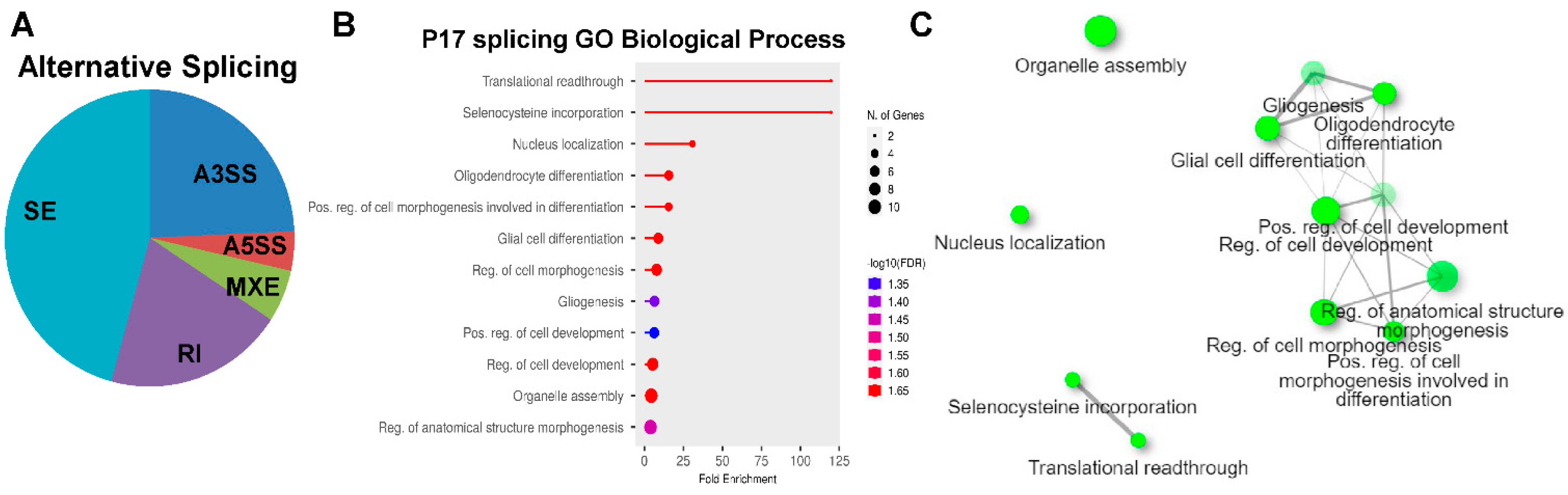
2.4. Alternative Splicing (AS) Analyses of RNAseq Dataset at P17
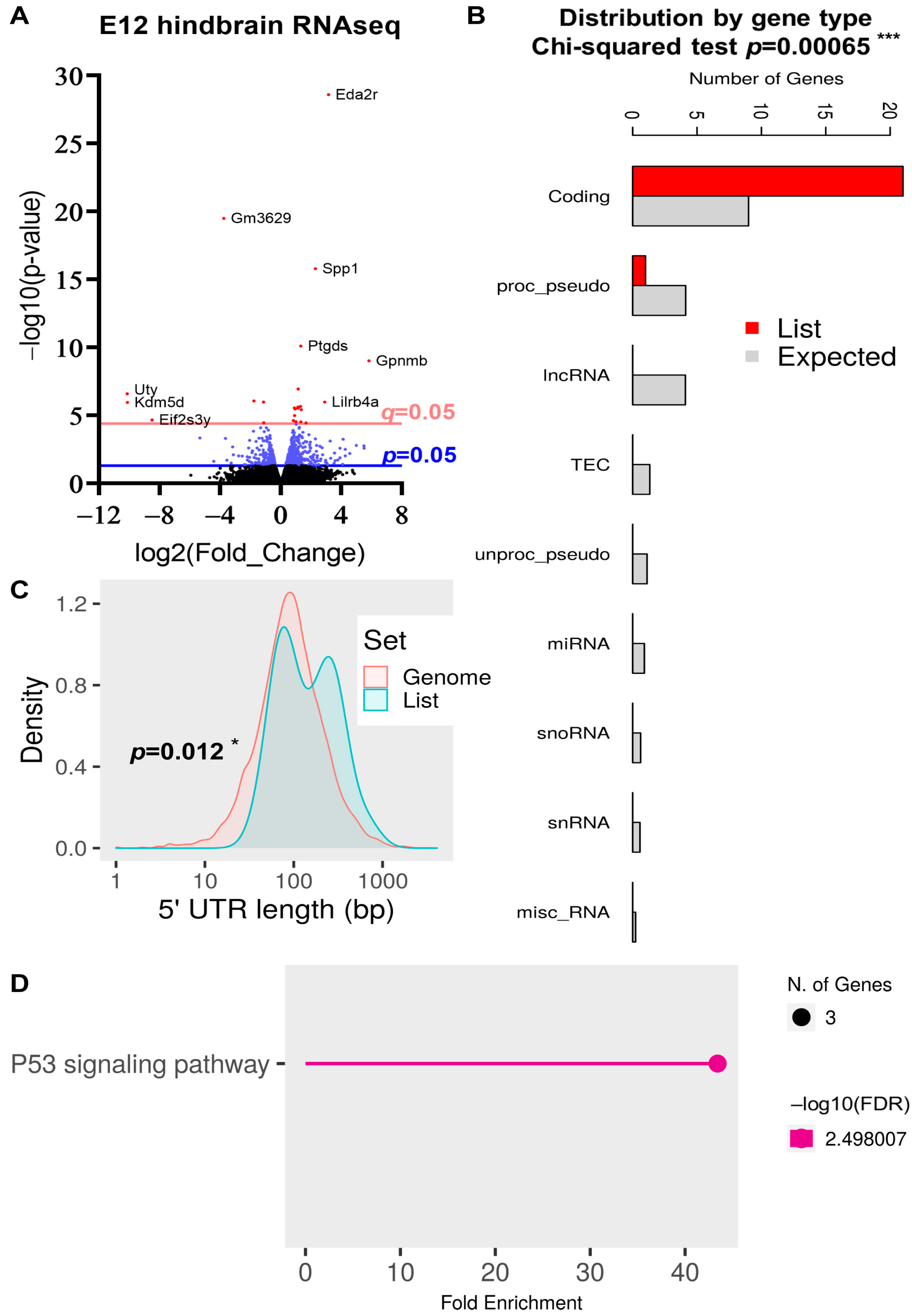
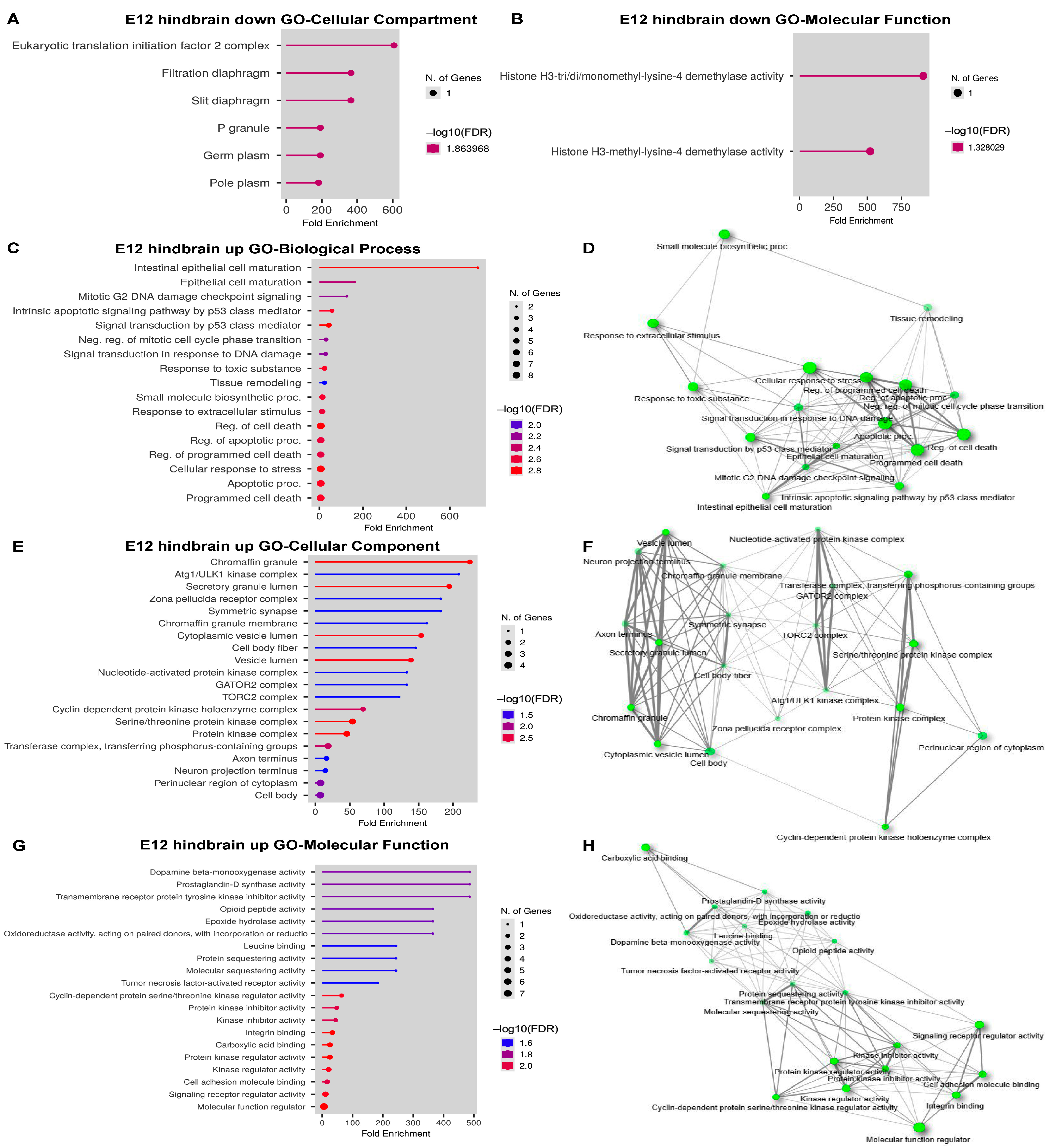
2.5. DEG Analysis in the E12 Hindbrain
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. RNA-Sequencing
4.3. Analysis of DEGs
4.4. Analysis of Enriched Gene Clusters
4.5. Alternative Splicing Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salicioni, A.M.; Xi, M.; Vanderveer, L.A.; Balsara, B.; Testa, J.R.; Dunbrack, R.L., Jr.; Godwin, A.K. Identification and structural analysis of human RBM8A and RBM8B: Two highly conserved RNA-binding motif proteins that interact with OVCA1, a candidate tumor suppressor. Genomics 2000, 69, 54–62. [Google Scholar] [CrossRef]
- Le Hir, H.; Saulière, J.; Wang, Z. The exon junction complex as a node of post-transcriptional networks. Nat. Rev. Mol. Cell Biol. 2016, 17, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Singh, A.K.; McLeod, T.; Blanchette, M.; Jang, B.; Badenhorst, P.; Kanhere, A.; Brogna, S. Exon junction complex proteins bind nascent transcripts independently of pre-mRNA splicing in Drosophila melanogaster. eLife 2016, 5, e19881. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.L.; Leeds, K.E.; Hwang, H.W.; Miller, E.E.; Pavan, W.J. The EJC component Magoh regulates proliferation and expansion of neural crest-derived melanocytes. Dev. Biol. 2013, 375, 172–181. [Google Scholar] [CrossRef]
- Obrdlik, A.; Lin, G.; Haberman, N.; Ule, J.; Ephrussi, A. The transcriptome-wide landscape and modalities of EJC binding in adult Drosophila. Cell Rep. 2019, 28, 1219–1236.e1211. [Google Scholar] [CrossRef]
- Gangras, P.; Gallagher, T.L.; Parthun, M.A.; Yi, Z.X.; Patton, R.D.; Tietz, K.T.; Deans, N.C.; Bundschuh, R.; Amacher, S.L.; Singh, G. Zebrafish rbm8a and magoh mutants reveal EJC developmental functions and new 3′UTR intron-containing NMD targets. PLoS Genet. 2020, 16, e1008830. [Google Scholar] [CrossRef]
- Guan, Q.; Zheng, W.; Tang, S.; Liu, X.; Zinkel, R.A.; Tsui, K.W.; Yandell, B.S.; Culbertson, M.R. Impact of nonsense-mediated mRNA decay on the global expression profile of budding yeast. PLoS Genet. 2006, 2, e203. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Das, B.; Sherman, F.; Maquat, L.E. Cap-binding protein 1-mediated and eukaryotic translation initiation factor 4E-mediated pioneer rounds of translation in yeast. Proc. Natl. Acad. Sci. USA 2005, 102, 4258–4263. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Yeo, G.W.; Stone, M.E.; Katz, D.B.; Burge, C.; Turrigiano, G.; Moore, M.J. The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell 2007, 130, 179–191. [Google Scholar] [CrossRef]
