
Glucocorticoid-Responsive Tissue Plasminogen Activator (tPA) and Its Inhibitor Plasminogen Activator Inhibitor-1 (PAI-1): Relevance in Stress-Related Psychiatric Disorders


Abstract
1. Introduction
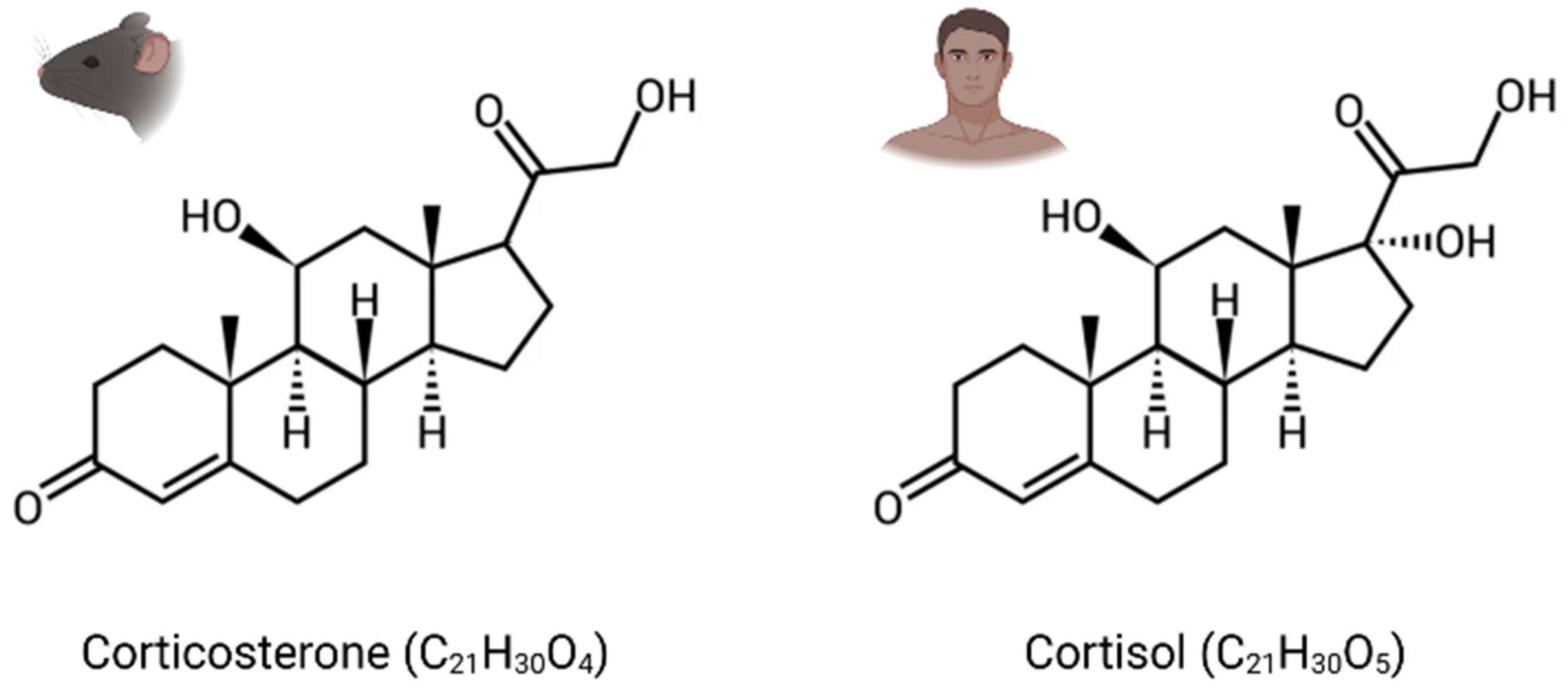
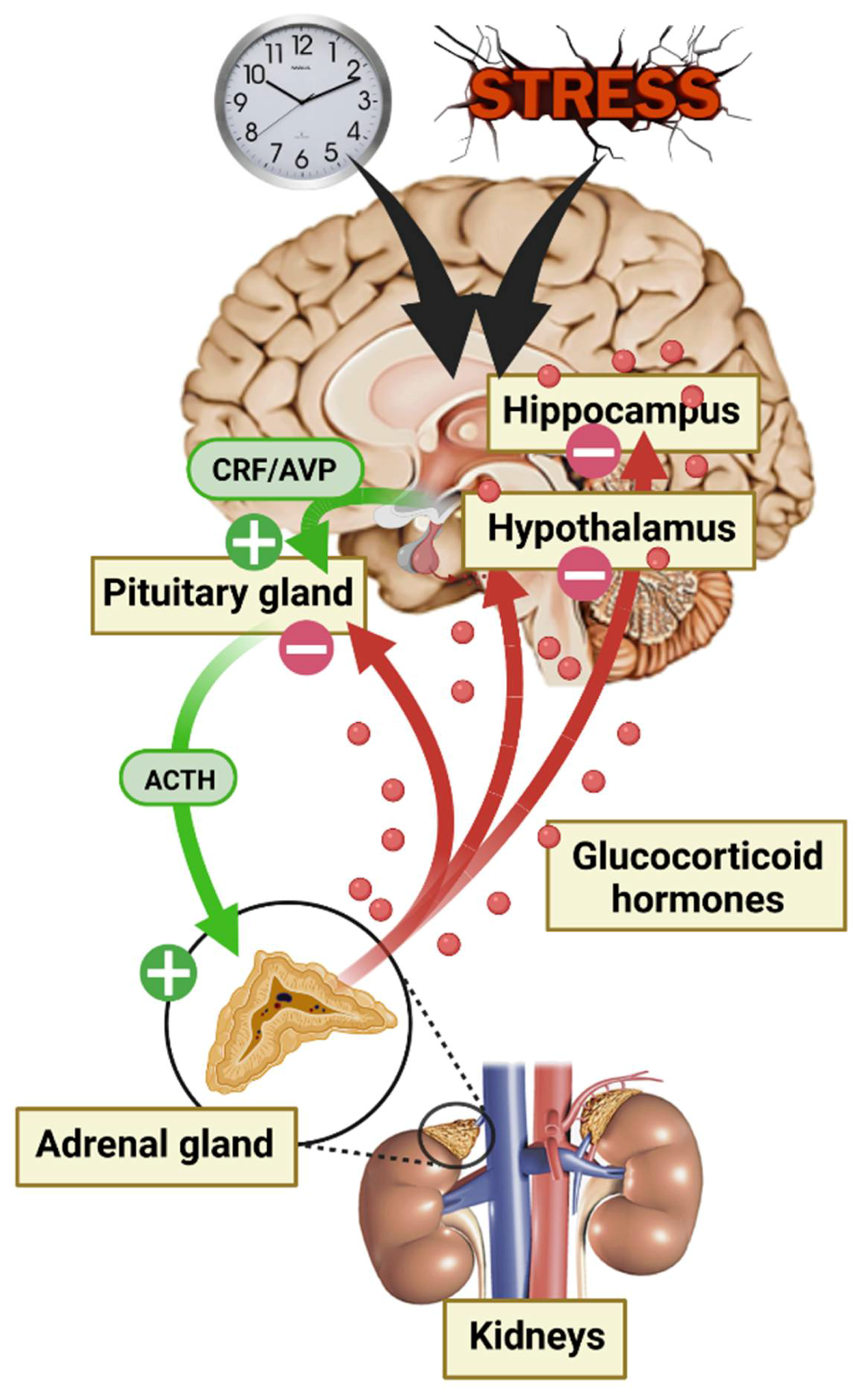
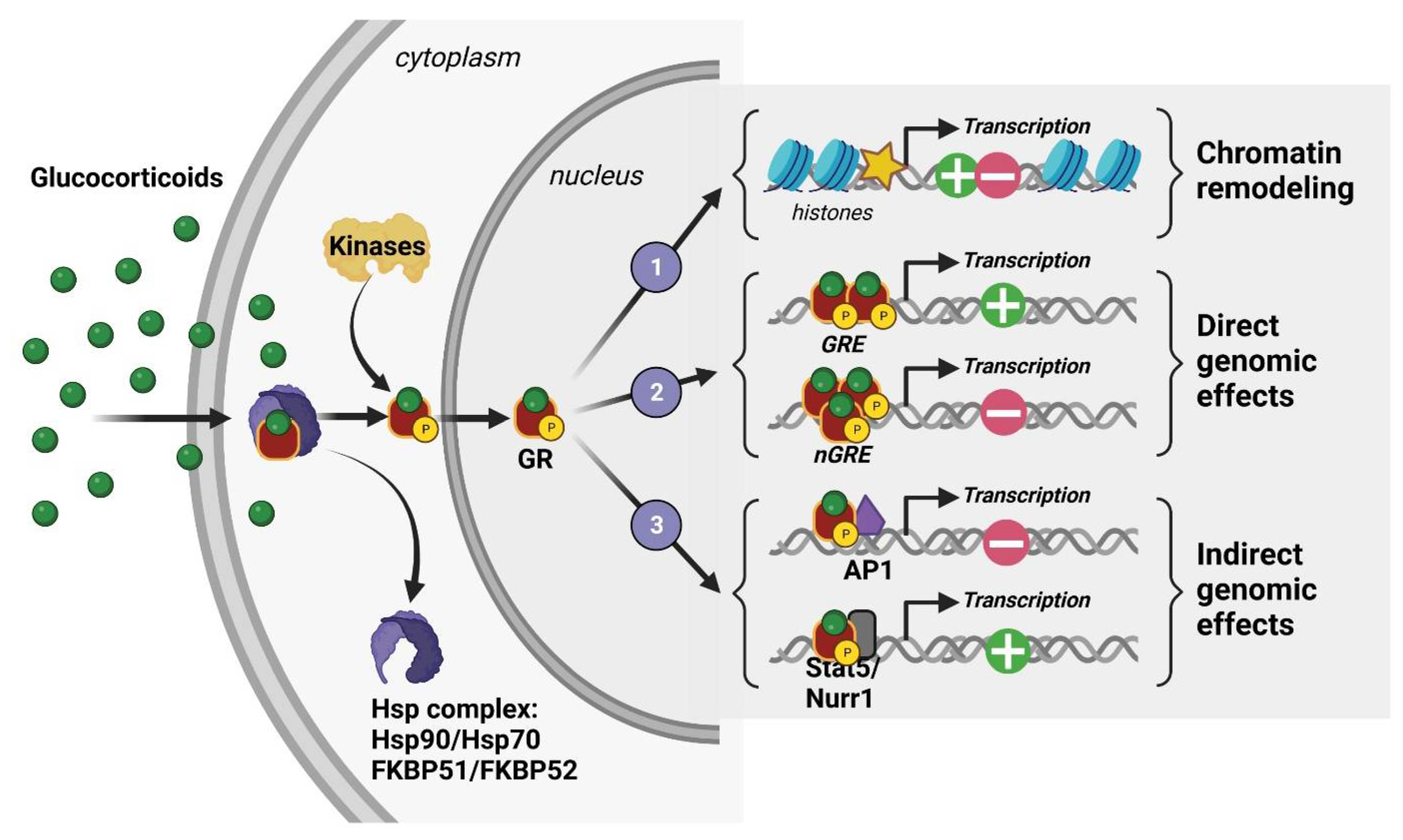
2. Glucocorticoid Hormones (GC), Their Receptors, and Mechanism of Action
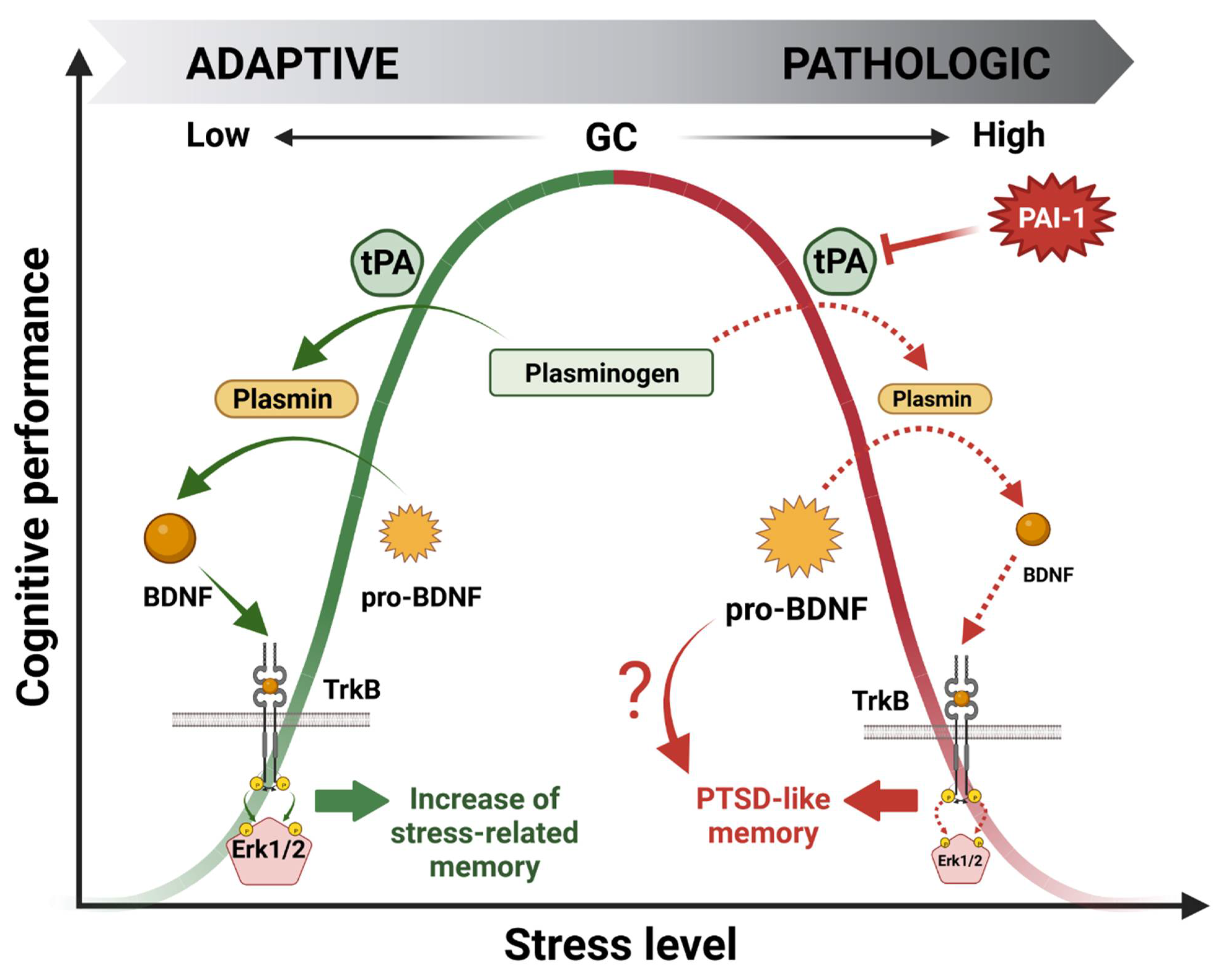
3. GMES Signaling Cascade in the HPC and Bell-Shaped Pattern of GC Effects on Cognitive Processes
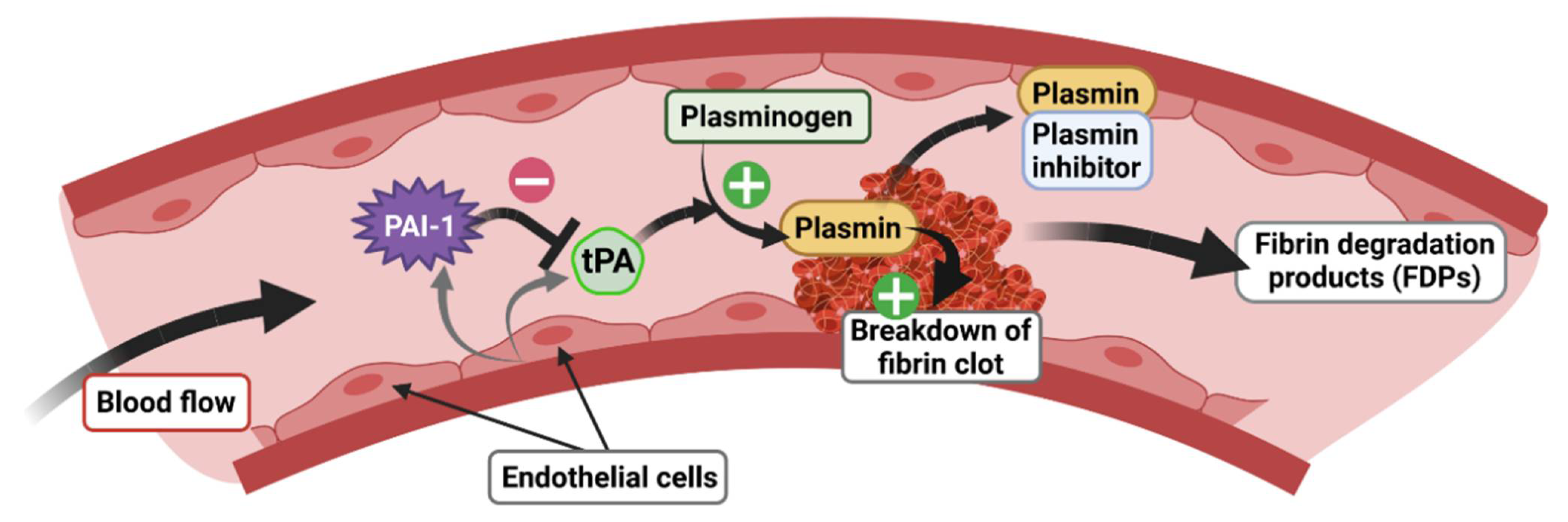
4. The tPA/PAI-1 System in Health and Diseases
4.1. tPA and Its Inhibitor PAI-1: Structure, Function, and Expression Pattern
4.2. The tPA/PAI-1 System Outside the CNS
4.3. Preclinical Studies Highlighting the tPA/PAI-1 System in the CNS
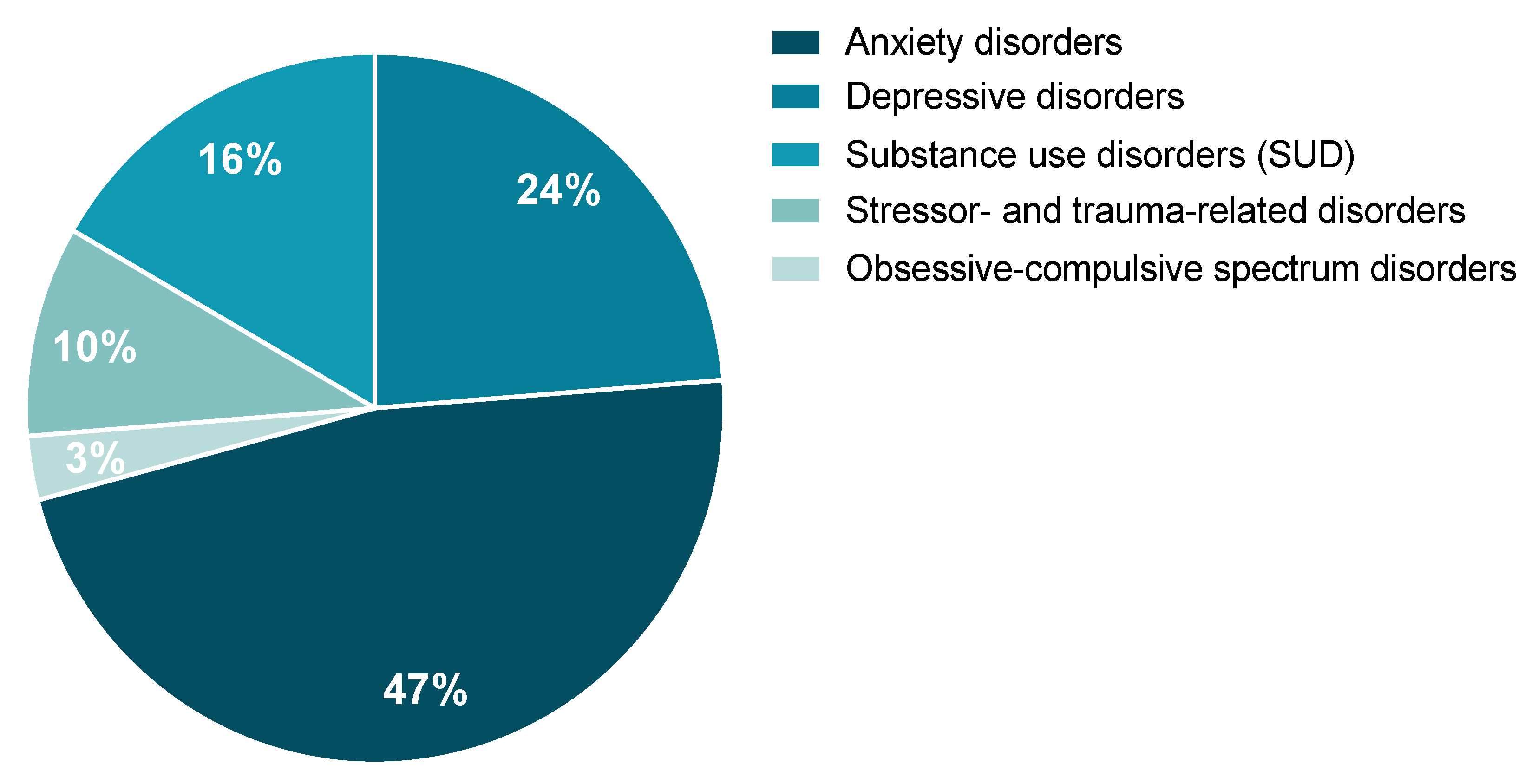
4.4. Clinical Studies Highlighting the Involvement of tPA/PAI-1 System in Stress-Related Psychiatric Disorders
4.4.1. Anxiety Disorders
4.4.2. Depressive Disorders
4.4.3. Substance Use Disorders (SUD)
4.4.4. Obsessive-Compulsive Spectrum Disorders
4.4.5. Stressor- and Trauma-Related Disorders

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorders | tPA | PAI-1 | BDNF | Cohorts Tested | n (n/Sex) | Age (Years) | Population | References |
|---|---|---|---|---|---|---|---|---|
| Anxiety | ↗ | ↗ | nt | PD (with agoraphobia) & SP | 29 (21 W/8 M) | 38.1 ± 10.5 | Germany | [129] Geiser et al., 2008 |
| → | → | → | PD | 30 (15 W/15 M) | 35.5 ± 12.38 | Chinese | [130] Chen et al., 2017 | |
| Depression | nt | ↗ | nt | MDD | 45 (45 W) | 37 ± 6.8 | American | [138] Eskandari et al., 2005 |
| → | ↗ | nt | Depressive men | 49 (49 M) | 40–65 | French | [140] Lahlou-Laforet et al., 2006 | |
| nt | ↗ | ↘ | MDD | 17 (14 W/3 M) | 48 ± 12 | Chinese | [135] Jiang et al., 2017 | |
| → | → | → | MDD | 30 (25 W/5 M) | 41.60 ± 12.43 | Chinese | [130] Chen et al., 2017 | |
| → | ↗ | nt | First-episode depression | 44 (21 W/23 M) | 25.89 ± 1.116 | Chinese | [139] Han et al., 2019 | |
| SUD | ↗ | ↗ | nt | Alcoholic before vs. after withdrawal | 10 (10 M) | 27–55 | French | [145] Delahousse et al., 2001 |
| PTSD | nt | ↗ | nt | PTSD (high severity score) | 40 (27 W/13 M) | 44.9 ± 3.5 | American | [155] Farr et al., 2015 |
| ↗ | nt | ↘ | PTSD sexually abused child | 45 (44 W/45 M) | 14.7 ± 2.5 | Turkish | [153] Aksu et al., 2018 | |
| ↗ | → | → | PTSD | 20 (11 W/9 M) | 41.5 ± 11 | Ireland | [154] Maguire et al., 2021 |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Konkle, B.A.; Schuster, S.J.; Kelly, M.D.; Harjes, K.; Hassett, D.E.; Bohrer, M.; Tavassoli, M. Plasminogen Activator Inhibitor-1 Messenger RNA Expression Is Induced in Rat Hepatocytes in Vivo by Dexamethasone. Blood 1992, 79, 2636–2642. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takeshita, K.; Shimokawa, T.; Yi, H.; Isobe, K.; Loskutoff, D.J.; Saito, H. Plasminogen Activator Inhibitor-1 Is a Major Stress-Regulated Gene: Implications for Stress-Induced Thrombosis in Aged Individuals. Proc. Natl. Acad. Sci. USA 2002, 99, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Bruzdzinski, C.J.; Riordan-Johnson, M.; Nordby, E.C.; Suter, S.M.; Gelehrter, T.D. Isolation and Characterization of the Rat Plasminogen Activator Inhibitor-1 Gene. J. Biol. Chem. 1990, 265, 2078–2085. [Google Scholar] [CrossRef] [PubMed]
- Bruzdzinski, C.J.; Johnson, M.R.; Goble, C.A.; Winograd, S.S.; Gelehrter, T.D. Mechanism of Glucocorticoid Induction of the Rat Plasminogen Activator Inhibitor-1 Gene in HTC Rat Hepatoma Cells: Identification of Cis-Acting Regulatory Elements. Mol. Endocrinol. 1993, 7, 1169–1177. [Google Scholar] [CrossRef][Green Version]
- Bulens, F.; Merchiers, P.; Ibañez-Tallon, I.; De Vriese, A.; Nelles, L.; Claessens, F.; Belayew, A.; Collen, D. Identification of a Multihormone Responsive Enhancer Far Upstream from the Human Tissue-Type Plasminogen Activator Gene. J. Biol. Chem. 1997, 272, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Kathju, S.; Heaton, J.H.; Bruzdzinski, C.J.; Gelehrter, T.D. Synergistic Induction of Tissue-Type Plasminogen Activator Gene Expression by Glucocorticoids and Cyclic Nucleotides in Rat HTC Hepatoma Cells. Endocrinology 1994, 135, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Xu, L.; Yu, S.; Hong, W.; Huang, M.; Xu, P. Therapeutics Targeting the Fibrinolytic System. Exp. Mol. Med. 2020, 52, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Wittchen, H.U.; Jacobi, F.; Rehm, J.; Gustavsson, A.; Svensson, M.; Jönsson, B.; Olesen, J.; Allgulander, C.; Alonso, J.; Faravelli, C.; et al. The Size and Burden of Mental Disorders and Other Disorders of the Brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 655–679. [Google Scholar] [CrossRef]
- Gustavsson, A.; Svensson, M.; Jacobi, F.; Allgulander, C.; Alonso, J.; Beghi, E.; Dodel, R.; Ekman, M.; Faravelli, C.; Fratiglioni, L.; et al. Cost of Disorders of the Brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 718–779. [Google Scholar] [CrossRef]
- Chrousos, G.P. Stress and Disorders of the Stress System. Nat. Rev. Endocrinol. 2009, 5, 374–381. [Google Scholar] [CrossRef]
