
N-Acetylcysteine Suppresses Microglial Inflammation and Induces Mortality Dose-Dependently via Tumor Necrosis Factor-α Signaling

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
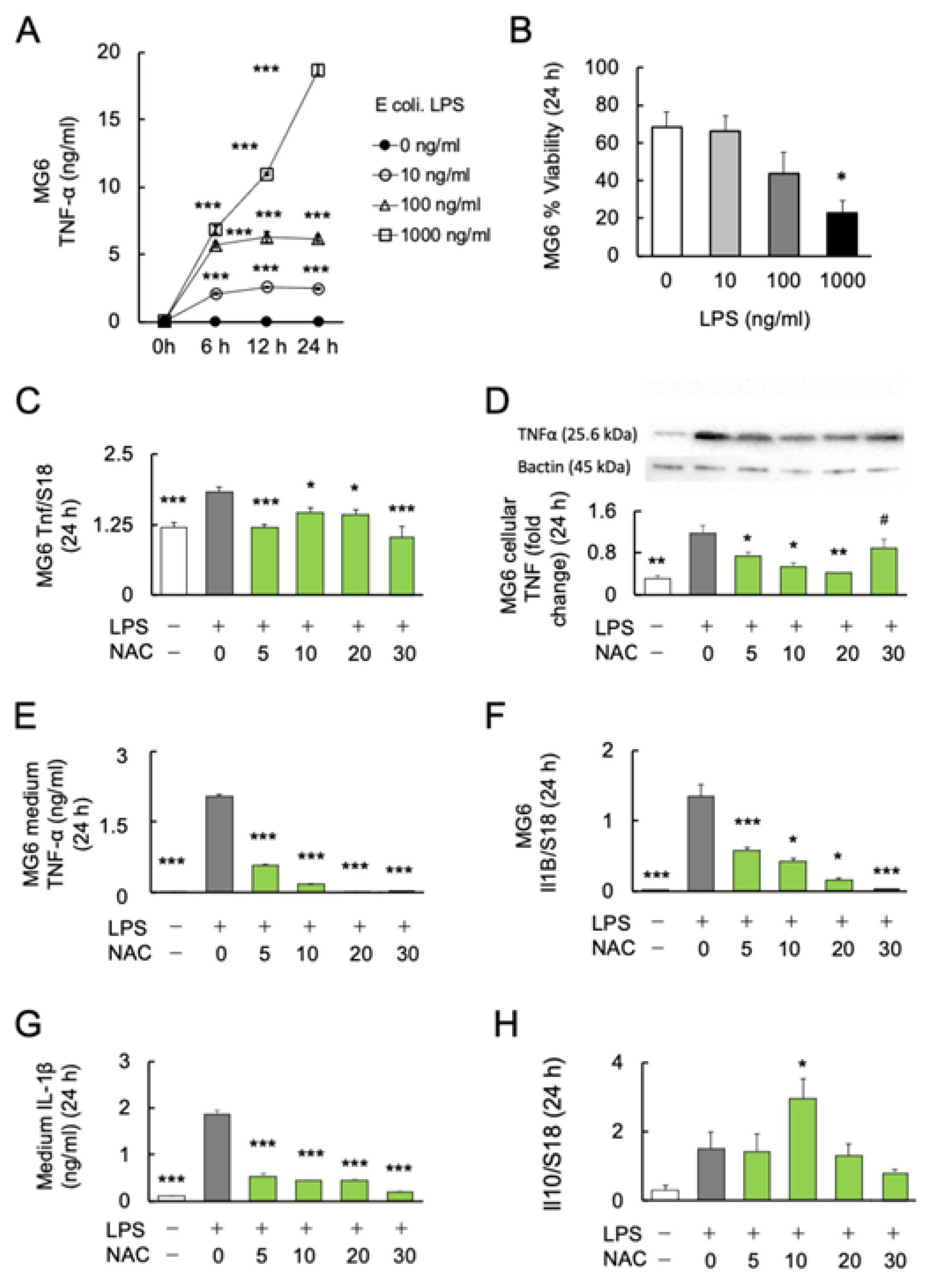
2.1. Effect of LPS on Microglial Activation and Viability
2.2. Effects of NAC on LPS-Induced Cytokine Production in MG6 Cells
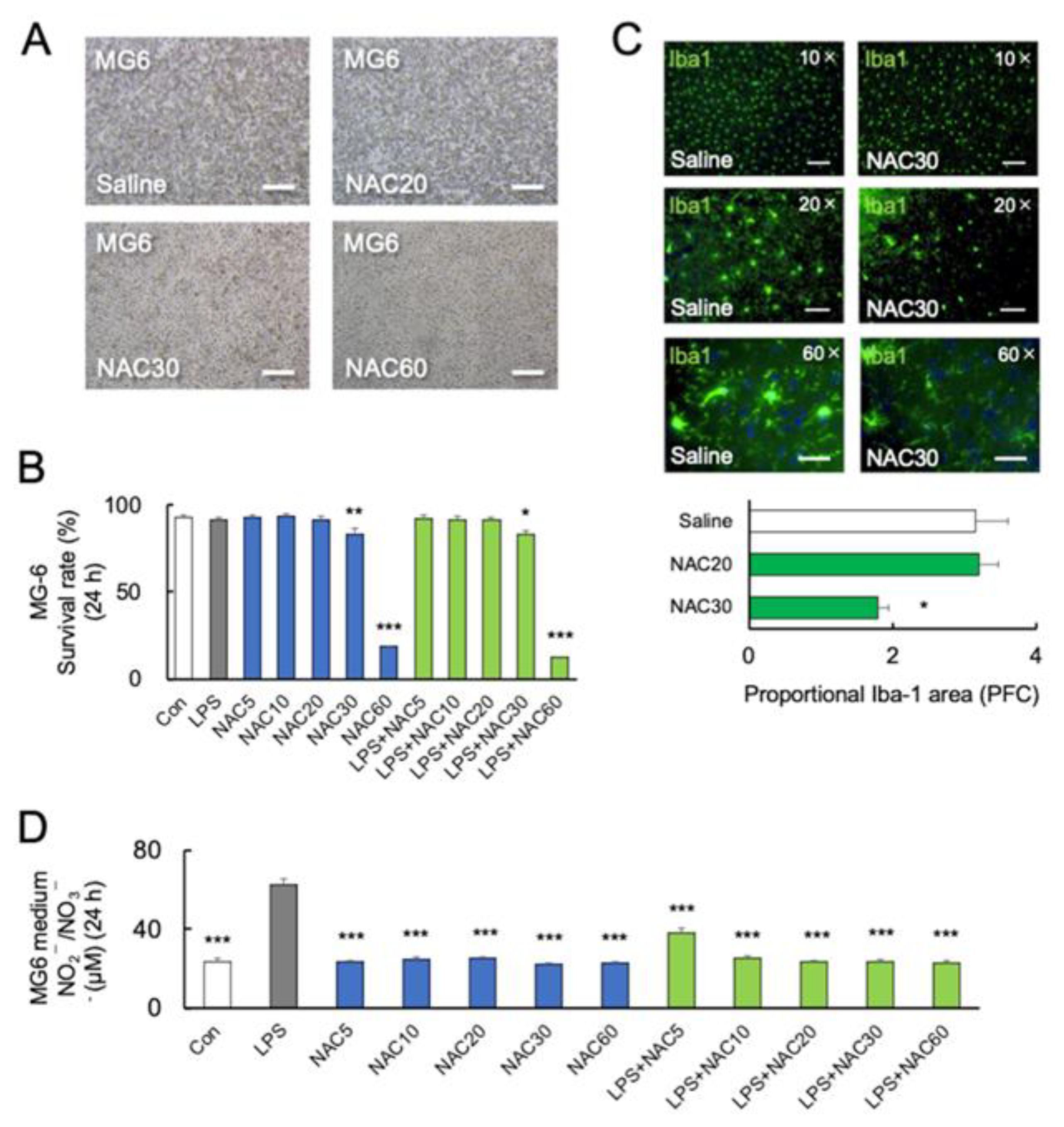
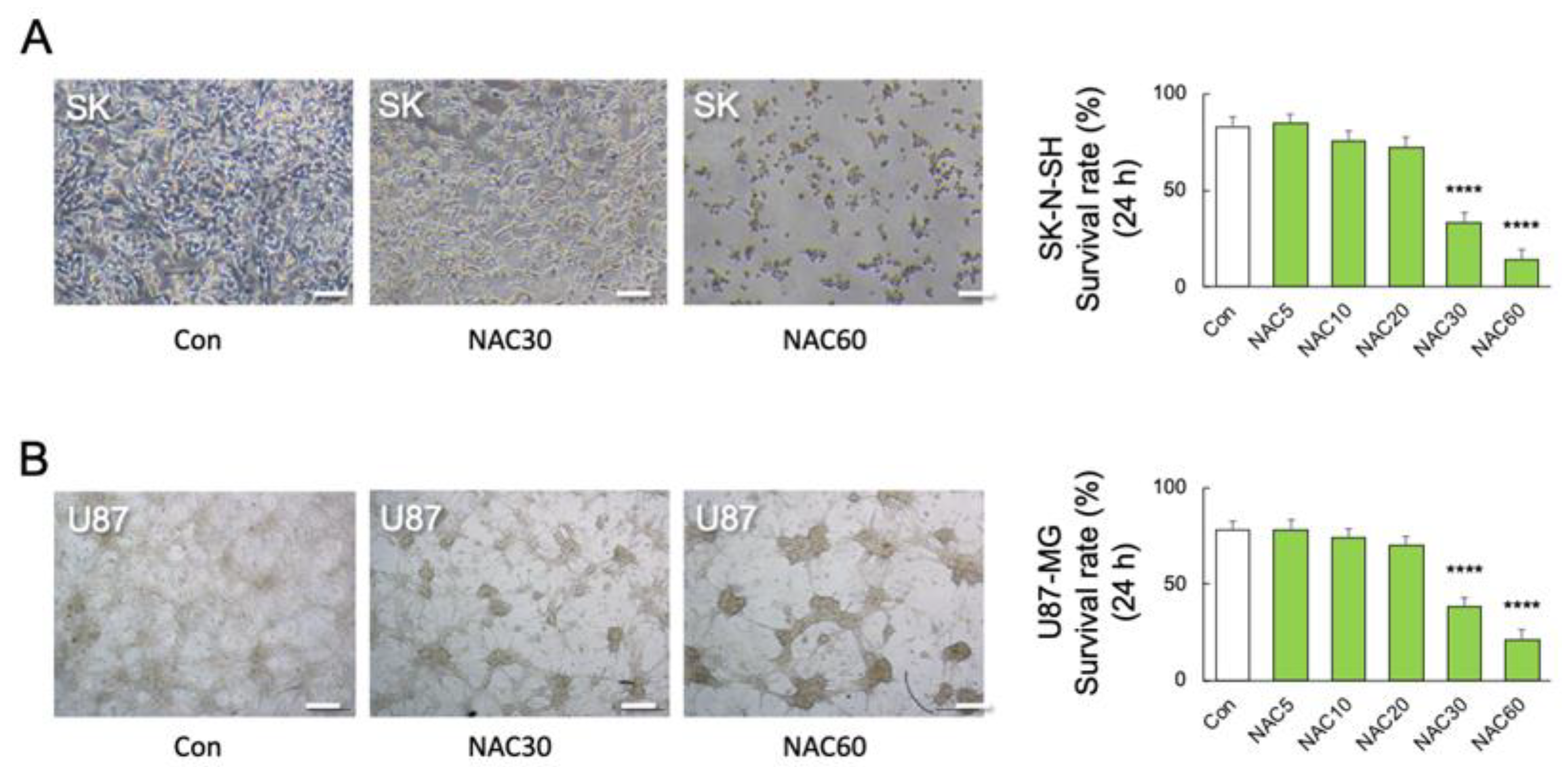
2.3. High Concentration of NAC Increased Microglial Mortality without LPS Challenge
2.4. Role of Nitric Oxide in NAC-Induced Microglial Mortality
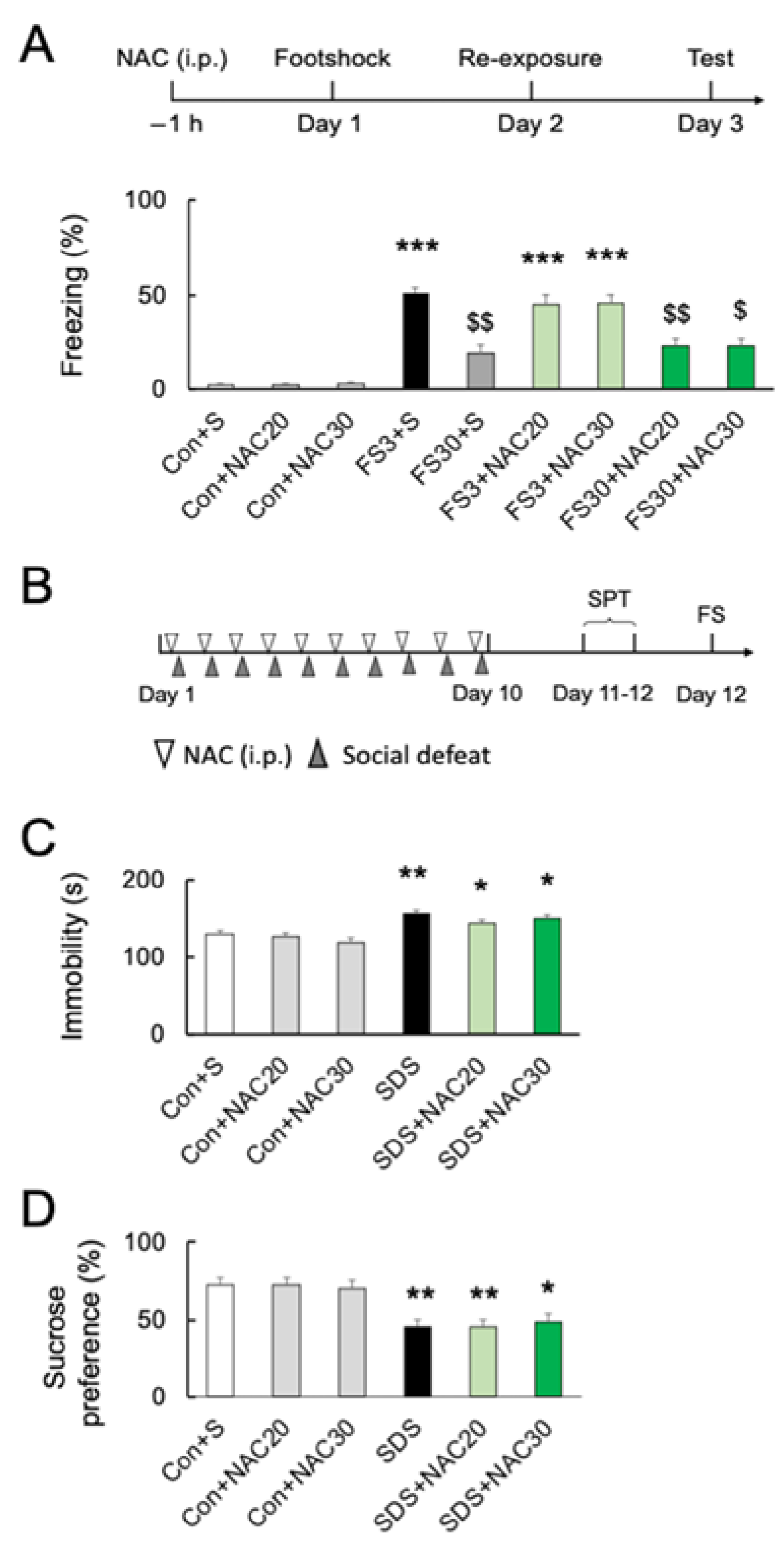
2.5. Effects of NAC on Acute and Chronic Stress-Induced Behavior
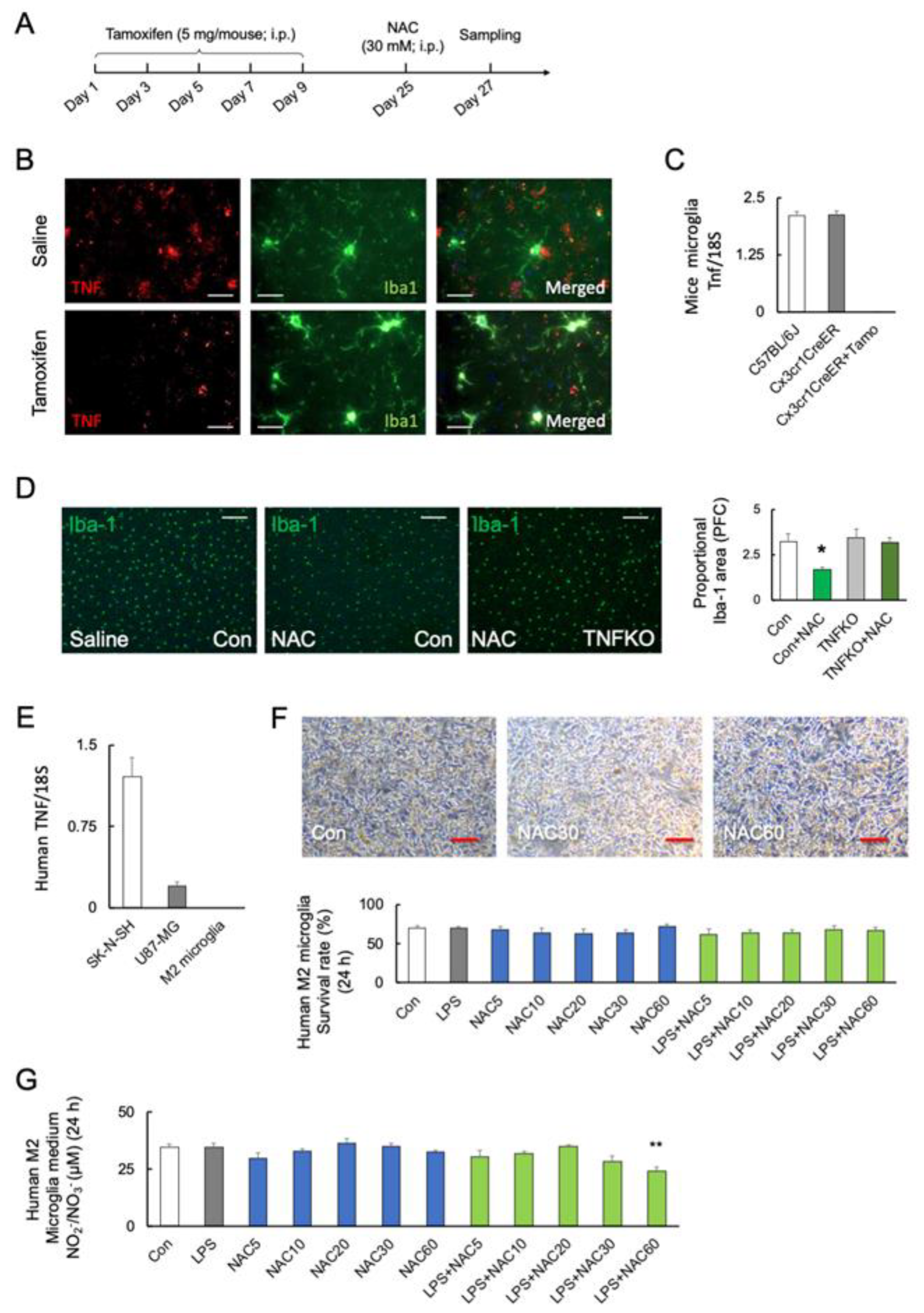
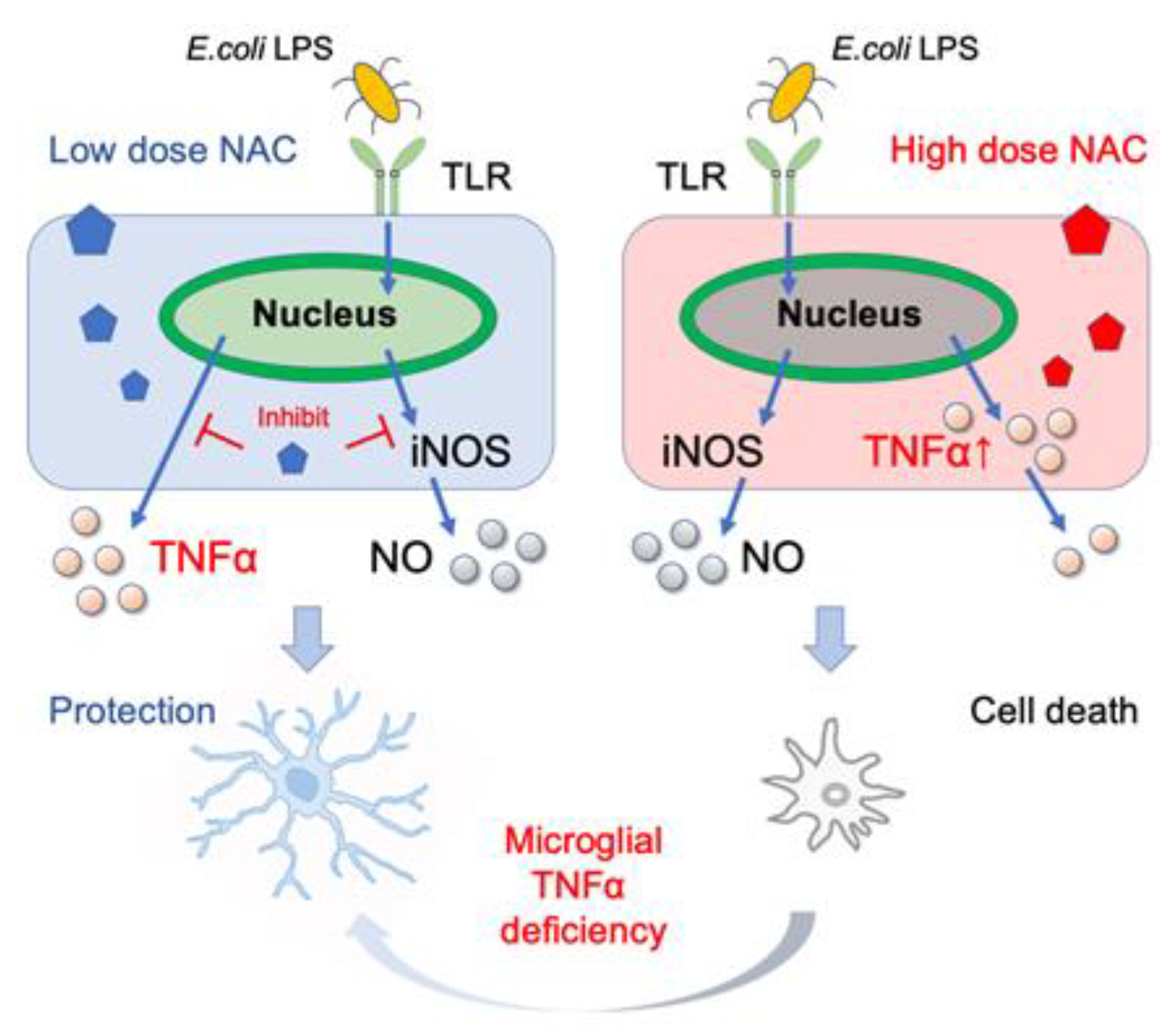
2.6. Role of Microglial TNF-α in NAC-Induced Cell Mortality
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cytokine Release
4.2. Chemicals and Treatment
4.3. Animals
4.4. Acute Stress (Contextual Fear Conditioning Tests)
4.5. Chronic Social Defeat Stress (SDS)
4.6. Sucrose Preference Test (SPT)
4.7. Forced Swim Test (FST)
4.8. Measurement Protein of TNF-α, IL-1, IL-10, Nitric Oxide, and Cell Viability
4.9. Isolation of Microglia
4.10. RNA Extraction and Quantitative Real-Time PCR