- Lin, Y.; Liang, R.; Qiu, Y.; Lv, Y.F.; Zhang, J.Y.; Qin, G.; Yuan, C.; Liu, Z.H.; Li, Y.Q.; Zou, D.H.; et al. Expression and gene regulation network of RBM8A in hepatocellular carcinoma based on data mining. Aging 2019, 11, 423–447. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Li, R.; Huang, X.; Chen, G.; Liu, Y.; Meng, Y.; Wang, Y.; Wu, Y.; Mao, Y. Identification of molecular correlations of RBM8A with autophagy in Alzheimer’s disease. Aging 2019, 11, 11673–11685. [Google Scholar] [CrossRef]
- Sauliere, J.; Haque, N.; Harms, S.; Barbosa, I.; Blanchette, M.; Le Hir, H. The exon junction complex differentially marks spliced junctions. Nat. Struct. Mol. Biol. 2010, 17, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Buhler, M.; Steiner, S.; Mohn, F.; Paillusson, A.; Muhlemann, O. EJC-independent degradation of nonsense immunoglobulin-mu mRNA depends on 3′ UTR length. Nat. Struct. Mol. Biol. 2006, 13, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Asthana, S.; Martin, H.; Rupkey, J.; Patel, S.; Yoon, J.; Keegan, A.; Mao, Y. The Physiological Roles of the Exon Junction Complex in Development and Diseases. Cells 2022, 11, 1192. [Google Scholar] [CrossRef] [PubMed]
- Tassano, E.; Gimelli, S.; Divizia, M.T.; Lerone, M.; Vaccari, C.; Puliti, A.; Gimelli, G. Thrombocytopenia-absent radius (TAR) syndrome due to compound inheritance for a 1q21.1 microdeletion and a low-frequency noncoding RBM8A SNP: A new familial case. Mol. Cytogenet. 2015, 8, 87. [Google Scholar] [CrossRef]
- Boussion, S.; Escande, F.; Jourdain, A.S.; Smol, T.; Brunelle, P.; Duhamel, C.; Alembik, Y.; Attie-Bitach, T.; Baujat, G.; Bazin, A.; et al. TAR syndrome: Clinical and molecular characterization of a cohort of 26 patients and description of novel noncoding variants of RBM8A. Hum. Mutat. 2020, 41, 1220–1225. [Google Scholar] [CrossRef]
- Albers, C.A.; Paul, D.S.; Schulze, H.; Freson, K.; Stephens, J.C.; Smethurst, P.A.; Jolley, J.D.; Cvejic, A.; Kostadima, M.; Bertone, P.; et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 2012, 44, 435–439. [Google Scholar] [CrossRef]