- Agorastos, A.; Chrousos, G.P. The Neuroendocrinology of Stress: The Stress-Related Continuum of Chronic Disease Development. Mol. Psychiatry 2022, 27, 502–513. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; de Jong, I.E.; Oitzl, M.S. Neuropharmacology of Glucocorticoids: Focus on Emotion, Cognition and Cocaine. Eur. J. Pharmacol. 2008, 585, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Finsterwald, C.; Alberini, C.M. Stress and Glucocorticoid Receptor-Dependent Mechanisms in Long-Term Memory: From Adaptive Responses to Psychopathologies. Neurobiol. Learn. Mem. 2014, 112, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Holsboer, F. The Corticosteroid Receptor Hypothesis of Depression. Neuropsychopharmacology 2000, 23, 477–501. [Google Scholar] [CrossRef]
- Kaouane, N.; Porte, Y.; Vallée, M.; Brayda-Bruno, L.; Mons, N.; Calandreau, L.; Marighetto, A.; Piazza, P.V.; Desmedt, A. Glucocorticoids Can Induce PTSD-like Memory Impairments in Mice. Science 2012, 335, 1510–1513. [Google Scholar] [CrossRef] [PubMed]
- Barik, J.; Marti, F.; Morel, C.; Fernandez, S.P.; Lanteri, C.; Godeheu, G.; Tassin, J.P.; Mombereau, C.; Faure, P.; Tronche, F. Chronic Stress Triggers Social Aversion via Glucocorticoid Receptor in Dopaminoceptive Neurons. Science 2013, 339, 332–335. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. The Neurobiology of Stress: From Serendipity to Clinical Relevance. Brain Res. 2000, 886, 172–189. [Google Scholar] [CrossRef]
- Piazza, P.V.; Le Moal, M. The Role of Stress in Drug Self-Administration. Trends Pharmacol. Sci. 1998, 19, 67–74. [Google Scholar] [CrossRef]
- McGaugh, J.L.; Roozendaal, B. Role of Adrenal Stress Hormones in Forming Lasting Memories in the Brain. Curr. Opin. Neurobiol. 2002, 12, 205–210. [Google Scholar] [CrossRef]
- Dalm, S.; Enthoven, L.; Meijer, O.C.; van der Mark, M.H.; Karssen, A.M.; De Kloet, E.R.; Oitzl, M.S. Age-Related Changes in Hypothalamic-Pituitary-Adrenal Axis Activity of Male C57BL/6J Mice. Neuroendocrinology 2005, 81, 372–380. [Google Scholar] [CrossRef]
- Deppermann, S.; Storchak, H.; Fallgatter, A.J.; Ehlis, A.C. Stress-Induced Neuroplasticity: (Mal)Adaptation to Adverse Life Events in Patients with PTSD—A Critical Overview. Neuroscience 2014, 283, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Joels, M. Corticosteroid Effects in the Brain: U-Shape It. Trends Pharmacol. Sci. 2006, 27, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Diamond, D.M.; Campbell, A.M.; Park, C.R.; Halonen, J.; Zoladz, P.R. The Temporal Dynamics Model of Emotional Memory Processing: A Synthesis on the Neurobiological Basis of Stress-Induced Amnesia, Flashbulb and Traumatic Memories, and the Yerkes-Dodson Law. Neural Plast. 2007. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, L.; Joëls, M.; Roozendaal, B.; Wolf, O.T.; Oitzl, M.S. Stress Effects on Memory: An Update and Integration. Neurosci. Biobehav. Rev. 2012, 36, 1740–1749. [Google Scholar] [CrossRef]
- Revest, J.-M.; Di Blasi, F.; Kitchener, P.; Rougé-Pont, F.; Desmedt, A.; Turiault, M.; Tronche, F.; Piazza, P.V. The MAPK Pathway and Egr-1 Mediate Stress-Related Behavioral Effects of Glucocorticoids. Nat. Neurosci. 2005, 8, 664–672. [Google Scholar] [CrossRef]
- Revest, J.-M.; Kaouane, N.; Mondin, M.; Le Roux, A.; Rougé-Pont, F.; Vallée, M.; Barik, J.; Tronche, F.; Desmedt, A.; Piazza, P.V. The Enhancement of Stress-Related Memory by Glucocorticoids Depends on Synapsin-Ia/Ib. Mol. Psychiatry 2010, 15, 1125, 1140–1151. [Google Scholar] [CrossRef]
- Revest, J.-M.; Le Roux, A.; Roullot-Lacarrière, V.; Kaouane, N.; Vallée, M.; Kasanetz, F.; Rougé-Pont, F.; Tronche, F.; Desmedt, A.; Piazza, P.V. BDNF-TrkB Signaling through Erk1/2 MAPK Phosphorylation Mediates the Enhancement of Fear Memory Induced by Glucocorticoids. Mol. Psychiatry 2014, 19, 1001–1009. [Google Scholar] [CrossRef]
- Sarrazin, N.; Di Blasi, F.; Roullot-Lacarrière, V.; Rougé-Pont, F.; Le Roux, A.; Costet, P.; Revest, J.-M.; Piazza, P.V. Transcriptional Effects of Glucocorticoid Receptors in the Dentate Gyrus Increase Anxiety-Related Behaviors. PLoS ONE 2009, 4, e7704. [Google Scholar] [CrossRef]
- Bouarab, C.; Roullot-Lacarrière, V.; Vallée, M.; Le Roux, A.; Guette, C.; Mennesson, M.; Marighetto, A.; Desmedt, A.; Piazza, P.V.; Revest, J.M. PAI-1 Protein Is a Key Molecular Effector in the Transition from Normal to PTSD-like Fear Memory. Mol. Psychiatry 2021, 26, 4968–4981. [Google Scholar] [CrossRef]
- McEwen, B.S.; De Kloet, E.R.; Rostene, W. Adrenal Steroid Receptors and Actions in the Nervous System. Physiol. Rev. 1986, 66, 1121–1188. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joëls, M. Brain Corticosteroid Receptor Balance in Health and Disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.M.; Sutanto, W.; van Eekelen, J.A.; Rothuizen, J.; de Kloet, E.R. Central Action of Adrenal Steroids during Stress and Adaptation. Adv. Exp. Med. Biol. 1990, 274, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Dallman, M.F.; Akana, S.F.; Cascio, C.S.; Darlington, D.N.; Jacobson, L.; Levin, N. Regulation of ACTH Secretion: Variations on a Theme of B. Recent Prog. Horm. Res. 1987, 43, 113–173. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Mietus, L.J. Transport of Steroid Hormones through the Rat Blood-Brain Barrier. Primary Role of Albumin-Bound Hormone. J. Clin. Investig. 1979, 64, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M. Glucocorticoids and Hippocampal Atrophy in Neuropsychiatric Disorders. Arch. Gen. Psychiatry 2000, 57, 925–935. [Google Scholar] [CrossRef]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid Receptor Control of Transcription: Precision and Plasticity via Allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, E.R.; Joëls, M. The Cortisol Switch between Vulnerability and Resilience. Mol. Psychiatry, 2023; online ahead of print. [Google Scholar] [CrossRef]