4.11. Cellular Protein Extraction and Western Blotting
4.12. Immunohistochemical Analysis of Mouse Microglia
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajendran, P.; Chen, Y.; Chen, Y.; Chung, L.; Tamilselvi, S.; Shen, C.; Day, C.H.; Chen, R.; Viswanadha, V.P.; Kuo, W.; et al. The multifaceted link between inflammation and human diseases. J. Cell. Physiol. 2018, 233, 6458–6471. [Google Scholar] [CrossRef] [PubMed]
- Réus, G.; Fries, G.; Stertz, L.; Badawy, M.; Passos, I.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Issels, R.D.; Nagele, A.; Eckert, K.-G.; Wllmanns, W. Promotion of cystine uptake and its utilization for glutathione biosynthesis induced by cysteamine and N-acetylcysteine. Biochem. Pharmacol. 1988, 37, 881–888. [Google Scholar] [CrossRef]
- Karalija, A.; Novikova, L.; Kingham, P.; Wiberg, M. The effects of N-acetyl-cysteine and acetyl-l-carnitine on neural survival, neuroinflammation and regeneration following spinal cord injury. Neuroscience 2014, 269, 143–151. [Google Scholar] [CrossRef]
- Amin, A.F.; Shaaban, O.M.; Bediawy, M.A. N-acetyl cysteine for treatment of recurrent unexplained pregnancy loss. Reprod. Biomed. Online 2008, 17, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef] [PubMed]