- Greenhalgh, K.L.; Howell, R.T.; Bottani, A.; Ancliff, P.J.; Brunner, H.G.; Verschuuren-Bemelmans, C.C.; Vernon, E.; Brown, K.W.; Newbury-Ecob, R.A. Thrombocytopenia-absent radius syndrome: A clinical genetic study. J. Med. genet. 2002, 39, 876–881. [Google Scholar] [CrossRef]
- Menghsol, S.C.; Harris, R.D.; Ornvold, K. Thrombocytopenia and absent radii, TAR syndrome: Report of cerebellar dysgenesis and newly identified cardiac and renal anomalies. Am. J. Med. Genet. A 2003, 123A, 193–196. [Google Scholar] [CrossRef]
- Skorka, A.; Bielicka-Cymermann, J.; Gieruszczak-Bialek, D.; Korniszewski, L. Thrombocytopenia-absent radius (tar) syndrome: A case with agenesis of corpus callosum, hypoplasia of cerebellar vermis and horseshoe kidney. Genet. Couns. 2005, 16, 377–382. [Google Scholar]
- Homans, A.C.; Cohen, J.L.; Mazur, E.M. Defective megakaryocytopoiesis in the syndrome of thrombocytopenia with absent radii. Br. J. Haematol. 1988, 70, 205–210. [Google Scholar] [CrossRef]
- Bernier, R.; Steinman, K.J.; Reilly, B.; Wallace, A.S.; Sherr, E.H.; Pojman, N.; Mefford, H.C.; Gerdts, J.; Earl, R.; Hanson, E.; et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med. 2016, 18, 341–349. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Berg, J.S.; Scaglia, F.; Belmont, J.; Bacino, C.A.; Sahoo, T.; Lalani, S.R.; Graham, B.; Lee, B.; Shinawi, M. Recurrent reciprocal 1q21. 1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008, 40, 1466–1471. [Google Scholar] [CrossRef]
- Dolcetti, A.; Silversides, C.K.; Marshall, C.R.; Lionel, A.C.; Stavropoulos, D.J.; Scherer, S.W.; Bassett, A.S. 1q21.1 Microduplication expression in adults. Genet. Med. 2013, 15, 282–289. [Google Scholar] [CrossRef]
- Mefford, H.C.; Sharp, A.J.; Baker, C.; Itsara, A.; Jiang, Z.; Buysse, K.; Huang, S.; Maloney, V.K.; Crolla, J.A.; Baralle, D. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008, 359, 1685–1699. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Tewes, A.-C.; Rall, K.K.; Römer, T.; Hucke, J.; Kapczuk, K.; Brucker, S.; Wieacker, P.; Ledig, S. Variations in RBM8A and TBX6 are associated with disorders of the müllerian ducts. Fertil. Steril. 2015, 103, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Ledig, S.; Wieacker, P. Clinical and genetic aspects of Mayer–Rokitansky–Küster–Hauser syndrome. Med. Genet. 2018, 30, 3–11. [Google Scholar] [CrossRef]
