
Design and Validation of a Custom Next-Generation Sequencing Panel in Pediatric Acute Lymphoblastic Leukemia

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
2.1. ALLseq Design
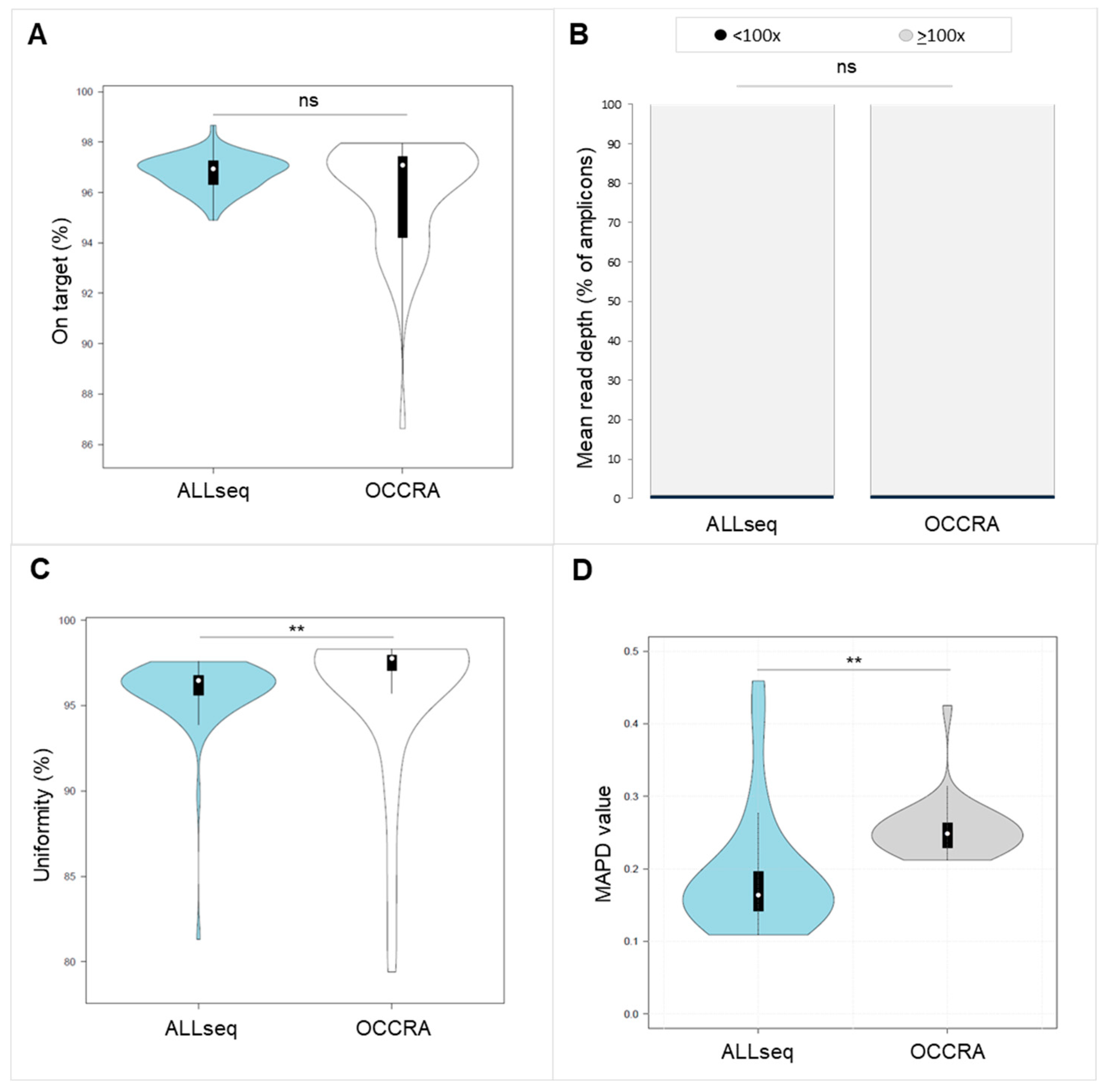
2.2. ALLseq Set-Up and Sequencing Metrics
