
Small RNA and Degradome Sequencing in Floral Bud Reveal Roles of miRNAs in Dormancy Release of Chimonanthus praecox

Abstract
1. Introduction
2. Results
2.1. Overview of sRNA Sequencing
2.2. Identification of Known and Novel miRNAs
2.3. Differential Expression of miRNAs during Dormancy Release

2.4. Summary of Degradome Sequencing
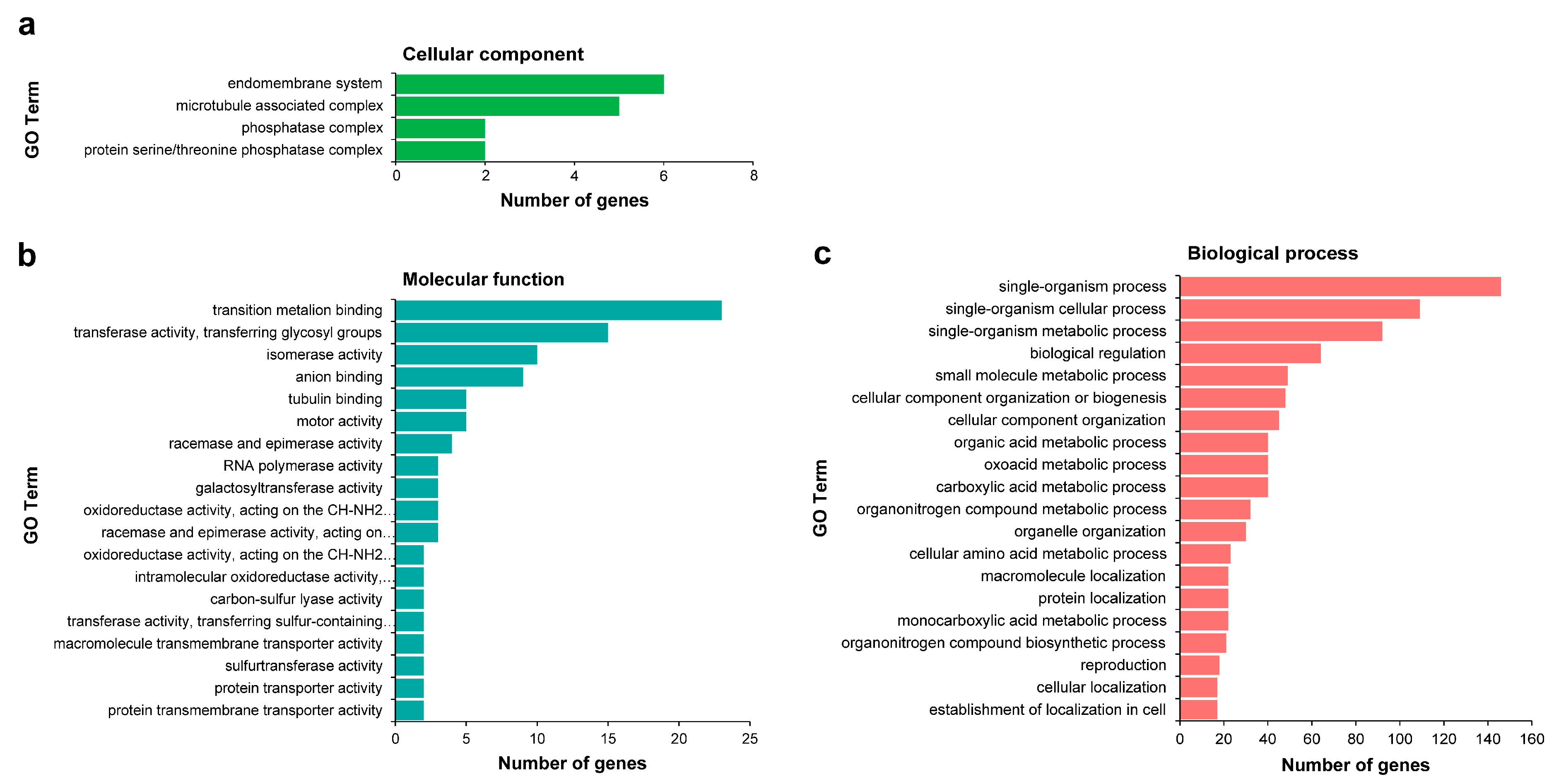
2.5. Identification of miRNA Targets
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Total RNA Extraction
4.2. sRNA Library Construction and Sequencing
4.3. Degradome Library Construction and Sequencing
4.4. Analysis of sRNA Sequencing Data
4.5. Analysis of Differentially Expressed miRNAs
4.6. Analysis of Degradome Sequencing Data and Identification of miRNA Targets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spanudakis, E.; Jackson, S. The role of microRNAs in the control of flowering time. J. Exp. Bot. 2014, 65, 365–380. [Google Scholar] [CrossRef]
- Rogers, K.; Chen, X. Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Zhu, Q.H.; Spriggs, A.; Matthew, L.; Fan, L.J.; Kennedy, G.; Gubler, F.; Helliwell, C. A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res. 2008, 18, 1456–1465. [Google Scholar] [CrossRef]
- Zhang, L.F.; Chia, J.M.; Kumari, S.; Stein, J.C.; Liu, Z.J.; Narechania, A.; Maher, C.A.; Guill, K.; McMullen, M.D.; Ware, D. A Genome-Wide Characterization of MicroRNA Genes in Maize. PLoS Genet. 2009, 5, e1000716. [Google Scholar] [CrossRef]
- Barakat, A.; Wall, P.K.; Diloreto, S.; Depamphilis, C.W.; Carlson, J.E. Conservation and divergence of microRNAs in Populus. BMC Genom. 2007, 8, 481. [Google Scholar] [CrossRef]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef]
- Pantaleo, V.; Szittya, G.; Moxon, S.; Miozzi, L.; Moulton, V.; Dalmay, T.; Burgyan, J. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010, 62, 960–976. [Google Scholar]
- Barakat, A.; Sriram, A.; Park, J.; Zhebentyayeva, T.; Main, D.; Abbott, A. Genome wide identification of chilling responsive microRNAs in Prunus persica. BMC Genom. 2012, 13, 481. [Google Scholar] [CrossRef]
- Li, Y.F.; Wei, K.; Wang, M.; Wang, L.; Cui, J.; Zhang, D.; Guo, J.; Zhao, M.; Zheng, Y. Identification and Temporal Expression Analysis of Conserved and Novel MicroRNAs in the Leaves of Winter Wheat Grown in the Field. Front. Genet. 2019, 10, 779. [Google Scholar] [CrossRef]
- Ye, Y.; Wang, J.; Ni, Z.; Meng, X.; Feng, Y.; Yang, Z.; Xu, L.-A. Small RNA and degradome sequencing reveal roles of miRNAs in strobilus development in masson pine (Pinus massoniana). Ind. Crops Prod. 2020, 154, 112724. [Google Scholar] [CrossRef]
- Zhang, B.H. MicroRNA: A new target for improving plant tolerance to abiotic stress. J. Exp. Bot. 2015, 66, 1749–1761. [Google Scholar] [CrossRef]