- Pujols, L.; Mullol, J.; Roca-Ferrer, J.; Torrego, A.; Xaubet, A.; Cidlowski, J.A.; Picado, C. Expression of Glucocorticoid Receptor Alpha- and Beta-Isoforms in Human Cells and Tissues. Am. J. Physiol. Cell Physiol. 2002, 283, C1324–C1331. [Google Scholar] [CrossRef]
- Robertson, D.A.; Beattie, J.E.; Reid, I.C.; Balfour, D.J. Regulation of Corticosteroid Receptors in the Rat Brain: The Role of Serotonin and Stress. Eur. J. Neurosci. 2005, 21, 1511–1520. [Google Scholar] [CrossRef]
- Segal, M.; Richter-Levin, G.; Maggio, N. Stress-Induced Dynamic Routing of Hippocampal Connectivity: A Hypothesis. Hippocampus 2010, 20, 1332–1338. [Google Scholar] [CrossRef]
- Sala, M.; Perez, J.; Soloff, P.; Ucelli di Nemi, S.; Caverzasi, E.; Soares, J.C.; Brambilla, P. Stress and Hippocampal Abnormalities in Psychiatric Disorders. Eur. Neuropsychopharmacol. 2004, 14, 393–405. [Google Scholar] [CrossRef]
- Hsiao, P.-W.; Deroo, B.J.; Archer, T.K. Chromatin Remodeling and Tissue-Selective Responses of Nuclear Hormone Receptors. Biochem. Cell Biol. 2002, 80, 343–351. [Google Scholar] [CrossRef]
- Thomassin, H. Glucocorticoid-Induced DNA Demethylation and Gene Memory during Development. EMBO J. 2001, 20, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Beato, M. Gene Regulation by Steroid Hormones. Cell 1989, 56, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Beato, M.; Herrlich, P.; Schutz, G. Steroid Hormone Receptors: Many Actors in Search of a Plot. Cell 1995, 83, 851–857. [Google Scholar] [CrossRef]
- Meaney, M.J. Maternal Care, Gene Expression, and the Transmission of Individual Differences in Stress Reactivity Across Generations. Annu. Rev. Neurosci. 2001, 24, 1161–1192. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. The Good Side of “Stress”. Stress 2019, 22, 524–525. [Google Scholar] [CrossRef] [PubMed]
- Roozendaal, B. Stress and Memory: Opposing Effects of Glucocorticoids on Memory Consolidation and Memory Retrieval. Neurobiol. Learn. Mem. 2002, 78, 578–595. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, E.R.; Oitzl, M.S.; Joëls, M. Stress and Cognition: Are Corticosteroids Good or Bad Guys? Trends Neurosci. 1999, 22, 422–426. [Google Scholar] [CrossRef]
- Yehuda, R.; Boisoneau, D.; Lowy, M.T.; Giller, E.L. Dose-Response Changes in Plasma Cortisol and Lymphocyte Glucocorticoid Receptors Following Dexamethasone Administration in Combat Veterans with and without Posttraumatic Stress Disorder. Arch. Gen. Psychiatry 1995, 52, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Finsterwald, C.; Steinmetz, A.B.; Travaglia, A.; Alberini, C.M. From Memory Impairment to Posttraumatic Stress Disorder-Like Phenotypes: The Critical Role of an Unpredictable Second Traumatic Experience. J. Neurosci. 2015, 35, 15903–15915. [Google Scholar] [CrossRef]
- Elzinga, B.M.; Schmahl, C.G.; Vermetten, E.; van Dyck, R.; Bremner, J.D. Higher Cortisol Levels Following Exposure to Traumatic Reminders in Abuse-Related PTSD. Neuropsychopharmacology 2003, 28, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.-Y.; Lee, F.S.; et al. ProBDNF Induces Neuronal Apoptosis via Activation of a Receptor Complex of P75NTR and Sortilin. J. Neurosci. 2005, 25, 5455–5463. [Google Scholar] [CrossRef] [PubMed]
- Woo, N.H.; Teng, H.K.; Siao, C.-J.; Chiaruttini, C.; Pang, P.T.; Milner, T.A.; Hempstead, B.L.; Lu, B. Activation of P75NTR by ProBDNF Facilitates Hippocampal Long-Term Depression. Nat. Neurosci. 2005, 8, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Pang, P.T.; Teng, H.K.; Zaitsev, E.; Woo, N.T.; Sakata, K.; Zhen, S.; Teng, K.K.; Yung, W.-H.; Hempstead, B.L.; Lu, B. Cleavage of ProBDNF by TPA/Plasmin Is Essential for Long-Term Hippocampal Plasticity. Science 2004, 306, 487–491. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Kim, J.-K.; Park, J.-S.; Byun, Y.; Kim, C.-K. The Use of PEGylated Liposomes to Prolong Circulation Lifetimes of Tissue Plasminogen Activator. Biomaterials 2009, 30, 5751–5756. [Google Scholar] [CrossRef]
- Sheehan, J.J.; Tsirka, S.E. Fibrin-Modifying Serine Proteases Thrombin, TPA, and Plasmin in Ischemic Stroke: A Review. Glia 2005, 50, 340–350. [Google Scholar] [CrossRef]
- Matys, T.; Pawlak, R.; Strickland, S. Tissue Plasminogen Activator in the Bed Nucleus of Stria Terminalis Regulates Acoustic Startle. Neuroscience 2005, 135, 715–722. [Google Scholar] [CrossRef]
- Zheng, S.; Yin, Z.Q.; Zeng, Y.X. Developmental Profile of Tissue Plasminogen Activator in Postnatal Long Evans Rat Visual Cortex. Mol. Vis. 2008, 14, 975–982. [Google Scholar]
- Mou, X.; Peterson, C.B.; Prosser, R.A. Tissue-Type Plasminogen Activator-Plasmin-BDNF Modulate Glutamate-Induced Phase-Shifts of the Mouse Suprachiasmatic Circadian Clock in Vitro. Eur. J. Neurosci. 2009, 30, 1451–1460. [Google Scholar] [CrossRef]