- Zafarullah, M.; Li, W.Q.; Sylvester, J.; Ahmad, M. Molecular mechanisms of N-acetylcysteine actions. Cell. Mol. Life Sci. 2003, 60, 6–20. [Google Scholar] [CrossRef]
- Pahan, K.; Sheikh, F.G.; Namboodiri, A.M.; Singh, I. N-Acetyl Cysteine Inhibits Induction of No Production by Endotoxin or Cytokine Stimulated Rat Peritoneal Macrophages, C6 Glial Cells and Astrocytes. Free Radic. Biol. Med. 1998, 24, 39–48. [Google Scholar] [CrossRef]
- Yu, G.; Zheng, S.; Zhang, H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces experimental autoimmune encephalomyelitis-induced injury and promotes oligodendrocyte regeneration and neurogenesis in a murine model of progressive multiple sclerosis. Neuroreport 2018, 29, 208–213. [Google Scholar] [CrossRef]
- Stanislaus, R.; Gilg, A.G.; Singh, A.K.; Singh, I. N-acetyl-L-cysteine ameliorates the inflammatory disease process in experimental autoimmune encephalomyelitis in Lewis rats. J. Autoimmune Dis. 2005, 2, 4. [Google Scholar] [CrossRef]
- Moreira, P.I.; Smith, M.A.; Zhu, X.; Nunomura, A.; Castellani, R.J.; Perry, G. Oxidative stress and neurodegeneration. Ann. N. Y. Acad. Sci. 2005, 1043, 545–552. [Google Scholar] [CrossRef]