- Chen, L.; Ren, Y.Y.; Zhang, Y.Y.; Xu, J.C.; Zhang, Z.Y.; Wang, Y.W. Genome-wide profiling of novel and conserved Populus microRNAs involved in pathogen stress response by deep sequencing. Planta 2012, 235, 873–883. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and Functional Diversification of MIRNA Genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef]
- Lang, G.A.; Early, J.D.; Martin, G.C.; Darnell, R.L. Endodormancy, Paradormancy, and Ecodormancy—Physiological Terminology and Classification for Dormancy Research. Hortscience 1987, 22, 371–377. [Google Scholar] [CrossRef]
- Samish, R.M. Dormancy in Woody Plants. Annu. Rev. Plant Phys. 1954, 5, 183–204. [Google Scholar] [CrossRef]
- Yang, Q.; Gao, Y.; Wu, X.; Moriguchi, T.; Bai, S.; Teng, Y. Bud endodormancy in deciduous fruit trees: Advances and prospects. Hortic. Res. 2021, 8, 139. [Google Scholar] [CrossRef]
- Arora, R.; Rowland, L.J.; Tanino, K. Induction and release of bud dormancy in woody perennials: A science comes of age. Hortscience 2003, 38, 911–921. [Google Scholar] [CrossRef]
- Chuine, I.; Beaubien, E.G. Phenology is a major determinant of tree species range. Ecol. Lett. 2001, 4, 500–510. [Google Scholar] [CrossRef]
- Niu, Q.; Li, J.; Cai, D.; Qian, M.; Jia, H.; Bai, S.; Hussain, S.; Liu, G.; Teng, Y.; Zheng, X. Dormancy-associated MADS-box genes and microRNAs jointly control dormancy transition in pear (Pyrus pyrifolia white pear group) flower bud. J. Exp. Bot. 2016, 67, 239–257. [Google Scholar] [CrossRef]
- Moser, M.; Asquini, E.; Miolli, G.V.; Weigl, K.; Hanke, M.V.; Flachowsky, H.; Si-Ammour, A. The MADS-Box Gene MdDAM1 Controls Growth Cessation and Bud Dormancy in Apple. Front. Plant Sci. 2020, 11, 1003. [Google Scholar] [CrossRef]
- Wu, R.; Wang, T.; Warren, B.A.W.; Allan, A.C.; Macknight, R.C.; Varkonyi-Gasic, E. Kiwifruit SVP2 gene prevents premature budbreak during dormancy. J. Exp. Bot. 2017, 68, 1071–1082. [Google Scholar] [CrossRef]
- Wu, R.; Tomes, S.; Karunairetnam, S.; Tustin, S.D.; Hellens, R.P.; Allan, A.C.; Macknight, R.C.; Varkonyi-Gasic, E. SVP-like MADS Box Genes Control Dormancy and Budbreak in Apple. Front. Plant Sci. 2017, 8, 477. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, P.-Y.; Zhong, S.; Dardick, C.; Callahan, A.; An, Y.-Q.; van Knocker, S.; Yang, Y.; Zhong, G.-Y.; Abbott, A.; et al. Thermal-responsive genetic and epigenetic regulation of DAM cluster controlling dormancy and chilling requirement in peach floral buds. Hortic. Res. 2020, 7, 114. [Google Scholar] [CrossRef]
- Singh, R.K.; Maurya, J.P.; Azeez, A.; Miskolczi, P.; Tylewicz, S.; Stojkovic, K.; Delhomme, N.; Busov, V.; Bhalerao, R.P. A genetic network mediating the control of bud break in hybrid aspen. Nat. Commun. 2018, 9, 4173. [Google Scholar] [CrossRef]
- Sasaki, R.; Yamane, H.; Ooka, T.; Jotatsu, H.; Kitamura, Y.; Akagi, T.; Tao, R. Functional and expressional analyses of PmDAM genes associated with endodormancy in Japanese apricot. Plant Physiol. 2011, 157, 485–497. [Google Scholar] [CrossRef]