- Carroll, P.M.; Tsirka, S.E.; Richards, W.G.; Frohman, M.A.; Strickland, S. The Mouse Tissue Plasminogen Activator Gene 5’ Flanking Region Directs Appropriate Expression in Development and a Seizure-Enhanced Response in the CNS. Development 1994, 120, 3173–3183. [Google Scholar] [CrossRef]
- Sappino, A.P.; Madani, R.; Huarte, J.; Belin, D.; Kiss, J.Z.; Wohlwend, A.; Vassalli, J.D. Extracellular Proteolysis in the Adult Murine Brain. J. Clin. Investig. 1993, 92, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Lochner, J.E.; Honigman, L.S.; Grant, W.F.; Gessford, S.K.; Hansen, A.B.; Silverman, M.A.; Scalettar, B.A. Activity-Dependent Release of Tissue Plasminogen Activator from the Dendritic Spines of Hippocampal Neurons Revealed by Live-Cell Imaging. J. Neurobiol. 2006, 66, 564–577. [Google Scholar] [CrossRef]
- Salles, F.J.; Strickland, S. Localization and Regulation of the Tissue Plasminogen Activator-Plasmin System in the Hippocampus. J. Neurosci. 2002, 22, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Teesalu, T.; Kulla, A.; Asser, T.; Koskiniemi, M.; Vaheri, A. Tissue Plasminogen Activator as a Key Effector in Neurobiology and Neuropathology. Biochem. Soc. Trans. 2002, 30, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Li, Y.; Shen, L.H.; Liu, X.; Wang, X.; Zhang, J.; Pourabdollah-Nejad, D.S.; Zhang, C.; Zhang, L.; Jiang, H.; et al. Increasing TPA Activity in Astrocytes Induced by Multipotent Mesenchymal Stromal Cells Facilitate Neurite Outgrowth after Stroke in the Mouse. PLoS ONE 2010, 5, e9027. [Google Scholar] [CrossRef]
- Louessard, M.; Lacroix, A.; Martineau, M.; Mondielli, G.; Montagne, A.; Lesept, F.; Lambolez, B.; Cauli, B.; Mothet, J.-P.; Vivien, D.; et al. Tissue Plasminogen Activator Expression Is Restricted to Subsets of Excitatory Pyramidal Glutamatergic Neurons. Mol. Neurobiol. 2016, 53, 5000–5012. [Google Scholar] [CrossRef]
- Potempa, J.; Korzus, E.; Travis, J. The Serpin Superfamily of Proteinase Inhibitors: Structure, Function, and Regulation. J. Biol. Chem. 1994, 269, 15957–15960. [Google Scholar] [CrossRef]
- Gettins, P.G.W. Serpin Structure, Mechanism, and Function. Chem. Rev. 2002, 102, 4751–4804. [Google Scholar] [CrossRef]
- Lee, T.W.; Tsang, V.W.K.; Birch, N.P. Physiological and Pathological Roles of Tissue Plasminogen Activator and Its Inhibitor Neuroserpin in the Nervous System. Front. Cell Neurosci. 2015, 9, 396. [Google Scholar] [CrossRef]
- Jankun, J.; Keck, R.; Selman, S.H.; Skrzypczak-Jankun, E. Systemic or Topical Application of Plasminogen Activator Inhibitor with Extended Half-Life (VLHL PAI-1) Reduces Bleeding Time and Total Blood Loss. Int. J. Mol. Med. 2010, 26, 501–504. [Google Scholar] [CrossRef][Green Version]
- Zhou, A.; Huntington, J.A.; Pannu, N.S.; Carrell, R.W.; Read, R.J. How Vitronectin Binds PAI-1 to Modulate Fibrinolysis and Cell Migration. Nat. Struct. Biol. 2003, 10, 541–544. [Google Scholar] [CrossRef]
- Sawdey, M.S.; Loskutoff, D.J. Regulation of Murine Type 1 Plasminogen Activator Inhibitor Gene Expression in Vivo. Tissue Specificity and Induction by Lipopolysaccharide, Tumor Necrosis Factor-Alpha, and Transforming Growth Factor-Beta. J. Clin. Investig. 1991, 88, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Ennas, M.G.; Torelli, S.; Ragnotti, G.; Gremo, F. Synthesis of Urokinase-Type Plasminogen Activator and of Type-1 Plasminogen Activator Inhibitor in Neuronal Cultures of Human Fetal Brain: Stimulation by Phorbol Ester. J. Neurochem. 1990, 55, 1647–1654. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Lu, D.; Paddy, M.R.; Lenk, S.E.; Raizada, M.K. Angiotensin II Regulation of Plasminogen Activator Inhibitor-1 Gene Expression in Neurons of Normotensive and Spontaneously Hypertensive Rat Brains. Endocrinology 1996, 137, 2503–2513. [Google Scholar] [CrossRef]
- Hino, H.; Akiyama, H.; Iseki, E.; Kato, M.; Kondo, H.; Ikeda, K.; Kosaka, K. Immunohistochemical Localization of Plasminogen Activator Inhibitor-1 in Rat and Human Brain Tissues. Neurosci. Lett. 2001, 297, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Docagne, F.; Nicole, O.; Marti, H.H.; MacKenzie, E.T.; Buisson, A.; Vivien, D. Transforming Growth Factor-Beta1 as a Regulator of the Serpins/t-PA Axis in Cerebral Ischemia. FASEB J. 1999, 13, 1315–1324. [Google Scholar] [CrossRef]
- Rydzewski, B.; Zelezna, B.; Tang, W.; Sumners, C.; Raizada, M.K. Angiotensin II Stimulation of Plasminogen Activator Inhibitor-1 Gene Expression in Astroglial Cells from the Brain. Endocrinology 1992, 130, 1255–1262. [Google Scholar] [CrossRef]
- Norris, E.H.; Strickland, S. Modulation of NR2B-Regulated Contextual Fear in the Hippocampus by the Tissue Plasminogen Activator System. Proc. Natl. Acad. Sci. USA 2007, 104, 13473–13478. [Google Scholar] [CrossRef]
- Nagai, T.; Kamei, H.; Ito, M.; Hashimoto, K.; Takuma, K.; Nabeshima, T.; Yamada, K. Modification by the Tissue Plasminogen Activator-Plasmin System of Morphine-Induced Dopamine Release and Hyperlocomotion, but Not Anti-Nociceptive Effect in Mice. J. Neurochem. 2005, 93, 1272–1279. [Google Scholar] [CrossRef]
- Singletary, J.H.; Chan, D.; Samani, N.J.; Chong, N.W. The Canonical E-Box Motif: A Target for Glucocorticoid Action That Drives Rhythmic Mouse Pai-1 Transcription in Vitro. Gene 2008, 420, 42–47. [Google Scholar] [CrossRef]