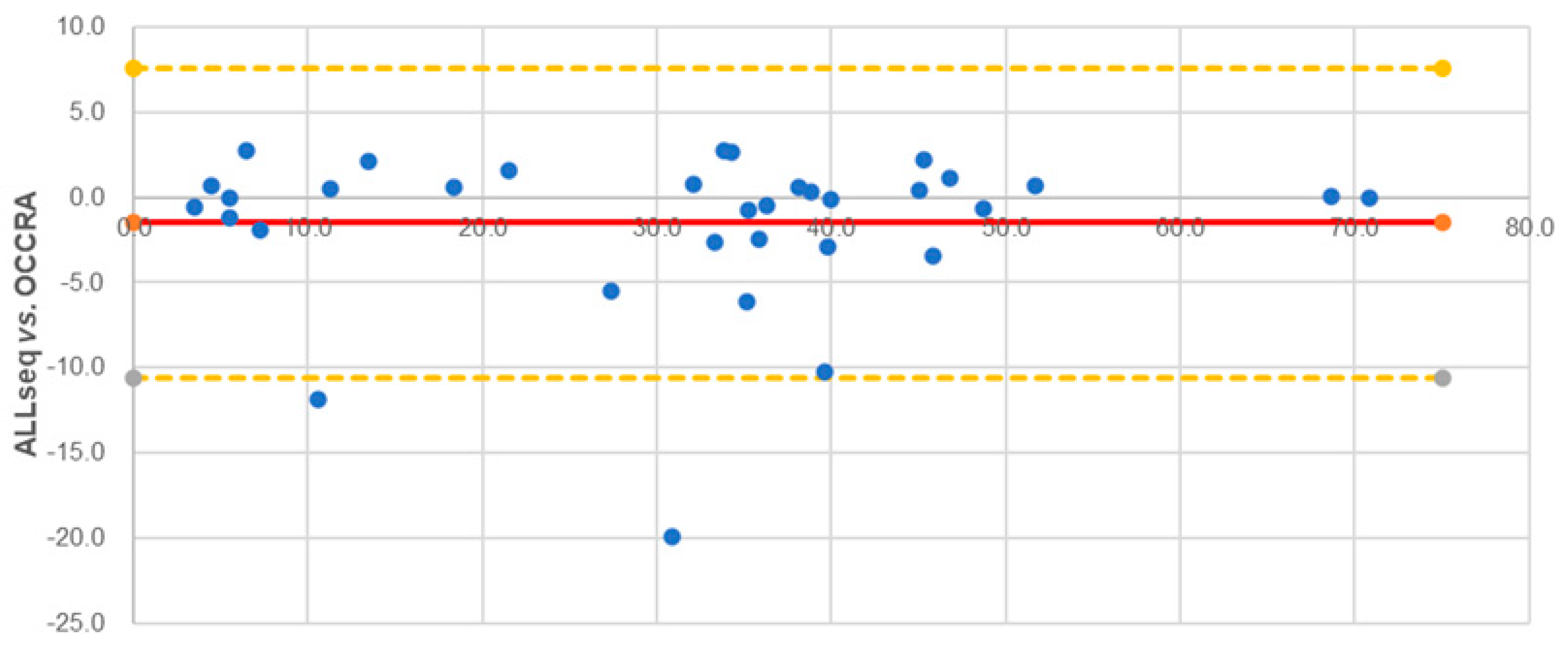
2.3. Technical Validation Strategy and ALLseq Performance
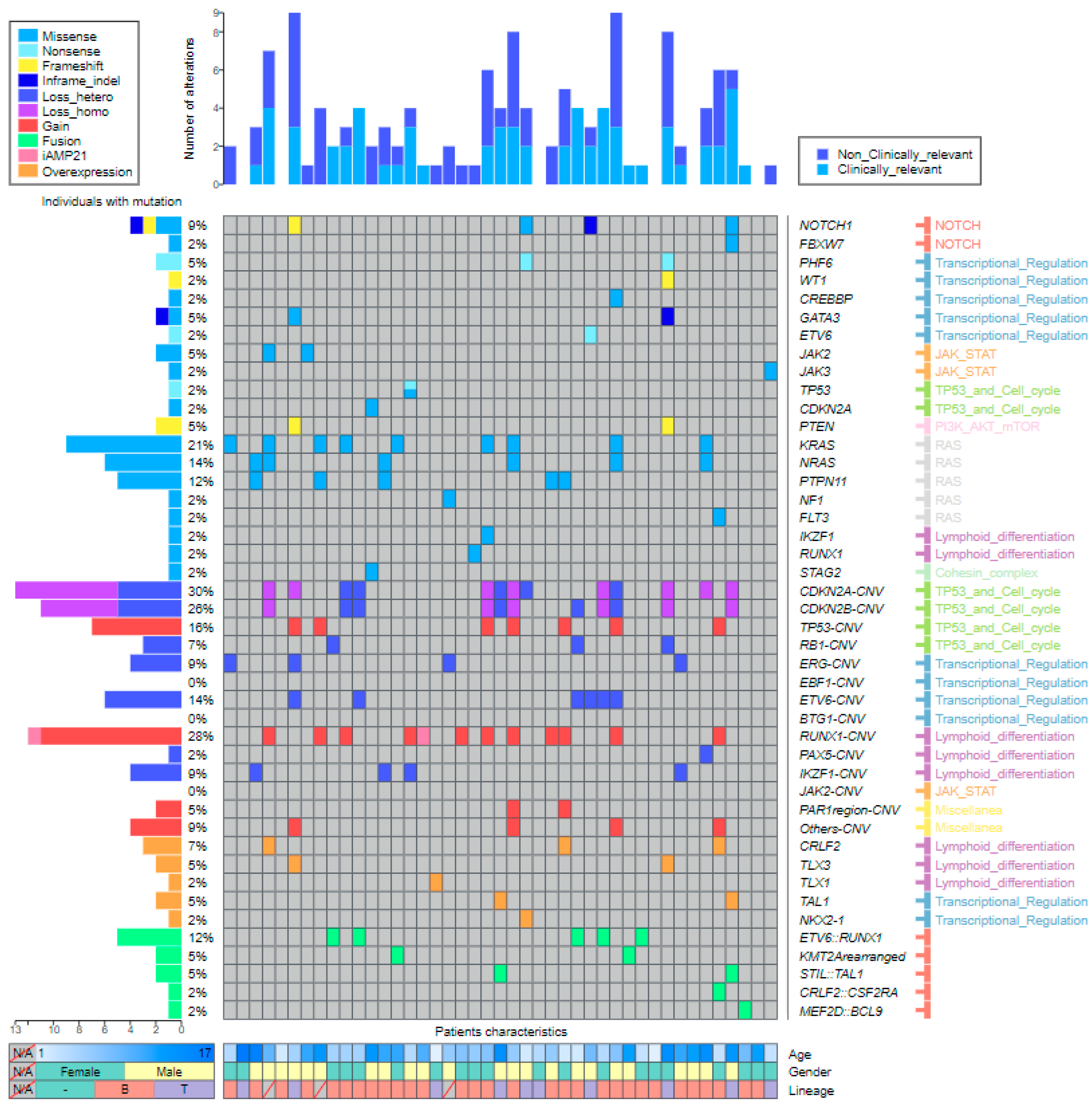
2.3.1. SNVs and Indels
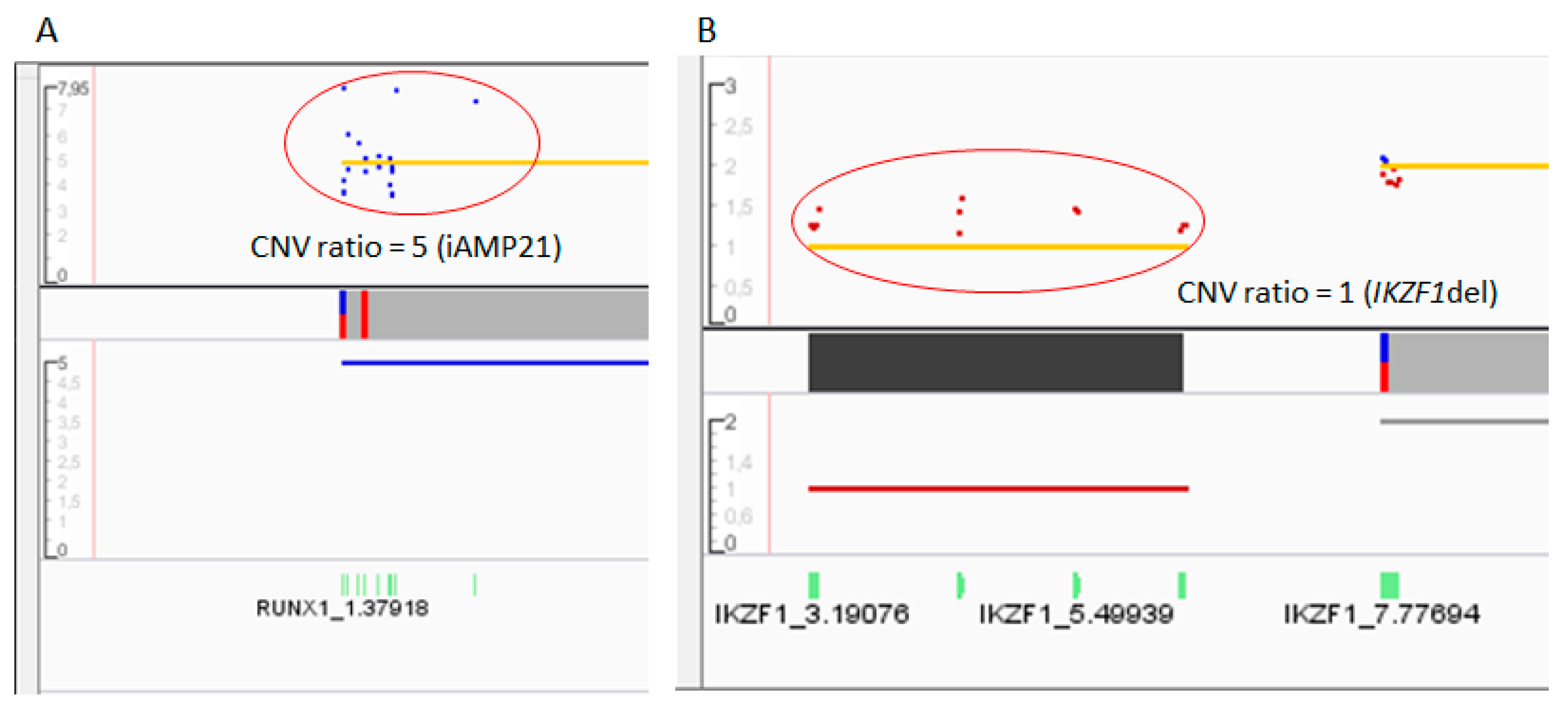
2.3.2. CNVs
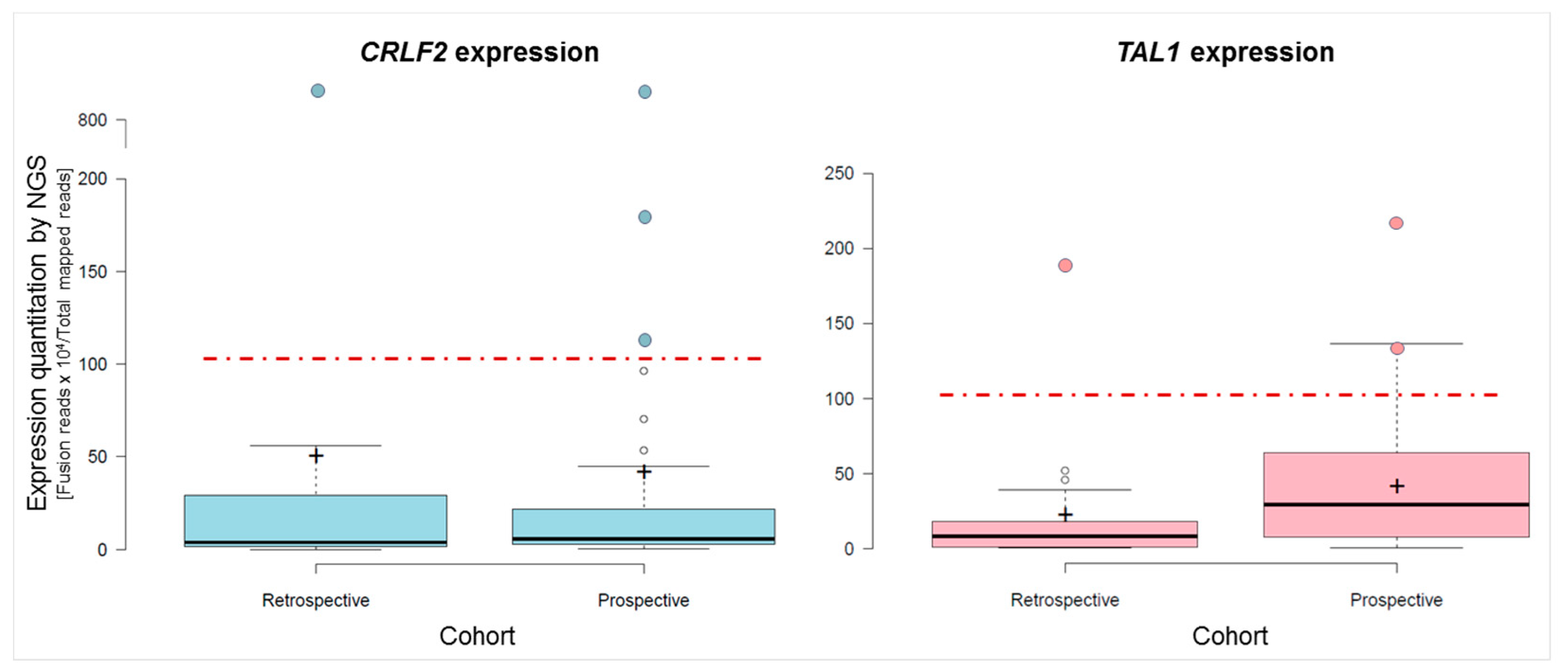