- Zou, D.; McSweeney, C.; Sebastian, A.; Reynolds, D.J.; Dong, F.; Zhou, Y.; Deng, D.; Wang, Y.; Liu, L.; Zhu, J.; et al. A critical role of RBM8a in proliferation and differentiation of embryonic neural progenitors. Neural Dev. 2015, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Alachkar, A.; Jiang, D.; Harrison, M.; Zhou, Y.; Chen, G.; Mao, Y. An EJC factor RBM8a Regulates Anxiety Behaviors. Curr. Mol. Med. 2013, 13, 887–899. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, C.; Dong, F.; Chen, M.; Vitale, J.; Xu, L.; Crowley, N.; Luscher, B.; Zou, D.; Mao, Y. Full function of exon junction complex factor, Rbm8a, is critical for interneuron development. Trans. Psychiatr. 2020, 10, 379. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Pilaz, L.-J.; McMahon, J.J.; Golzio, C.; Wu, D.; Shi, L.; Katsanis, N.; Silver, D.L. Rbm8a haploinsufficiency disrupts embryonic cortical development resulting in microcephaly. J. Neurosci. 2015, 35, 7003–7018. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-H.; Liao, W.-J.; Ke, W.-C.; Yang, R.-B.; Tarn, W.-Y. The Y14-p53 regulatory circuit in megakaryocyte differentiation and thrombocytopenia. iScience 2021, 24, 103368. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; McMahon, J.J.; Tsai, Y.-H.; Wang, Z.; Silver, D.L. Haploinsufficiency for core exon junction complex components disrupts embryonic neurogenesis and causes p53-mediated microcephaly. PLoS Genet. 2016, 12, e1006282. [Google Scholar] [CrossRef]
- Sheehan, C.J.; McMahon, J.J.; Serdar, L.D.; Silver, D.L. Dosage-dependent requirements of Magoh for cortical interneuron generation and survival. Development 2020, 147, dev182295. [Google Scholar] [CrossRef]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schütz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2019, 36, 2628–2629. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2018, 47, D330–D338. [Google Scholar] [CrossRef]
- Consortium, T.G.O. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2020, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pleasure, S.J.; Collins, A.E.; Noebels, J.L.; Naya, F.J.; Tsai, M.-J.; Lowenstein, D.H. Loss of BETA2/NeuroD leads to malformation of the dentate gyrus and epilepsy. Proc. Natl. Acad. Sci. USA 2000, 97, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pereira, F.A.; Price, S.D.; Chu, M.-j.; Shope, C.; Himes, D.; Eatock, R.A.; Brownell, W.E.; Lysakowski, A.; Tsai, M.-J. Essential role of BETA2/NeuroD1 in development of the vestibular and auditory systems. Genes Dev. 2000, 14, 2839–2854. [Google Scholar] [CrossRef] [PubMed]