- Rouch, A.; Vanucci-Bacqué, C.; Bedos-Belval, F.; Baltas, M. Small Molecules Inhibitors of Plasminogen Activator Inhibitor-1—An Overview. Eur. J. Med. Chem. 2015, 92, 619–636. [Google Scholar] [CrossRef]
- Izuhara, Y.; Takahashi, S.; Nangaku, M.; Takizawa, S.; Ishida, H.; Kurokawa, K.; van Ypersele de Strihou, C.; Hirayama, N.; Miyata, T. Inhibition of Plasminogen Activator Inhibitor-1: Its Mechanism and Effectiveness on Coagulation and Fibrosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 672–677. [Google Scholar] [CrossRef]
- Izuhara, Y.; Yamaoka, N.; Kodama, H.; Dan, T.; Takizawa, S.; Hirayama, N.; Meguro, K.; van Ypersele de Strihou, C.; Miyata, T. A Novel Inhibitor of Plasminogen Activator Inhibitor-1 Provides Antithrombotic Benefits Devoid of Bleeding Effect in Nonhuman Primates. J. Cereb. Blood Flow Metab. 2010, 30, 904–912. [Google Scholar] [CrossRef]
- Pelisch, N.; Dan, T.; Ichimura, A.; Sekiguchi, H.; Vaughan, D.E.; van Ypersele de Strihou, C.; Miyata, T. Plasminogen Activator Inhibitor-1 Antagonist TM5484 Attenuates Demyelination and Axonal Degeneration in a Mice Model of Multiple Sclerosis. PLoS ONE 2015, 10, e0124510. [Google Scholar] [CrossRef]
- Sillen, M.; Declerck, P.J. Targeting PAI-1 in Cardiovascular Disease: Structural Insights Into PAI-1 Functionality and Inhibition. Front. Cardiovasc. Med. 2020, 7, 622473. [Google Scholar] [CrossRef]
- Al-Horani, R.A. Serpin Regulation of Fibrinolytic System: Implications for Therapeutic Applications in Cardiovascular Diseases. Cardiovasc. Hematol. Agents Med. Chem. 2014, 12, 91–125. [Google Scholar] [CrossRef] [PubMed]
- Morrow, G.B.; Whyte, C.S.; Mutch, N.J. A Serpin with a Finger in Many PAIs: PAI-1’s Central Function in Thromboinflammation and Cardiovascular Disease. Front. Cardiovasc. Med. 2021, 8, 653655. [Google Scholar] [CrossRef] [PubMed]
- Batiha, G.E.-S.; Al-Kuraishy, H.M.; Al-Maiahy, T.J.; Al-Buhadily, A.K.; Saad, H.M.; Al-Gareeb, A.I.; Simal-Gandara, J. Plasminogen Activator Inhibitor 1 and Gestational Diabetes: The Causal Relationship. Diabetol. Metab. Syndr. 2022, 14, 127. [Google Scholar] [CrossRef] [PubMed]
- Altalhi, R.; Pechlivani, N.; Ajjan, R.A. PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 3170. [Google Scholar] [CrossRef] [PubMed]
- Rabieian, R.; Boshtam, M.; Zareei, M.; Kouhpayeh, S.; Masoudifar, A.; Mirzaei, H. Plasminogen Activator Inhibitor Type-1 as a Regulator of Fibrosis. J. Cell Biochem. 2018, 119, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in Tissue Fibrosis. J. Cell Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.; Kwaan, H.C. The Plasminogen Activator System and Cancer. Pathophysiol. Haemost. Thromb. 2008, 36, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.A.; Shaker, B.T.; Bajou, K. The Plasminogen-Activator Plasmin System in Physiological and Pathophysiological Angiogenesis. Int. J. Mol. Sci. 2021, 23, 337. [Google Scholar] [CrossRef]
- Kubala, M.H.; DeClerck, Y.A. The Plasminogen Activator Inhibitor-1 Paradox in Cancer: A Mechanistic Understanding. Cancer Metastasis Rev. 2019, 38, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Goolaerts, A.; Lafargue, M.; Song, Y.; Miyazawa, B.; Arjomandi, M.; Carlès, M.; Roux, J.; Howard, M.; Parks, D.A.; Iles, K.E.; et al. PAI-1 Is an Essential Component of the Pulmonary Host Response during Pseudomonas Aeruginosa Pneumonia in Mice. Thorax 2011, 66, 788–796. [Google Scholar] [CrossRef]
- Lim, J.H.; Woo, C.-H.; Li, J.-D. Critical Role of Type 1 Plasminogen Activator Inhibitor (PAI-1) in Early Host Defense against Nontypeable Haemophilus Influenzae (NTHi) Infection. Biochem. Biophys. Res. Commun. 2011, 414, 67–72. [Google Scholar] [CrossRef][Green Version]
- Huang, P.; Zuo, Q.; Li, Y.; Oduro, P.K.; Tan, F.; Wang, Y.; Liu, X.; Li, J.; Wang, Q.; Guo, F.; et al. A Vicious Cycle: In Severe and Critically Ill COVID-19 Patients. Front. Immunol. 2022, 13, 930673. [Google Scholar] [CrossRef]
- Bliss, T.V.P.; Collingridge, G.L. A Synaptic Model of Memory: Long-Term Potentiation in the Hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Samson, A.L.; Medcalf, R.L. Tissue-Type Plasminogen Activator: A Multifaceted Modulator of Neurotransmission and Synaptic Plasticity. Neuron 2006, 50, 673–678. [Google Scholar] [CrossRef]
- Qian, Z.; Gilbert, M.E.; Colicos, M.A.; Kandel, E.R.; Kuhl, D. Tissue-Plasminogen Activator Is Induced as an Immediate-Early Gene during Seizure, Kindling and Long-Term Potentiation. Nature 1993, 361, 453–457. [Google Scholar] [CrossRef]
- Baranes, D.; Lederfein, D.; Huang, Y.Y.; Chen, M.; Bailey, C.H.; Kandel, E.R. Tissue Plasminogen Activator Contributes to the Late Phase of LTP and to Synaptic Growth in the Hippocampal Mossy Fiber Pathway. Neuron 1998, 21, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Madani, R.; Hulo, S.; Toni, N.; Madani, H.; Steimer, T.; Muller, D.; Vassalli, J.D. Enhanced Hippocampal Long-Term Potentiation and Learning by Increased Neuronal Expression of Tissue-Type Plasminogen Activator in Transgenic Mice. EMBO J. 1999, 18, 3007–3012. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Bach, M.E.; Lipp, H.P.; Zhuo, M.; Wolfer, D.P.; Hawkins, R.D.; Schoonjans, L.; Kandel, E.R.; Godfraind, J.M.; Mulligan, R.; et al. Mice Lacking the Gene Encoding Tissue-Type Plasminogen Activator Show a Selective Interference with Late-Phase Long-Term Potentiation in Both Schaffer Collateral and Mossy Fiber Pathways. Proc. Natl. Acad. Sci. USA 1996, 93, 8699–8704. [Google Scholar] [CrossRef] [PubMed]