- Dong, Y.; Khalil-Ur-Rehman, M.; Liu, X.; Wang, X.; Yang, L.; Tao, J.; Zheng, H. Functional characterisation of five SVP genes in grape bud dormancy and flowering. Plant Growth Regul. 2022, 97, 511–522. [Google Scholar] [CrossRef]
- Liu, J.; Sherif, S.M. Hormonal Orchestration of Bud Dormancy Cycle in Deciduous Woody Perennials. Front. Plant Sci. 2019, 10, 1136. [Google Scholar] [CrossRef]
- Leida, C.; Conesa, A.; Llacer, G.; Badenes, M.L.; Rios, G. Histone modifications and expression of DAM6 gene in peach are modulated during bud dormancy release in a cultivar-dependent manner. New Phytol. 2012, 193, 67–80. [Google Scholar] [CrossRef]
- Wu, R.; Wang, T.; Richardson, A.C.; Allan, A.C.; Macknight, R.C.; Varkonyi-Gasic, E. Histone modification and activation by SOC1-like and drought stress-related transcription factors may regulate AcSVP2 expression during kiwifruit winter dormancy. Plant Sci. Int. J. Exp. Plant Biol. 2019, 281, 242–250. [Google Scholar] [CrossRef]
- Santamaria, M.E.; Hasbun, R.; Valera, M.J.; Meijon, M.; Valledor, L.; Rodriguez, J.L.; Toorop, P.E.; Canal, M.J.; Rodriguez, R. Acetylated H4 histone and genomic DNA methylation patterns during bud set and bud burst in Castanea sativa. J. Plant Physiol. 2009, 166, 1360–1369. [Google Scholar] [CrossRef]
- Kumar, G.; Rattan, U.K.; Singh, A.K. Chilling-Mediated DNA Methylation Changes during Dormancy and Its Release Reveal the Importance of Epigenetic Regulation during Winter Dormancy in Apple (Malus × domestica Borkh.). PLoS ONE 2016, 11, e0149934. [Google Scholar] [CrossRef]
- Prudencio, A.S.; Werner, O.; Martinez-Garcia, P.J.; Dicenta, F.; Ros, R.M.; Martinez-Gomez, P. DNA Methylation Analysis of Dormancy Release in Almond (Prunus dulcis) Flower Buds Using Epi-Genotyping by Sequencing. Int. J. Mol. Sci. 2018, 19, 3542. [Google Scholar] [CrossRef]
- Rothkegel, K.; Sanchez, E.; Montes, C.; Greve, M.; Tapia, S.; Bravo, S.; Prieto, H.; Almeida, A.M. DNA methylation and small interference RNAs participate in the regulation of MADS-box genes involved in dormancy in sweet cherry (Prunus avium L.). Tree Physiol. 2017, 37, 1739–1751. [Google Scholar] [CrossRef]
- Chen, W.; Tamada, Y.; Yamane, H.; Matsushita, M.; Osako, Y.; Gao-Takai, M.; Luo, Z.; Tao, R. H3K4me3 plays a key role in establishing permissive chromatin states during bud dormancy and bud break in apple. Plant J. Cell Mol. Biol. 2022, 11, 1015–1031. [Google Scholar] [CrossRef] [PubMed]