- Boutin, C.; Hardt, O.; de Chevigny, A.; Coré, N.; Goebbels, S.; Seidenfaden, R.; Bosio, A.; Cremer, H. NeuroD1 induces terminal neuronal differentiation in olfactory neurogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhang, L.; Wu, Z.; Chen, Y.; Wang, F.; Chen, G. In Vivo Direct Reprogramming of Reactive Glial Cells into Functional Neurons after Brain Injury and in an Alzheimer’s Disease Model. Cell Stem Cell 2014, 14, 188–202. [Google Scholar] [CrossRef]
- Puls, B.; Ding, Y.; Zhang, F.; Pan, M.; Lei, Z.; Pei, Z.; Jiang, M.; Bai, Y.; Forsyth, C.; Metzger, M.; et al. Regeneration of Functional Neurons After Spinal Cord Injury via in situ NeuroD1-Mediated Astrocyte-to-Neuron Conversion. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Boschian, C.; Messina, A.; Bozza, A.; Castellini, M.E.; Provenzano, G.; Bozzi, Y.; Casarosa, S. Impaired Neuronal Differentiation of Neural Stem Cells Lacking the Engrailed-2 Gene. Neuroscience 2018, 386, 137–149. [Google Scholar] [CrossRef]
- Millen, K.J.; Wurst, W.; Herrup, K.; Joyner, A.L. Abnormal embryonic cerebellar development and patterning of postnatal foliation in two mouse Engrailed-2 mutants. Development 1994, 120, 695–706. [Google Scholar] [CrossRef]
- Hanks, M.; Wurst, W.; Anson-Cartwright, L.; Auerbach, A.B.; Joyner, A.L. Rescue of the En-1 Mutant Phenotype by Replacement of En-1 with En-2. Science 1995, 269, 679–682. [Google Scholar] [CrossRef]
- Richardson, S.J.; Wijayagunaratne, R.C.; D’Souza, D.G.; Darras, V.M.; Van Herck, S.L.J. Transport of thyroid hormones via the choroid plexus into the brain: The roles of transthyretin and thyroid hormone transmembrane transporters. Front. Neurosci. 2015, 9, 66. [Google Scholar] [CrossRef]
- Park, G.Y.; Jamerlan, A.; Shim, K.H.; An, S.S.A. Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases. Int. J. Mol. Sci. 2019, 20, 2982. [Google Scholar] [CrossRef] [PubMed]
- Gennet, N.; Tamburini, C.; Nan, X.; Li, M. FolR1: A novel cell surface marker for isolating midbrain dopamine neural progenitors and nascent dopamine neurons. Sci. Rep. 2016, 6, 32488. [Google Scholar] [CrossRef] [PubMed]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Fujii, T.; Imao, T.; Ishihara, K.; Kuba, H.; Nagata, S. Calcium-dependent Phospholipid Scramblase Activity of TMEM16 Protein Family Members. J. Biol. Chem. 2013, 288, 13305–13316. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Yumoto, T.; Sakakibara, S.