- Seeds, N.W.; Williams, B.L.; Bickford, P.C. Tissue Plasminogen Activator Induction in Purkinje Neurons after Cerebellar Motor Learning. Science 1995, 270, 1992–1994. [Google Scholar] [CrossRef]
- Pawlak, R.; Nagai, N.; Urano, T.; Napiorkowska-Pawlak, D.; Ihara, H.; Takada, Y.; Collen, D.; Takada, A. Rapid, Specific and Active Site-Catalyzed Effect of Tissue-Plasminogen Activator on Hippocampus-Dependent Learning in Mice. Neuroscience 2002, 113, 995–1001. [Google Scholar] [CrossRef]
- Neuhoff, H.; Roeper, J.; Schweizer, M. Activity-Dependent Formation of Perforated Synapses in Cultured Hippocampal Neurons. Eur. J. Neurosci. 1999, 11, 4241–4250. [Google Scholar] [CrossRef]
- Haile, W.B.; Wu, J.; Echeverry, R.; Wu, F.; An, J.; Yepes, M. Tissue-Type Plasminogen Activator Has a Neuroprotective Effect in the Ischemic Brain Mediated by Neuronal TNF-α. J. Cereb. Blood Flow Metab. 2012, 32, 57–69. [Google Scholar] [CrossRef]
- Wu, F.; Wu, J.; Nicholson, A.D.; Echeverry, R.; Haile, W.B.; Catano, M.; An, J.; Lee, A.K.; Duong, D.; Dammer, E.B.; et al. Tissue-Type Plasminogen Activator Regulates the Neuronal Uptake of Glucose in the Ischemic Brain. J. Neurosci. 2012, 32, 9848–9858. [Google Scholar] [CrossRef]
- Wu, F.; Echeverry, R.; Wu, J.; An, J.; Haile, W.B.; Cooper, D.S.; Catano, M.; Yepes, M. Tissue-Type Plasminogen Activator Protects Neurons from Excitotoxin-Induced Cell Death via Activation of the ERK1/2-CREB-ATF3 Signaling Pathway. Mol. Cell Neurosci. 2013, 52, 9–19. [Google Scholar] [CrossRef]
- Tsirka, S.E.; Gualandris, A.; Amaral, D.G.; Strickland, S. Excitotoxin-Induced Neuronal Degeneration and Seizure Are Mediated by Tissue Plasminogen Activator. Nature 1995, 377, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Tsirka, S.E.; Strickland, S.; Stieg, P.E.; Soriano, S.G.; Lipton, S.A. Tissue Plasminogen Activator (TPA) Increases Neuronal Damage after Focal Cerebral Ischemia in Wild-Type and TPA-Deficient Mice. Nat. Med. 1998, 4, 228–231. [Google Scholar] [CrossRef]
- Nicole, O.; Docagne, F.; Ali, C.; Margaill, I.; Carmeliet, P.; MacKenzie, E.T.; Vivien, D.; Buisson, A. The Proteolytic Activity of Tissue-Plasminogen Activator Enhances NMDA Receptor-Mediated Signaling. Nat. Med. 2001, 7, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Flavin, M.P.; Zhao, G. Tissue Plasminogen Activator Protects Hippocampal Neurons from Oxygen-Glucose Deprivation Injury. J. Neurosci. Res. 2001, 63, 388–394. [Google Scholar] [CrossRef]
- Pawlak, R.; Magarinos, A.M.; Melchor, J.; McEwen, B.; Strickland, S. Tissue Plasminogen Activator in the Amygdala Is Critical for Stress-Induced Anxiety-like Behavior. Nat. Neurosci. 2003, 6, 168–174. [Google Scholar] [CrossRef]
- Matys, T.; Pawlak, R.; Matys, E.; Pavlides, C.; McEwen, B.S.; Strickland, S. Tissue Plasminogen Activator Promotes the Effects of Corticotropin-Releasing Factor on the Amygdala and Anxiety-like Behavior. Proc. Natl. Acad. Sci. USA 2004, 101, 16345–16350. [Google Scholar] [CrossRef]
- Pawlak, R.; Rao, B.S.S.; Melchor, J.P.; Chattarji, S.; McEwen, B.; Strickland, S. Tissue Plasminogen Activator and Plasminogen Mediate Stress-Induced Decline of Neuronal and Cognitive Functions in the Mouse Hippocampus. Proc. Natl. Acad. Sci. USA 2005, 102, 18201–18206. [Google Scholar] [CrossRef] [PubMed]
- Bennur, S.; Shankaranarayana Rao, B.S.; Pawlak, R.; Strickland, S.; McEwen, B.S.; Chattarji, S. Stress-Induced Spine Loss in the Medial Amygdala Is Mediated by Tissue-Plasminogen Activator. Neuroscience 2007, 144, 8–16. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takeshita, K.; Kojima, T.; Takamatsu, J.; Saito, H. Aging and Plasminogen Activator Inhibitor-1 (PAI-1) Regulation: Implication in the Pathogenesis of Thrombotic Disorders in the Elderly. Cardiovasc. Res. 2005, 66, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; de Leon, M.; de Santi, S.; Convit, A.; Tarshish, C.; Nair, N.P.; Thakur, M.; McEwen, B.S.; Hauger, R.L.; Meaney, M.J. Cortisol Levels during Human Aging Predict Hippocampal Atrophy and Memory Deficits. Nat. Neurosci. 1998, 1, 69–73. [Google Scholar] [CrossRef]
- Lupien, S.J.; Lepage, M. Stress, Memory, and the Hippocampus: Can’t Live with It, Can’t Live without It. Behav. Brain Res. 2001, 127, 137–158. [Google Scholar] [CrossRef]
- Räikkönen, K.; Lassila, R.; Keltikangas-Järvinen, L.; Hautanen, A. Association of Chronic Stress with Plasminogen Activator Inhibitor-1 in Healthy Middle-Aged Men. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Mausbach, B.T.; von Känel, R.; Patterson, T.L.; Dimsdale, J.E.; Depp, C.A.; Aschbacher, K.; Mills, P.J.; Ancoli-Israel, S.; Grant, I. The Moderating Effect of Personal Mastery and the Relations between Stress and Plasminogen Activator Inhibitor-1 (PAI-1) Antigen. Health Psychol. 2008, 27, S172–S179. [Google Scholar] [CrossRef] [PubMed]
- Gibert, L.; Verdonk, C.; Tarquinio, C.; Falissard, B.; El Hage, W.; Trousselard, M. 2015 Paris Terrorist Attacks: Care Guidance for the Massive Influx of Psychologically Traumatized Civilian Casualties. Helping Victims to Develop Their Capacity to Create a Safe and Protective Environment by Leveraging Social Resources like Family, and Inner Resources like Mindfulness Should Optimize Global Resilience. Eur. J. Trauma Dissociation 2020, 4, 100079. [Google Scholar] [CrossRef]