- Garighan, J.; Dvorak, E.; Estevan, J.; Loridon, K.; Huettel, B.; Sarah, G.; Farrera, I.; Leclercq, J.; Grynberg, P.; Coiti Togawa, R.; et al. The Identification of Small RNAs Differentially Expressed in Apple Buds Reveals a Potential Role of the Mir159-MYB Regulatory Module during Dormancy. Plants 2021, 10, 2665. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Wang, Y.Y.; Gao, X.K.; Liu, C.Y.; Gai, S.P. Identification and characterization of microRNAs in tree peony during chilling induced dormancy release by high-throughput sequencing. Sci. Rep. 2018, 8, 4537. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.L.; Bennett, D.; Dardick, C.; Zhebentyayeva, T.; Abbott, A.G.; Liu, Z.R.; Staton, M.E. Genome-Wide Changes of Regulatory Non-Coding RNAs Reveal Pollen Development Initiated at Ecodormancy in Peach. Front. Mol. Biosci. 2021, 8, 612881. [Google Scholar] [CrossRef]
- Smita, S.; Robben, M.; Deuja, A.; Accerbi, M.; Green, P.J.; Subramanian, S.; Fennell, A. Integrative Analysis of Gene Expression and miRNAs Reveal Biological Pathways Associated with Bud Paradormancy and Endodormancy in Grapevine. Plants 2021, 10, 669. [Google Scholar] [CrossRef]
- Li, Z.; Liu, N.; Zhang, W.; Wu, C.; Jiang, Y.; Ma, J.; Li, M.; Sui, S. Integrated transcriptome and proteome analysis provides insight into chilling-induced dormancy breaking in Chimonanthus praecox. Hortic. Res. 2020, 7, 198. [Google Scholar] [CrossRef]
- Li, S.J.; Yang, N.; Chen, L.Q. Paraffin section observation of flower bud differentiation of Chimonanthus praecox in Kunming and comparison of the differentiation processes in different regions, China. Hortic. Plant J. 2022, 8, 221–229. [Google Scholar] [CrossRef]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Xu, W.; Yuan, Y.; Yao, Q.; Zhao, Y.; Wang, Z.; Jiang, W.; Zhang, X. Genome-wide Investigation of microRNAs and Their Targets in Brassica rapa ssp. pekinensis Root with Plasmodiophora brassicae Infection. Hortic. Plant J. 2016, 2, 209–216. [Google Scholar] [CrossRef]
- Zeng, X.; Xu, Y.; Jiang, J.; Zhang, F.; Ma, L.; Wu, D.; Wang, Y.; Sun, W. Identification of cold stress responsive microRNAs in two winter turnip rape (Brassica rapa L.) by high throughput sequencing. BMC Plant Biol. 2018, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Ou, L.; Kang, L.; Liu, Y.; Lv, J.; Wei, G.; Yang, B.; Yang, S.; Chen, W.; et al. Identification and characterization of novel microRNAs for fruit development and quality in hot pepper (Capsicum annuum L.). Gene 2017, 608, 66–72. [Google Scholar] [CrossRef]
- Mi, S.; Cai, T.; Hu, Y.; Chen, Y.; Hodges, E.; Ni, F.; Wu, L.; Li, S.; Zhou, H.; Long, C.; et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.L. GraphPad prism, data analysis, and scientific graphing. J. Chem. Inf. Comput. Sci. 1997, 37, 411–412. [Google Scholar] [CrossRef]
- Lu, S.F.; Sun, Y.H.; Chiang, V.L. Stress-responsive microRNAs in Populus. Plant J. 2008, 55, 131–151. [Google Scholar] [CrossRef]
- Bao, H.; Chen, M.; Chen, H.; Du, L.; Wang, Y. Transcriptome-wide identification of miRNA targets and a TAS3-homologous gene in Populus by degradome sequencing. Genes Genom. 2019, 41, 849–861. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.Z.; Tian, J.P.; Cheng, H.H.; Yan, Q.M.; Li, L.; Jamal, A.; Xu, Z.P.; Xiang, L.; Saski, C.A.; Jin, S.X.; et al. The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 2020, 21, 200. [Google Scholar] [CrossRef]