- Rammal, H.; Bouayed, J.; Younos, C.; Soulimani, R. Evidence that oxidative stress is linked to anxiety-related behaviour in mice. Brain Behav. Immun. 2008, 22, 1156–1159. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Kapczinski, F.; Andreazza, A.; Dean, O.; Giorlando, F.; Maes, M.; Yücel, M.; Gama, C.; Dodd, S.; Dean, B.; et al. Pathways underlying neuroprogression in bipolar disorder: Focus on inflammation, oxidative stress and neurotrophic factors. Neurosci. Biobehav. Rev. 2011, 35, 804–817. [Google Scholar] [CrossRef]
- Skvarc, D.R.; Dean, O.M.; Byrne, L.K.; Gray, L.; Lane, S.; Lewis, M.; Fernandes, B.S.; Berk, M.; Marriott, A. The effect of N-acetylcysteine (NAC) on human cognition—A systematic review. Neurosci. Biobehav. Rev. 2017, 78, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.J.; Francis, L.; Dawood, M.; Lai, Z.-W.; Faraone, S.V.; Perl, A. Attention Deficit and Hyperactivity Disorder Scores Are Elevated and Respond toN-Acetylcysteine Treatment in Patients with Systemic Lupus Erythematosus. Arthritis Rheum. 2013, 65, 1313–1318. [Google Scholar] [CrossRef]
- Magalhães, P.; Dean, O.; Bush, A.; Copolov, D.; Malhi, G.; Kohlmann, K.; Jeavons, S.; Schapkaitz, I.; Anderson-Hunt, M.; Berk, M. N-acetyl cysteine add-on treatment for bipolar II disorder: A subgroup analysis of a randomized placebo-controlled trial. J. Affect. Disord. 2011, 129, 317–320. [Google Scholar] [CrossRef]