- Schein, J.; Childress, A.; Adams, J.; Cloutier, M.; Gagnon-Sanschagrin, P.; Maitland, J.; Bungay, R.; Guérin, A.; Lefebvre, P. Treatment Patterns among Adults with Attention-Deficit/Hyperactivity Disorder in the United States: A Retrospective Claims Study. Curr. Med. Res. Opin. 2021, 37, 2007–2014. [Google Scholar] [CrossRef] [PubMed]
- Mental Health and COVID-19: Early Evidence of the Pandemic’s Impact: Scientific Brief, 2 March 2022. Available online: https://www.who.int/publications-detail-redirect/WHO-2019-nCoV-Sci_Brief-Mental_health-2022.1 (accessed on 15 December 2022).
- Chamberlain, S.R.; Grant, J.E.; Trender, W.; Hellyer, P.; Hampshire, A. Post-Traumatic Stress Disorder Symptoms in COVID-19 Survivors: Online Population Survey. BJPsych Open 2021, 7, e47. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Philadelphia, PA, USA, 2013; ISBN 978-0-89042-555-8. [Google Scholar]
- Geiser, F.; Meier, C.; Wegener, I.; Imbierowicz, K.; Conrad, R.; Liedtke, R.; Oldenburg, J.; Harbrecht, U. Association between Anxiety and Factors of Coagulation and Fibrinolysis. Psychother. Psychosom. 2008, 77, 377–383. [Google Scholar] [CrossRef]
- Chen, S.; Jiang, H.; Liu, Y.; Hou, Z.; Yue, Y.; Zhang, Y.; Zhao, F.; Xu, Z.; Li, Y.; Mou, X.; et al. Combined Serum Levels of Multiple Proteins in TPA-BDNF Pathway May Aid the Diagnosis of Five Mental Disorders. Sci. Rep. 2017, 7, 6871. [Google Scholar] [CrossRef]
- Kessler, R.C.; Chiu, W.T.; Demler, O.; Merikangas, K.R.; Walters, E.E. Prevalence, Severity, and Comorbidity of 12-Month DSM-IV Disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 2005, 62, 617–627. [Google Scholar] [CrossRef]
- Flory, J.D.; Yehuda, R. Comorbidity between Post-Traumatic Stress Disorder and Major Depressive Disorder: Alternative Explanations and Treatment Considerations. Dialogues Clin. Neurosci. 2015, 17, 141–150. [Google Scholar] [CrossRef]
- Xu, L.; Nan, J.; Lan, Y. The Nucleus Accumbens: A Common Target in the Comorbidity of Depression and Addiction. Front. Neural Circuits 2020, 14, 37. [Google Scholar] [CrossRef]
- Party, H.; Dujarrier, C.; Hébert, M.; Lenoir, S.; Martinez de Lizarrondo, S.; Delépée, R.; Fauchon, C.; Bouton, M.-C.; Obiang, P.; Godefroy, O.; et al. Plasminogen Activator Inhibitor-1 (PAI-1) Deficiency Predisposes to Depression and Resistance to Treatments. Acta Neuropathol. Commun. 2019, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Chen, S.; Li, C.; Lu, N.; Yue, Y.; Yin, Y.; Zhang, Y.; Zhi, X.; Zhang, D.; Yuan, Y. The Serum Protein Levels of the TPA-BDNF Pathway Are Implicated in Depression and Antidepressant Treatment. Transl. Psychiatry 2017, 7, e1079. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Qin, Y.-Q.; Sun, Y.-C.; Yao, H.-J.; Cheng, X.-K.; Yu, Y.; Lu, S.-S. Electroacupuncture Ameliorates Depressive-Like Behaviors in Poststroke Rats via Activating the TPA/BDNF/TrkB Pathway. Neuropsychiatr. Dis. Treat. 2021, 17, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Luo, J.; Zhu, X. Ketamine Ameliorates Depressive-like Behaviors by TPA-Mediated Conversion of ProBDNF to MBDNF in the Hippocampus of Stressed Rats. Psychiatry Res. 2018, 269, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, F.; Mistry, S.; Martinez, P.E.; Torvik, S.; Kotila, C.; Sebring, N.; Drinkard, B.E.; Levy, C.; Reynolds, J.C.; Csako, G.; et al. Younger, Premenopausal Women with Major Depressive Disorder Have More Abdominal Fat and Increased Serum Levels of Prothrombotic Factors: Implications for Greater Cardiovascular Risk. Metabolism 2005, 54, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Dang, R.; Xu, P.; Li, G.; Zhou, X.; Chen, L.; Guo, Y.; Yang, M.; Chen, D.; Jiang, P. Altered Fibrinolytic System in Rat Models of Depression and Patients with First-Episode Depression. Neurobiol. Stress 2019, 11, 100188. [Google Scholar] [CrossRef] [PubMed]
- Lahlou-Laforet, K.; Alhenc-Gelas, M.; Pornin, M.; Bydlowski, S.; Seigneur, E.; Benetos, A.; Kierzin, J.-M.; Scarabin, P.-Y.; Ducimetiere, P.; Aiach, M.; et al. Relation of Depressive Mood to Plasminogen Activator Inhibitor, Tissue Plasminogen Activator, and Fibrinogen Levels in Patients with versus without Coronary Heart Disease. Am. J. Cardiol. 2006, 97, 1287–1291. [Google Scholar] [CrossRef]
- Tsai, S.-J. Role of Tissue-Type Plasminogen Activator and Plasminogen Activator Inhibitor-1 in Psychological Stress and Depression. Oncotarget 2017, 8, 113258–113268. [Google Scholar] [CrossRef]
- Piazza, P.V.; Le Moal, M. Glucocorticoids as a Biological Substrate of Reward: Physiological and Pathophysiological Implications. Brain Res. Brain Res. Rev. 1997, 25, 359–372. [Google Scholar] [CrossRef]
- Bachis, A.; Campbell, L.A.; Jenkins, K.; Wenzel, E.; Mocchetti, I. Morphine Withdrawal Increases Brain-Derived Neurotrophic Factor Precursor. Neurotox. Res. 2017, 32, 509–517. [Google Scholar] [CrossRef]