2.3.3. Fusions and Gene Expression
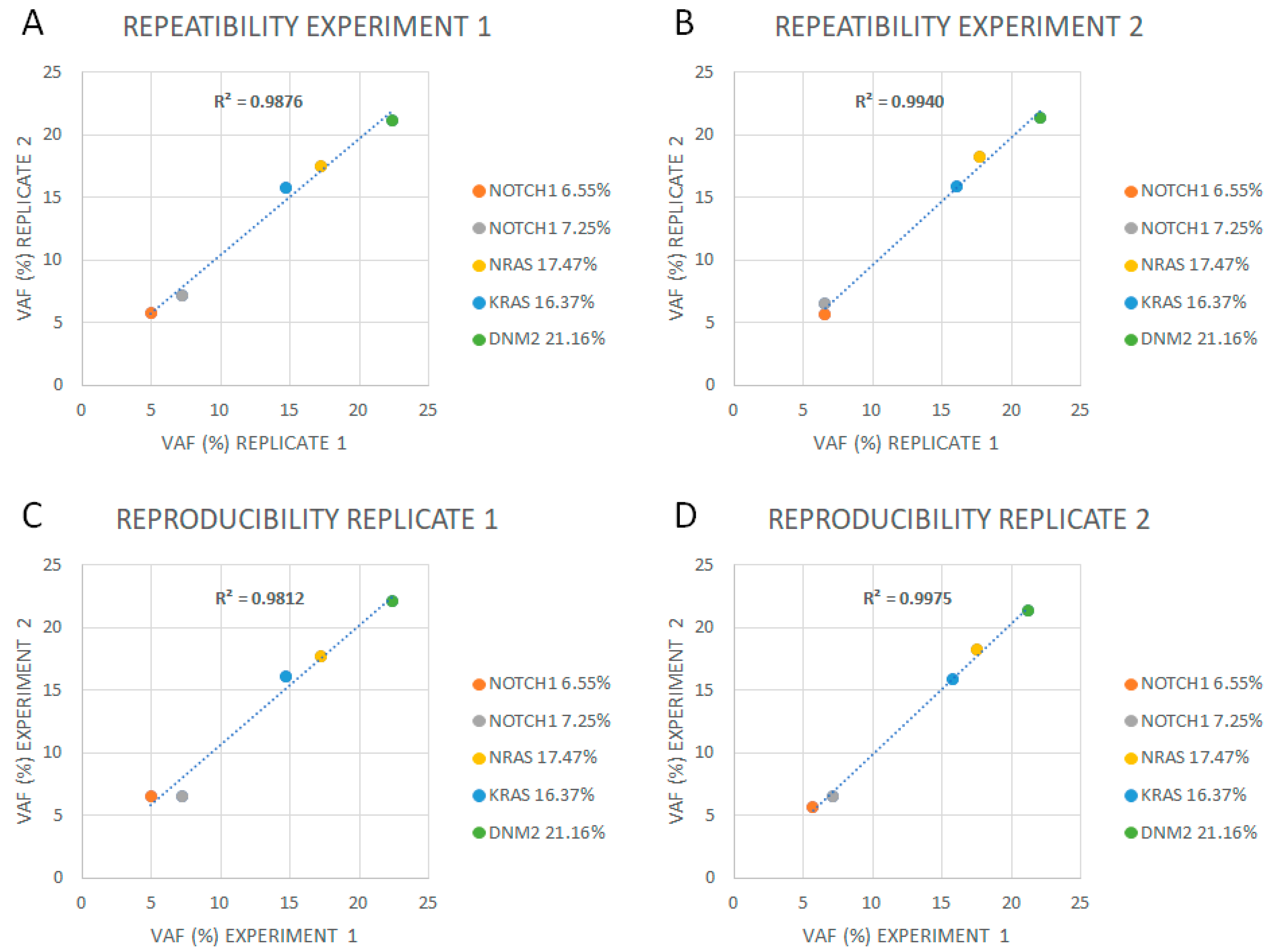
2.4. Limit of Detection, Reproducibility, Repeatability, and Linearity
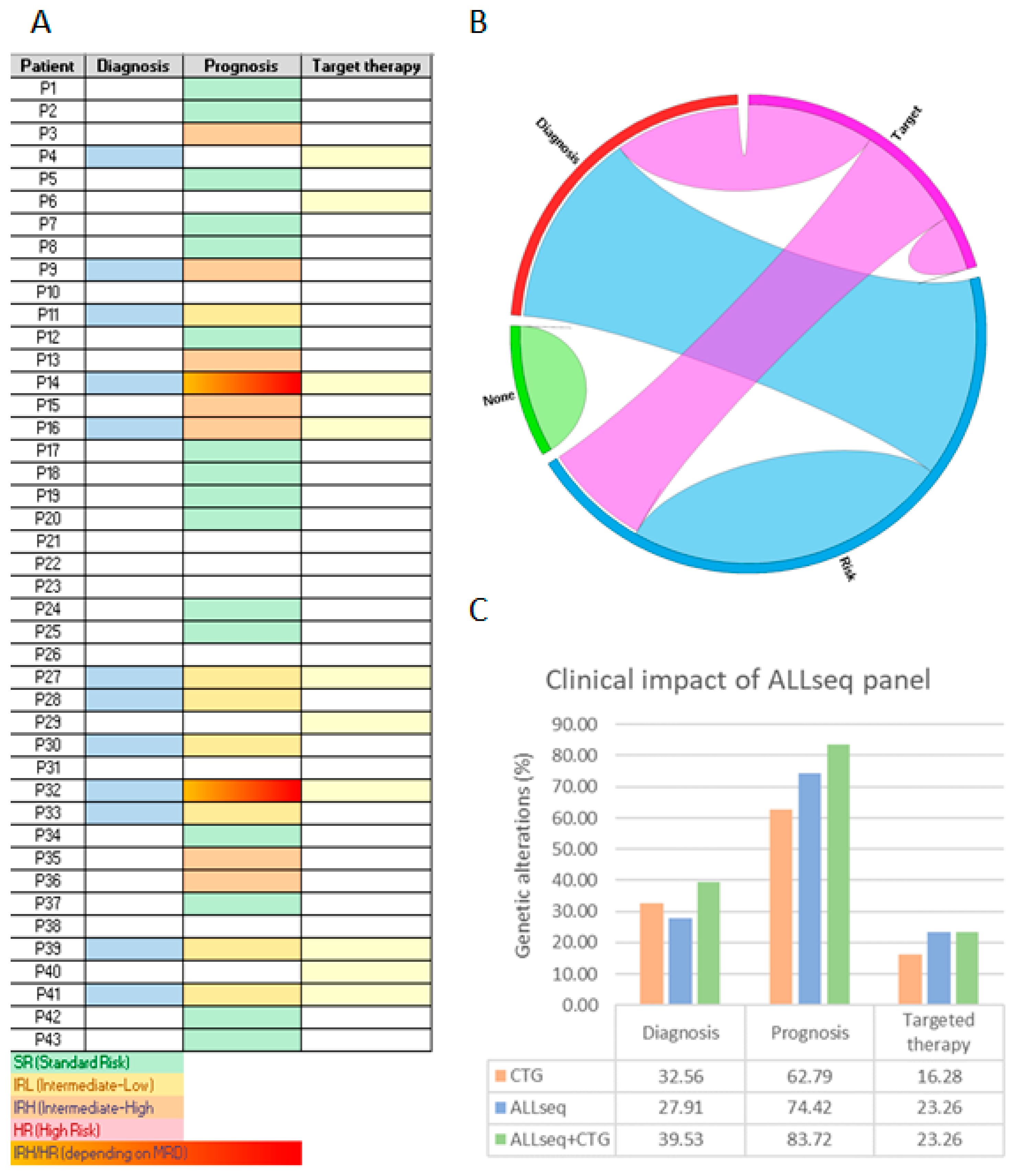
2.5. Clinical Validation: Prospective Sequencing
3. Discussion
4. Materials and Methods
4.1. Patient Samples and Inclusion Criteria
4.2. ALLseq: An ALL-Targeted Custom NGS Panel
- Level 1: clinical guidelines and clinical trials: (a) alterations included in the WHO classification of hematolymphoid tumors in force at the time of the start of the study [29]; (b) alterations defining genetic ALL subtypes [30]; (c) alterations used for risk stratification by international cooperative groups [31], NCT04307576]; (d) alterations used for potential targeted therapy [32].