-i. Expression profiles of inka2 in the murine nervous system. Gene Expr. Patterns 2015, 19, 83–97. [Google Scholar] [CrossRef]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.H.; Ikawa, M.; Ito, A.; Okabe, M.; Nojima, H. Cyclin G1 is involved in G2/M arrest in response to DNA damage and in growth control after damage recovery. Oncogene 2001, 20, 3290–3300. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Ohki, R.; Shibata, T.; Tsutsumi, S.; Kamimura, N.; Inazawa, J.; Ohta, T.; Ichikawa, H.; Aburatani, H.; Tashiro, F.; et al. PH Domain-Only Protein PHLDA3 Is a p53-Regulated Repressor of Akt. Cell 2009, 136, 535–550. [Google Scholar] [CrossRef]
- Ding, B.; Parmigiani, A.; Divakaruni, A.S.; Archer, K.; Murphy, A.N.; Budanov, A.V. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death. Sci. Rep. 2016, 6, 22538. [Google Scholar] [CrossRef]
- Brosh, R.; Sarig, R.; Natan, E.B.; Molchadsky, A.; Madar, S.; Bornstein, C.; Buganim, Y.; Shapira, T.; Goldfinger, N.; Paus, R.; et al. p53-dependent transcriptional regulation of EDA2R and its involvement in chemotherapy-induced hair loss. FEBS Lett. 2010, 584, 2473–2477. [Google Scholar] [CrossRef]
- Zhao, Y.; Marín, O.; Hermesz, E.; Powell, A.; Flames, N.; Palkovits, M.; Rubenstein, J.L.R.; Westphal, H. The LIM-homeobox gene Lhx8 is required for the development of many cholinergic neurons in the mouse forebrain. Proc. Natl. Acad. Sci. USA 2003, 100, 9005–9010. [Google Scholar] [CrossRef] [PubMed]
- Fragkouli, A.; van Wijk, N.V.; Lopes, R.; Kessaris, N.; Pachnis, V. LIM homeodomain transcription factor-dependent specification of bipotential MGE progenitors into cholinergic and GABAergic striatal interneurons. Development 2009, 136, 3841–3851. [Google Scholar] [CrossRef] [PubMed]
- Flandin, P.; Zhao, Y.; Vogt, D.; Jeong, J.; Long, J.; Potter, G.; Westphal, H.; Rubenstein, J.L.R. Lhx6 and Lhx8 Coordinately Induce Neuronal Expression of Shh that Controls the Generation of Interneuron Progenitors. Neuron 2011, 70, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.L.; Boddeke, H.W.G.M.; et al. Glioma-Associated Microglia/Macrophages Display an Expression Profile Different from M1 and M2 Polarization and Highly Express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Oishi, T.; Yamashita, A.; Murata, Y.; Yamamoto, T.; Takashima, I.; Isa, T.; Higo, N. Neuronal and microglial localization of secreted phosphoprotein 1 (osteopontin) in intact and damaged motor cortex of macaques. Brain Res. 2019, 1714, 52–64. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e1296. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Graham, L.C.; Reagan, A.M.; Grabowska, W.A.; Schott, W.H.; Howell, G.R. Transcriptome profiling of brain myeloid cells revealed activation of Itgal, Trem1, and Spp1 in western diet-induced obesity. J. Neuroinflamm. 2019, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Qiu, Y.; Wight, A.E.; Kim, H.-J.; Cantor, H. Definition of a mouse microglial subset that regulates neuronal development and proinflammatory responses in the brain. Proc. Natl. Acad. Sci. USA 2022, 119, e2116241119. [Google Scholar] [CrossRef]
- Zhang, Z.; Shively, J.E. Acceleration of Bone Repair in NOD/SCID Mice by Human Monoosteophils, Novel LL-37-Activated Monocytes. PLoS ONE 2013, 8, e67649. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McSweeney, C.; Chen, M.; Dong, F.; Sebastian, A.; Reynolds, D.J.; Mott, J.; Pei, Z.; Zou, J.; Shi, Y.; Mao, Y. Transcriptomic Analyses of Brains of RBM8A Conditional Knockout Mice at Different Developmental Stages Reveal Conserved Signaling Pathways Contributing to Neurodevelopmental Diseases. Int. J. Mol. Sci. 2023, 24, 4600. https://doi.org/10.3390/ijms24054600
McSweeney C, Chen M, Dong F, Sebastian A, Reynolds DJ, Mott J, Pei Z, Zou J, Shi Y, Mao Y. Transcriptomic Analyses of Brains of RBM8A Conditional Knockout Mice at Different Developmental Stages Reveal Conserved Signaling Pathways Contributing to Neurodevelopmental Diseases. International Journal of Molecular Sciences. 2023; 24(5):4600. https://doi.org/10.3390/ijms24054600
Chicago/Turabian StyleMcSweeney, Colleen, Miranda Chen, Fengping Dong, Aswathy Sebastian, Derrick James Reynolds, Jennifer Mott, Zifei Pei, Jizhong Zou, Yongsheng Shi, and Yingwei Mao. 2023. "Transcriptomic Analyses of Brains of RBM8A Conditional Knockout Mice at Different Developmental Stages Reveal Conserved Signaling Pathways Contributing to Neurodevelopmental Diseases" International Journal of Molecular Sciences 24, no. 5: 4600. https://doi.org/10.3390/ijms24054600
APA StyleMcSweeney, C., Chen, M., Dong, F., Sebastian, A., Reynolds, D. J., Mott, J., Pei, Z., Zou, J., Shi, Y., & Mao, Y. (2023). Transcriptomic Analyses of Brains of RBM8A Conditional Knockout Mice at Different Developmental Stages Reveal Conserved Signaling Pathways Contributing to Neurodevelopmental Diseases. International Journal of Molecular Sciences, 24(5), 4600. https://doi.org/10.3390/ijms24054600