- Shen, Z.G.; Li, W.Y.; Li, Y.L.; Liu, M.L.; Cao, H.P.; Provart, N.; Ding, X.; Sun, M.; Tang, Z.H.; Yue, C.P.; et al. The red flower wintersweet genome provides insights into the evolution of magnoliids and the molecular mechanism for tepal color development. Plant J. 2021, 108, 1662–1678. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, W.; Yang, L.; Su, X.; He, M. Identification and Expression Analysis of microRNAs in Response to Dormancy Release During Cold Storage of Lilium pumilum Bulbs. J. Plant Growth Regul. 2020, 40, 388–404. [Google Scholar] [CrossRef]
- Qu, H.; Liu, Y.; Jiang, H.; Liu, Y.; Song, W.; Chen, L. Identification and characterization of miRNAs associated with sterile flower buds in the tea plant based on small RNA sequencing. Hereditas 2021, 158, 26. [Google Scholar] [CrossRef] [PubMed]
- Song, C.N.A.; Wang, C.; Zhang, C.Q.; Korir, N.K.; Yu, H.P.; Ma, Z.Q.; Fang, J.G. Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genom. 2010, 11, 431. [Google Scholar] [CrossRef]
- Ling, L.-Z.; Zhang, S.-D.; Zhao, F.; Yang, J.-L.; Song, W.-H.; Guan, S.-M.; Li, X.-S.; Huang, Z.-J.; Cheng, L. Transcriptome-Wide Identification and Prediction of miRNAs and Their Targets in Paris polyphylla var. yunnanensis by High-Throughput Sequencing Analysis. Int. J. Mol. Sci. 2017, 18, 219. [Google Scholar] [CrossRef]
- Li, D.; Wang, L.; Liu, X.; Cui, D.; Chen, T.; Zhang, H.; Jiang, C.; Xu, C.; Li, P.; Li, S.; et al. Deep sequencing of maize small RNAs reveals a diverse set of microRNA in dry and imbibed seeds. PLoS ONE 2013, 8, e55107. [Google Scholar] [CrossRef]
- Bai, S.; Saito, T.; Ito, A.; Tuan, P.A.; Xu, Y.; Teng, Y.; Moriguchi, T. Small RNA and PARE sequencing in flower bud reveal the involvement of sRNAs in endodormancy release of Japanese pear (Pyrus pyrifolia ‘Kosui’). BMC Genom. 2016, 17, 230. [Google Scholar] [CrossRef]
- Axtell, M.J. Classification and Comparison of Small RNAs from Plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, Y.; Tang, R.; Qu, H.; Duan, X.; Jiang, Y. Banana sRNAome and degradome identify microRNAs functioning in differential responses to temperature stress. BMC Genom. 2019, 20, 33. [Google Scholar] [CrossRef]
- Zheng, C.L.; Halaly, T.; Acheampong, A.K.; Takebayashi, Y.; Jikumaru, Y.; Kamiya, Y.; Or, E. Abscisic acid (ABA) regulates grape bud dormancy, and dormancy release stimuli may act through modification of ABA metabolism. J. Exp. Bot. 2015, 66, 1527–1542. [Google Scholar] [CrossRef]
- Wang, D.L.; Gao, Z.Z.; Du, P.Y.; Xiao, W.; Tan, Q.P.; Chen, X.D.; Li, L.; Gao, D.S. Expression of ABA Metabolism-Related Genes Suggests Similarities and Differences Between Seed Dormancy and Bud Dormancy of Peach (Prunus persica). Front. Plant Sci. 2016, 6, 1248. [Google Scholar] [CrossRef]