- Berk, M.; Copolov, D.; Dean, O.; Lu, K.; Jeavons, S.; Schapkaitz, I.; Anderson-Hunt, M.; Judd, F.; Katz, F.; Katz, P.; et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—A double-blind, randomized, placebo-controlled trial. Biol Psychiatry 2008, 64, 361–368. [Google Scholar] [CrossRef]
- Berk, M.; Dean, O.M.; Cotton, S.M.; Jeavons, S.; Tanious, M.; Kohlmann, K.; Hewitt, K.; Moss, K.; Allwang, C.; Schapkaitz, I.; et al. The efficacy of adjunctive N-acetylcysteine in major depressive disorder: A double-blind, randomized, placebo-controlled trial. J. Clin. Psychiatry 2014, 75, 628–636. [Google Scholar] [CrossRef]
- Sakai, M.; Yu, Z.; Hirayama, R.; Nakasato, M.; Kikuchi, Y.; Ono, C.; Komatsu, H.; Nakanishi, M.; Yoshii, H.; Stellwagen, D.; et al. Deficient Autophagy in Microglia Aggravates Repeated Social Defeat Stress-Induced Social Avoidance. Neural Plast. 2022, 2022, 7503553. [Google Scholar] [CrossRef]
- Yu, Z.; Fukushima, H.; Ono, C.; Sakai, M.; Kasahara, Y.; Kikuchi, Y.; Gunawansa, N.; Takahashi, Y.; Matsuoka, H.; Kida, S.; et al. Microglial production of TNF-alpha is a key element of sustained fear memory. Brain Behav. Immun. 