- Level 2: other pathogenic alterations described in large cohorts: (a) variants that cluster into specific subtypes of ALL [15,33]; (b) variants associated with good or bad prognosis but not currently used for patient risk stratification [34]; (c) variants that confer resistance to specific drugs in vitro/in vivo experiments [35].
4.3. Data Analysis
4.4. ALLseq Validation Strategy
Complementary Molecular and Cytogenetic Methods
4.5. ALLseq Technical Validation
4.5.1. Run Metrics and Quality Criteria
4.5.2. Assessment of Analytical Performance
4.5.3. Limit of Detection, Reproducibility, and Repeatability
4.6. ALLseq Clinical Validation
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lejman, M.; Chałupnik, A.; Chilimoniuk, Z.; Dobosz, M. Genetic Biomarkers and Their Clinical Implications in B-Cell Acute Lymphoblastic Leukemia in Children. Int. J. Mol. Sci. 2022, 23, 2755. [Google Scholar] [CrossRef]
- Advani, A.S. Novel Strategies in the Treatment of Acute Lymphoblastic Leukaemia. Lancet Haematol. 2022, 9, e240–e241. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.D.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Hamadeh, L.; Enshaei, A.; Schwab, C.; Alonso, C.N.; Attarbaschi, A.; Barbany, G.; den Boer, M.L.; Boer, J.M.; Braun, M.; Dalla Pozza, L.; et al. Validation of the United Kingdom Copy-Number Alteration Classifier in 3239 Children with B-Cell Precursor ALL. Blood Adv. 2019, 3, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Pastorczak, A.; Sędek, Ł.; Taha, J.; Madzio, J.; Jatczak-Pawlik, I.; Wypyszczak, K.; Matysiak, M.; Derwich, K.; Lejman, M.; et al. Prognostic Significance of IKZF1 Deletions and IKZF1plus Profile in Children with B-Cell Precursor Acute Lymphoblastic Leukemia Treated According to the ALL-IC BFM 2009 Protocol. Hematol. Oncol. 2022, 40, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Lato, M.W.; Przysucha, A.; Grosman, S.; Zawitkowska, J.; Lejman, M. The New Therapeutic Strategies in Pediatric T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 4502. [Google Scholar] [CrossRef] [PubMed]
- Malczewska, M.; Kośmider, K.; Bednarz, K.; Ostapińska, K.; Lejman, M.; Zawitkowska, J. Recent Advances in Treatment Options for Childhood Acute Lymphoblastic Leukemia. Cancers 2022, 14, 2021. [Google Scholar] [CrossRef]
- Pervez, M.T.; Hasnain, M.J.U.; Abbas, S.H.; Moustafa, M.F.; Aslam, N.; Shah, S.S.M. A Comprehensive Review of Performance of Next-Generation Sequencing Platforms. Biomed Res. Int. 2022, 2022, 3457806. [Google Scholar] [CrossRef] [PubMed]
- Churchman, M.L.; Qian, M.; Te Kronnie, G.; Zhang, R.; Yang, W.; Zhang, H.; Lana, T.; Tedrick, P.; Baskin, R.; Verbist, K.; et al. Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell 2018, 33, 937–948.e8. [Google Scholar] [CrossRef]
- Luo, X.; Feurstein, S.; Mohan, S.; Porter, C.C.; Jackson, S.A.; Keel, S.; Chicka, M.; Brown, A.L.; Kesserwan, C.; Agarwal, A.; et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel Recommendations for Germline RUNX1 Variants. Blood Adv. 2019, 3, 2962–2979. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Garcés, C.; Esperanza-Cebollada, E.; Montesdeoca, S.; Torrebadell, M.; Rives, S.; Dapena, J.L.; Català, A.; Conde, N.; Camós, M.; Vega-García, N. Technical Validation and Clinical Utility of an NGS Targeted Panel to Improve Molecular Characterization of Pediatric Acute Leukemia. Front. Mol. Biosci. 2022, 9, 854098. [Google Scholar] [CrossRef] [PubMed]