- Tuan, P.A.; Bai, S.; Saito, T.; Ito, A.; Moriguchi, T. Dormancy-Associated MADS-Box (DAM) and the Abscisic Acid Pathway Regulate Pear Endodormancy Through a Feedback Mechanism. Plant Cell Physiol. 2017, 58, 1378–1390. [Google Scholar] [CrossRef]
- Li, J.Z.; Xu, Y.; Niu, Q.F.; He, L.F.; Teng, Y.W.; Bai, S.L. Abscisic Acid (ABA) Promotes the Induction and Maintenance of Pear (Pyrus pyrifolia White Pear Group) Flower Bud Endodormancy. Int. J. Mol. Sci. 2018, 19, 310. [Google Scholar] [CrossRef]
- Chmielewski, F.M.; Baldermann, S.; Götz, K.P.; Homann, T.; Gödeke, K.; Schumacher, F.; Huschek, G.; Rawel, H.M. Abscisic Acid Related Metabolites in Sweet Cherry Buds (Prunus avium L.). J. Hortic. 2018, 5, 1000221. [Google Scholar]
- Bai, S.L.; Saito, T.; Sakamoto, D.; Ito, A.; Fujii, H.; Moriguchi, T. Transcriptome Analysis of Japanese Pear (Pyrus pyrifolia Nakai) Flower Buds Transitioning Through Endodormancy. Plant Cell Physiol. 2013, 54, 1132–1151. [Google Scholar] [CrossRef]
- Nagar, P.K.; Sood, S. Changes in endogenous auxins during winter dormancy in tea (Camellia sinensis L.) O. Kuntze. Acta Physiol. Plant. 2006, 28, 165–169. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Zhuo, X.K.; Zhao, K.; Zheng, T.C.; Han, Y.; Yuan, C.Q.; Zhang, Q.X. Transcriptome Profiles Reveal the Crucial Roles of Hormone and Sugar in the Bud Dormancy of Prunus mume. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Zhong, W.J.; Gao, Z.H.; Zhuang, W.B.; Shi, T.; Zhang, Z.; Ni, Z.J. Genome-wide expression profiles of seasonal bud dormancy at four critical stages in Japanese apricot. Plant Mol. Biol. 2013, 83, 247–264. [Google Scholar] [CrossRef]
- Howe, G.T.; Horvath, D.P.; Dharmawardhana, P.; Priest, H.D.; Mockler, T.C.; Strauss, S.H. Extensive Transcriptome Changes During Natural Onset and Release of Vegetative Bud Dormancy in Populus. Front. Plant Sci. 2015, 6, 989. [Google Scholar] [CrossRef]
- Lee, D.J.; Park, J.W.; Lee, H.W.; Kim, J. Genome-wide analysis of the auxin-responsive transcriptome downstream of iaa1 and its expression analysis reveal the diversity and complexity of auxin-regulated gene expression. J. Exp. Bot. 2009, 60, 3935–3957. [Google Scholar] [CrossRef]
- Kepinski, S.; Leyser, O. The Arabidopsis F-box protein TIR1 is an auxin receptor. Nature 2005, 435, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Yu, Q.; Liu, J.; Wen, X.; Yan, Z.; Hu, K.; Li, H.; Kong, X.; Li, C.; Tian, H.; et al. Non-canonical AUX/IAA protein IAA33 competes with canonical AUX/IAA repressor IAA5 to negatively regulate auxin signaling. EMBO J. 2020, 39, e101515. [Google Scholar] [CrossRef]
- Luo, J.; Zhou, J.J.; Zhang, J.Z. Aux/IAA Gene Family in Plants: Molecular Structure, Regulation, and Function. Int. J. Mol. Sci. 2018, 19, 259. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Sasaki, T.; Ueda, M.; Sako, K.; Seki, M. Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front. Plant Sci. 2015, 6, 114. [Google Scholar] [CrossRef]