2017, 59, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Ohtaki, Y.; Kai, K.; Sasano, T.; Shimauchi, H.; Yokochi, T.; Takada, H.; Sugawara, S.; Kumagai, K.; Endo, Y. Critical roles of platelets in lipopolysaccharide-induced lethality: Effects of glycyrrhizin and possible strategy for acute respiratory distress syndrome. Int. Immunopharmacol. 2005, 5, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Chen, C.; Hou, J.; Xin, W.; Mori, A. Nitric oxide induces oxidative stress and apoptosis in neuronal cells. Biochim. Biophys. Acta 2000, 1498, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Ciotti, G.M.P.; Braun, D.; Kalinin, S.; Currò, D.; Dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef]
- Simpson, D.; Oliver, P. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Mayer, B.; John, M.; Heinzel, B.; Werner, E.R.; Wachter, H.; Schultz, G.; Böhme, E. Brain nitric oxide synthase is a biopterin- and flavin-containing multi-functional oxido-reductase. FEBS Lett. 1991, 288, 187–191. [Google Scholar] [CrossRef]
- Rettori, V.; Fernandez-Solari, J.; Mohn, C.; Zubilete, M.A.Z.; De La Cal, C.; Prestifilippo, J.P.; De Laurentiis, A. Nitric Oxide at the Crossroad of Immunoneuroendocrine Interactions. Ann. N. Y. Acad. Sci. 2009, 1153, 35–47. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Weitzberg, E. Nitric oxide signaling in health and disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef]