- Teo, S.M.; Pawitan, Y.; Ku, C.S.; Chia, K.S.; Salim, A. Statistical Challenges Associated with Detecting Copy Number Variations with Next-Generation Sequencing. Bioinformatics 2012, 28, 2711–2718. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Pandzic, T.; Ladenvall, C.; Engvall, M.; Mattsson, M.; Hermanson, M.; Cavelier, L.; Ljungström, V.; Baliakas, P. Five Percent Variant Allele Frequency Is a Reliable Reporting Threshold for TP53 Variants Detected by next Generation Sequencing in Chronic Lymphocytic Leukemia in the Clinical Setting. HemaSphere 2022, 6, e761. [Google Scholar] [CrossRef]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. JMD 2017, 19, 341–365. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-Rearranged Subtypes in Paediatric B-Cell Precursor Acute Lymphoblastic Leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef]
- Rehn, J.A.; O’Connor, M.J.; White, D.L.; Yeung, D.T. DUX Hunting—Clinical Features and Diagnostic Challenges Associated with DUX4-Rearranged Leukaemia. Cancers 2020, 12, 2815. [Google Scholar] [CrossRef] [PubMed]
- Palmi, C.; Silvestri, D.; Bronzini, I.; Cario, G.; Savino, A.; Paganin, M.; Buldini, B.; Galbiati, M.; Muckenthaler, M.U.; Arico, M.; et al. CRLF2 Over-Expression Is a Poor Prognostic Marker in Children with High Risk T-Cell Acute Lymphoblastic Leukemia. Blood 2014, 124, 1071. [Google Scholar] [CrossRef]
- Li, D.; Li, T.; Shang, Z.; Zhao, L.; Xu, Q.; Tan, J.; Qin, Y.; Zhang, Y.; Cao, Y.; Wang, N.; et al. Combined Inhibition of Notch and FLT3 Produces Synergistic Cytotoxic Effects in FLT3/ITD+ Acute Myeloid Leukemia. Signal Transduct. Target. Ther. 2020, 5, 21. [Google Scholar] [CrossRef]
- Felice, M.S.; Rubio, P.L.; Digiorge, J.; Barreda Frank, M.; Martínez, C.S.; Guitter, M.R.; Sajaroff, E.O.; Sánchez La Rosa, C.G.; Pennella, C.L.; Peruzzo, L.B.; et al. Impact of IKZF1 Deletions in the Prognosis of Childhood Acute Lymphoblastic Leukemia in Argentina. Cancers 2022, 14, 3283. [Google Scholar] [CrossRef]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Möricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; te Kronnie, G.; Bourquin, J.-P.; Bornhauser, B.; et al. IKZF1plus Defines a New Minimal Residual Disease–Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar] [CrossRef]
- Moorman, A.V.; Enshaei, A.; Schwab, C.; Wade, R.; Chilton, L.; Elliott, A.; Richardson, S.; Hancock, J.; Kinsey, S.E.; Mitchell, C.D.; et al. A Novel Integrated Cytogenetic and Genomic Classification Refines Risk Stratification in Pediatric Acute Lymphoblastic Leukemia. Blood 2014, 124, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Walker, H.; Oliver, F.; Wheatley, K.; Harrison, C.; Harrison, G.; Rees, J.; Hann, I.; Stevens, R.; Burnett, A.; et al. The Importance of Diagnostic Cytogenetics on Outcome in AML: Analysis of 1,612 Patients Entered into the MRC AML 10 Trial. Blood 1998, 92, 2322–2333. [Google Scholar] [CrossRef]
- Llop, M.; Sargas, C.; Barragán, E. The Role of Next-Generation Sequencing in Acute Myeloid Leukemia. Curr. Opin. Oncol. 2022, 34, 723–728. [Google Scholar] [CrossRef]
- Gulilat, M.; Lamb, T.; Teft, W.A.; Wang, J.; Dron, J.S.; Robinson, J.F.; Tirona, R.G.; Hegele, R.A.; Kim, R.B.; Schwarz, U.I. Targeted next Generation Sequencing as a Tool for Precision Medicine. BMC Med. Genom. 2019, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Plebani, M. Integrated Diagnostics. Biochem. Med. 2020, 30, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef]
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef]
- Mengxuan, S.; Fen, Z.; Runming, J. Novel Treatments for Pediatric Relapsed or Refractory Acute B-Cell Lineage Lymphoblastic Leukemia: Precision Medicine Era. Front. Pediatr. 2022, 10, 923419. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.W.; Roberts, K.G.; Gu, Z.; Shi, L.; Pounds, S.; Pei, D.; Cheng, C.; Dai, Y.; Devidas, M.; Qu, C.; et al. The Genomic Landscape of Pediatric Acute Lymphoblastic Leukemia. Nat. Genet. 2022, 54, 1376–1389. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Redefining ALL Classification: Toward Detecting High-Risk ALL and Implementing Precision Medicine. Blood 2015, 125, 3977–3987. [Google Scholar] [CrossRef]