- Dunoyer, P.; Himber, C.; Voinnet, O. DICER-LIKE 4 is required for RNA interference and produces the 21-nucleotide small interfering RNA component of the plant cell-to-cell silencing signal. Nat. Genet. 2005, 37, 1356–1360. [Google Scholar] [CrossRef]
- Liu, B.; Chen, Z.; Song, X.; Liu, C.; Cui, X.; Zhao, X.; Fang, J.; Xu, W.; Zhang, H.; Wang, X.; et al. Oryza sativa dicer-like4 reveals a key role for small interfering RNA silencing in plant development. Plant Cell 2007, 19, 2705–2718. [Google Scholar] [CrossRef]
- Vazquez, F.; Legrand, S.; Windels, D. The biosynthetic pathways and biological scopes of plant small RNAs. Trends Plant Sci. 2010, 15, 337–345. [Google Scholar] [CrossRef]
- Shen, D.; Wang, S.; Chen, H.; Zhu, Q.-H.; Helliwell, C.; Fan, L. Molecular phylogeny of miR390-guided trans-acting siRNA genes (TAS3) in the grass family. Plant Syst. Evol. 2009, 283, 125–132. [Google Scholar] [CrossRef]
- Richardson, E.A.; Seeley, S.D.; Walker, D.R. A model for estimating the completion of rest for ‘Redhaven’ and ‘Elberta’ peach trees. Hortscience 1974, 9, 331–332. [Google Scholar] [CrossRef]
- Hafner, M.; Landgraf, P.; Ludwig, J.; Rice, A.; Ojo, T.; Lin, C.; Holoch, D.; Lim, C.; Tuschl, T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 2008, 44, 3–12. [Google Scholar] [CrossRef]
- Fang, X.; Zhao, Y.; Ma, Q.; Huang, Y.; Wang, P.; Zhang, J.; Nian, H.; Yang, C. Identification and comparative analysis of cadmium tolerance-associated miRNAs and their targets in two soybean genotypes. PLoS ONE 2013, 8, e81471. [Google Scholar] [CrossRef] [PubMed]
- Xiaofang, X. Identification of miRNA Target Genes in Tea (Camellia sinensis) and Their Expression Patterns under Cold Stress; Anhui Agricultural University: Hefei, China, 2016. [Google Scholar]
- German, M.A.; Luo, S.; Schroth, G.; Meyers, B.C.; Green, P.J. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat. Protoc. 2009, 4, 356–362. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank: Update. Nucleic Acids Res. 2004, 32, D23–D26. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, Y.; Huang, N.; Liu, F.; Su, W.; Xu, L.; Ahmad, W.; Wu, Q.; Guo, J.; Que, Y. Small RNA sequencing reveals a role for sugarcane miRNAs and their targets in response to Sporisorium scitamineum infection. BMC Genom. 2017, 18, 325. [Google Scholar] [CrossRef]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.M.; Green, P.J.; et al. Criteria for Annotation of Plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Li, R.Q.; Li, Y.R.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Folkes, L.; Moxon, S.; Woolfenden, H.C.; Stocks, M.B.; Szittya, G.; Dalmay, T.; Moulton, V. PAREsnip: A tool for rapid genome-wide discovery of small RNA/target interactions evidenced through degradome sequencing. Nucleic Acids Res. 2012, 40, e103. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Count of Each Sample | Total | ||||||