- Yagci, G.; Gul, H.; Simsek, A.; Buyukdogan, V.; Onguru, O.; Zeybek, N.; Aydin, A.; Balkan, M.; Yildiz, O.; Sen, D. Beneficial effects of N-acetylcysteine on sodium taurocholate-induced pancreatitis in rats. J. Gastroenterol. 2004, 39, 268–276. [Google Scholar] [CrossRef]
- Jayasooriya, R.G.; Kang, C.-H.; Seo, M.-J.; Choi, Y.H.; Jeong, Y.-K.; Kim, G.-Y. Exopolysaccharide of Laetiporus sulphureus var. miniatus downregulates LPS-induced production of NO, PGE2, and TNF-α in BV2 microglia cells via suppression of the NF-κB pathway. Food Chem. Toxicol. 2011, 49, 2758–2764. [Google Scholar] [CrossRef] [PubMed]
- Min, K.J.; Jou, I.; Joe, E. Plasminogen-induced IL-1beta and TNF-alpha production in microglia is regulated by reactive oxygen species. Biochem. Biophys. Res. Commun. 2003, 312, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Kim, B.S.; Im, H.-I. Pathophysiological Role of Neuroinflammation in Neurodegenerative Diseases and Psychiatric Disorders. Int. Neurourol. J. 2016, 20 (Suppl. 1), S2–S7. [Google Scholar] [CrossRef]