- Zhou, Y.; Maiya, R.; Norris, E.H.; Kreek, M.J.; Strickland, S. Involvement of Tissue Plasminogen Activator in Stress Responsivity during Acute Cocaine Withdrawal in Mice. Stress 2010, 13, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Delahousse, B.; Maillot, F.; Gabriel, I.; Schellenberg, F.; Lamisse, F.; Gruel, Y. Increased Plasma Fibrinolysis and Tissue-Type Plasminogen Activator/Tissue-Type Plasminogen Activator Inhibitor Ratios after Ethanol Withdrawal in Chronic Alcoholics. Blood Coagul. Fibrinolysis 2001, 12, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Berta, T.; Liu, Y.-C.; Xu, Z.-Z.; Ji, R.-R. Tissue Plasminogen Activator Contributes to Morphine Tolerance and Induces Mechanical Allodynia via Astrocytic IL-1β and ERK Signaling in the Spinal Cord of Mice. Neuroscience 2013, 247, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Alonso, P.; López-Solà, C.; Real, E.; Segalàs, C.; Menchón, J.M. Animal Models of Obsessive-Compulsive Disorder: Utility and Limitations. Neuropsychiatr. Dis. Treat. 2015, 11, 1939–1955. [Google Scholar] [CrossRef] [PubMed]
- Szechtman, H.; Ahmari, S.E.; Beninger, R.J.; Eilam, D.; Harvey, B.H.; Edemann-Callesen, H.; Winter, C. Obsessive-Compulsive Disorder: Insights from Animal Models. Neurosci. Biobehav. Rev. 2017, 76, 254–279. [Google Scholar] [CrossRef]
- Luo, Y.; Chen, X.; Wei, C.; Zhang, H.; Zhang, L.; Han, L.; Sun, K.; Li, B.; Wen, S. BDNF Alleviates Microglial Inhibition and Stereotypic Behaviors in a Mouse Model of Obsessive-Compulsive Disorder. Front. Mol. Neurosci. 2022, 15, 926572. [Google Scholar] [CrossRef]
- Hao, L.-S.; Du, Y.; Chen, L.; Jiao, Y.-G.; Cheng, Y. Brain-Derived Neurotrophic Factor as a Biomarker for Obsessive-Compulsive Disorder: A Meta-Analysis. J. Psychiatr. Res. 2022, 151, 676–682. [Google Scholar] [CrossRef]
- Wentworth, B.A.; Stein, M.B.; Redwine, L.S.; Xue, Y.; Taub, P.R.; Clopton, P.; Nayak, K.R.; Maisel, A.S. Post-Traumatic Stress Disorder: A Fast Track to Premature Cardiovascular Disease? Cardiol. Rev. 2013, 21, 16–22. [Google Scholar] [CrossRef]
- Vaughan, D.E. PAI-1 and Atherothrombosis. J. Thromb. Haemost. 2005, 3, 1879–1883. [Google Scholar] [CrossRef]
- Aksu, S.; Unlu, G.; Kardesler, A.C.; Cakaloz, B.; Aybek, H. Altered Levels of Brain-Derived Neurotrophic Factor, ProBDNF and Tissue Plasminogen Activator in Children with Posttraumatic Stress Disorder. Psychiatry Res. 2018, 268, 478–483. [Google Scholar] [CrossRef]
- Maguire, D.; Watt, J.; Armour, C.; Milanak, M.; Lagdon, S.; Lamont, J.V.; Kurth, M.J.; Fitzgerald, P.; Moore, T.; Ruddock, M.W. Post-Traumatic Stress Disorder: A Biopsychosocial Case-Control Study Investigating Peripheral Blood Protein Biomarkers. Biomark. Neuropsychiatry 2021, 5, 100042–100050. [Google Scholar] [CrossRef]
- Farr, O.M.; Ko, B.J.; Joung, K.E.; Zaichenko, L.; Usher, N.; Tsoukas, M.; Thakkar, B.; Davis, C.R.; Crowell, J.A.; Mantzoros, C.S. Posttraumatic Stress Disorder, Alone or Additively with Early Life Adversity, Is Associated with Obesity and Cardiometabolic Risk. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 479–488. [Google Scholar] [CrossRef]
- Stratta, P.; Sanità, P.; Bonanni, R.L.; de Cataldo, S.; Angelucci, A.; Rossi, R.; Origlia, N.; Domenici, L.; Carmassi, C.; Piccinni, A.; et al. Clinical Correlates of Plasma Brain-Derived Neurotrophic Factor in Post-Traumatic Stress Disorder Spectrum after a Natural Disaster. Psychiatry Res. 2016, 244, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Trousselard, M.; Claverie, D.; Fromage, D.; Becker, C.; Houël, J.-G.; Benoliel, J.-J.; Canini, F. The Relationship between Allostasis and Mental Health Patterns in a Pre-Deployment French Military Cohort. Eur. J. Investig. Health Psychol. Educ. 2021, 11, 1239–1253. [Google Scholar] [CrossRef]
- Yehuda, R.; Hoge, C.W.; McFarlane, A.C.; Vermetten, E.; Lanius, R.A.; Nievergelt, C.M.; Hobfoll, S.E.; Koenen, K.C.; Neylan, T.C.; Hyman, S.E. Post-Traumatic Stress Disorder. Nat. Rev. Dis. Prim. 2015, 1, 15057. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor and Neuropsychiatric Disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [PubMed]
- Pang, P.T.; Lu, B. Regulation of Late-Phase LTP and Long-Term Memory in Normal and Aging Hippocampus: Role of Secreted Proteins TPA and BDNF. Ageing Res. Rev. 2004, 3, 407–430. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mennesson, M.; Revest, J.-M. Glucocorticoid-Responsive Tissue Plasminogen Activator (tPA) and Its Inhibitor Plasminogen Activator Inhibitor-1 (PAI-1): Relevance in Stress-Related Psychiatric Disorders. Int. J. Mol. Sci. 2023, 24, 4496. https://doi.org/10.3390/ijms24054496
Mennesson M, Revest J-M. Glucocorticoid-Responsive Tissue Plasminogen Activator (tPA) and Its Inhibitor Plasminogen Activator Inhibitor-1 (PAI-1): Relevance in Stress-Related Psychiatric Disorders. International Journal of Molecular Sciences. 2023; 24(5):4496. https://doi.org/10.3390/ijms24054496
Chicago/Turabian StyleMennesson, Marie, and Jean-Michel Revest. 2023. "Glucocorticoid-Responsive Tissue Plasminogen Activator (tPA) and Its Inhibitor Plasminogen Activator Inhibitor-1 (PAI-1): Relevance in Stress-Related Psychiatric Disorders" International Journal of Molecular Sciences 24, no. 5: 4496. https://doi.org/10.3390/ijms24054496
APA StyleMennesson, M., & Revest, J.-M. (2023). Glucocorticoid-Responsive Tissue Plasminogen Activator (tPA) and Its Inhibitor Plasminogen Activator Inhibitor-1 (PAI-1): Relevance in Stress-Related Psychiatric Disorders. International Journal of Molecular Sciences, 24(5), 4496. https://doi.org/10.3390/ijms24054496