|---|---|---|---|---|---|---|---|---|
| FB.Nov | nF16 | FB150 | FB300 | FB450 | IB570 | Count | Percent (%) | |
| Clean tags | 59,744,323 | 48,982,971 | 60,793,889 | 54,640,035 | 54,201,813 | 46,146,283 | 324,509,314 | 100.00 |
| rRNA | 5,502,518 | 7,399,048 | 5,744,220 | 5,189,114 | 5,899,625 | 5,018,955 | 34,753,480 | 10.71 |
| snRNA | 100,945 | 91,590 | 65,898 | 58,395 | 66,318 | 108,220 | 491,366 | 0.15 |
| snoRNA | 39,384 | 58,075 | 51,357 | 58,198 | 60,944 | 50,743 | 318,701 | 0.10 |
| tRNA | 219,683 | 183,278 | 307,234 | 312,200 | 285,118 | 276,611 | 1,584,124 | 0.49 |
| scRNA | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.00 |
| Repeat | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.00 |
| Transcriptome | 13,462,284 | 11,230,416 | 13,953,551 | 12,197,013 | 13,505,105 | 9,878,030 | 74,226,399 | 22.87 |
| Known miRNAs | 10,771,326 | 9,898,793 | 12,640,126 | 13,364,950 | 11,784,001 | 15,704,868 | 74,164,064 | 22.85 |
| Novel miRNAs | 167,975 | 59,549 | 226,927 | 155,615 | 244,083 | 160,774 | 1,014,923 | 0.31 |
| Unannotated | 29,480,208 | 20,062,222 | 27,804,576 | 23,304,550 | 22,356,619 | 14,948,082 | 137,956,257 | 42.51 |
| Category | Total Tags | Unique Tags | ||
|---|---|---|---|---|
| Count | Percent (%) | Count | Percent (%) | |
| Clean tags | 37,913,833 | 100.00 | 12,290,544 | 100.00 |
| rRNA | 542,685 | 1.43 | 21,435 | 0.17 |
| tRNA | 488 | 0.00 | 140 | 0.00 |
| snRNA | 762 | 0.00 | 320 | 0.00 |
| snoRNA | 26,782 | 0.07 | 371 | 0.00 |
| polyN | 65,227 | 0.17 | 32,771 | 0.27 |
| cDNA_sense | 14,139,730 | 37.29 | 3,850,879 | 31.33 |
| cDNA_antisense | 12,076,616 | 31.85 | 3,581,528 | 29.14 |
| Unannotated | 11,061,543 | 29.18 | 4,803,100 | 39.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, N.; Jiang, Y.; Zhu, T.; Li, Z.; Sui, S. Small RNA and Degradome Sequencing in Floral Bud Reveal Roles of miRNAs in Dormancy Release of Chimonanthus praecox. Int. J. Mol. Sci. 2023, 24, 4210. https://doi.org/10.3390/ijms24044210
Liu N, Jiang Y, Zhu T, Li Z, Sui S. Small RNA and Degradome Sequencing in Floral Bud Reveal Roles of miRNAs in Dormancy Release of Chimonanthus praecox. International Journal of Molecular Sciences. 2023; 24(4):4210. https://doi.org/10.3390/ijms24044210
Chicago/Turabian StyleLiu, Ning, Yingjie Jiang, Ting Zhu, Zhineng Li, and Shunzhao Sui. 2023. "Small RNA and Degradome Sequencing in Floral Bud Reveal Roles of miRNAs in Dormancy Release of Chimonanthus praecox" International Journal of Molecular Sciences 24, no. 4: 4210. https://doi.org/10.3390/ijms24044210
APA StyleLiu, N., Jiang, Y., Zhu, T., Li, Z., & Sui, S. (2023). Small RNA and Degradome Sequencing in Floral Bud Reveal Roles of miRNAs in Dormancy Release of Chimonanthus praecox. International Journal of Molecular Sciences, 24(4), 4210. https://doi.org/10.3390/ijms24044210