- Frismantas, V.; Dobay, M.P.; Rinaldi, A.; Tchinda, J.; Dunn, S.H.; Kunz, J.; Richter-Pechanska, P.; Marovca, B.; Pail, O.; Jenni, S.; et al. Ex Vivo Drug Response Profiling Detects Recurrent Sensitivity Patterns in Drug-Resistant Acute Lymphoblastic Leukemia. Blood 2017, 129, e26–e37. [Google Scholar] [CrossRef]
- Gabert, J.; Beillard, E.; van der Velden, V.H.J.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cavé, H.; et al. Standardization and Quality Control Studies of “Real-Time” Quantitative Reverse Transcriptase Polymerase Chain Reaction of Fusion Gene Transcripts for Residual Disease Detection in Leukemia—A Europe against Cancer Program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Jia, W.; Li, H.; Li, S.; Chen, L.; Li, S.C. Oviz-Bio: A Web-Based Platform for Interactive Cancer Genomics Data Visualization. Nucleic Acids Res. 2020, 48, 8203–8204. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA (2 Primer Pools): 96,7 kb; 1138 Amplicons, 97.91% Coverage | RNA (2 Primer Pools) | ||
|---|---|---|---|
| HotSpots (SNVs and Indels; 22 Genes) | Whole Coding Sequence (SNVs and Indels; 32 Genes) | Fusions (271 Fusions, 634 Isoforms) | Expression Quantitation (7 Genes) |
| CREBBP *, CRLF2, DNMT3A, EP300, EZH2, FBXW7, FLT3, IDH1/2, IL7R, JAK1/2/3, NOTCH1, PAX5, PIK3CA, PTPN11, K/NRAS, SETD2, STAT5B, and SH2B3 * | AKT, BCL11B, BTG1, CDKN2A/B *, DNM2, EBF1, EED, ERG *, ETV6 *, GATA3, IKZF1 *, IL2RB, KDM6A, LEF1, NF1, NT5C, PAX5 *, PHF6, PTEN, PTPN2, RB1 *, RUNX1 *, STAG2, SUZ12, TET2, TP53, and WT1 | Main drivers: ABL1/2, KMT2A, TCF3, ETV6, EPOR, CSF1R, FLT3, JAK2, PDGFRA/B, LYN, NTRK1/2/3, TYK2, FGFR1, IL2RB, TSLP, PAX5, NOTCH1, MEF2D, ZNF384, and MYB/L1. Main fusions: P2RY8::CRLF2, SET::NUP214, PICALM::MLLT10, and STIL::TAL | CRLF2, HOXA, LMO2, NKX2, TAL1, TLX1, and TLX3 |
| Alteration Type | Sensitivity | Specificity | PPV | NPV | Precision |
|---|---|---|---|---|---|
| SNVs and indels | 100% | 100% | 100% | 100% | 100% |
| CNVs (Cohen kappa coefficient = 0.88) | 88.87% | 97.92% | 92.85% | 95.91% | 96.45% |
| Fusions | 100% | 100% | 100% | 100% | 100% |
| Gene expression | 100% | 100% | 100% | 100% | 100% |
| Copy Number Ratio | ||||
|---|---|---|---|---|
| CDKN2A/B | IKZF1 | |||
| Expected | Observed | Expected | Observed | |
| Dil1 (3:1) | 0.5 | <1 | 0.25 | ND 1 |
| Dil2 (1:1) | 1 | 1 | 0.5 | <1 |
| Dil3 (1:2) | 0.66 | <1 | 0.66 | <1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil, J.V.; Such, E.; Sargas, C.; Simarro, J.; Miralles, A.; Pérez, G.; de Juan, I.; Palanca, S.; Avetisyan, G.; Santiago, M.; et al. Design and Validation of a Custom Next-Generation Sequencing Panel in Pediatric Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2023, 24, 4440. https://doi.org/10.3390/ijms24054440
Gil JV, Such E, Sargas C, Simarro J, Miralles A, Pérez G, de Juan I, Palanca S, Avetisyan G, Santiago M, et al. Design and Validation of a Custom Next-Generation Sequencing Panel in Pediatric Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2023; 24(5):4440. https://doi.org/10.3390/ijms24054440
Chicago/Turabian StyleGil, José Vicente, Esperanza Such, Claudia Sargas, Javier Simarro, Alberto Miralles, Gema Pérez, Inmaculada de Juan, Sarai Palanca, Gayane Avetisyan, Marta Santiago, and et al. 2023. "Design and Validation of a Custom Next-Generation Sequencing Panel in Pediatric Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 24, no. 5: 4440. https://doi.org/10.3390/ijms24054440
APA StyleGil, J. V., Such, E., Sargas, C., Simarro, J., Miralles, A., Pérez, G., de Juan, I., Palanca, S., Avetisyan, G., Santiago, M., Fuentes, C., Fernández, J. M., Vicente, A. I., Romero, S., Llop, M., & Barragán, E. (2023). Design and Validation of a Custom Next-Generation Sequencing Panel in Pediatric Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences, 24(5), 4440. https://doi.org/10.3390/ijms24054440