- Yu, Z.; Sakai, M.; Fukushima, H.; Ono, C.; Kikuchi, Y.; Koyama, R.; Matsui, K.; Furuyashiki, T.; Kida, S.; Tomita, H. Contextual fear conditioning regulates synapse-related gene transcription in mouse microglia. Brain Res. Bull. 2022, 189, 57–68. [Google Scholar] [CrossRef]
- Gill, J.; Vythilingam, M.; Page, G.G. Low cortisol, high DHEA, and high levels of stimulated TNF-alpha, and IL-6 in women with PTSD. J. Trauma. Stress 2008, 21, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Fukushima, H.; Yu, Z.; Tomita, H.; Kida, S. Tumor necrosis factor α negatively regulates the retrieval and reconsolidation of hippocampus-dependent memory. Brain Behav. Immun. 2021, 94, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Himmerich, H.; Berthold-Losleben, M.; Pollmächer, T. The relevance of the TNF-alpha system in psychiatric disorders. Fortschr. Neurol. Psychiatr. 2009, 77, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.-Y.; Wu, T.-C.; Yang, S.-F.; Chang, J.T. Effects of NAC and Gallic Acid on the Proliferation Inhibition and Induced Death of Lung Cancer Cells with Different Antioxidant Capacities. Molecules 2021, 27, 75. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Wu, M.-S.; Lien, G.-S.; Shen, S.-C.; Yang, L.-Y.; Chen, Y.-C. N-acetyl-L-cysteine enhances fisetin-induced cytotoxicity via induction of ROS-independent apoptosis in human colonic cancer cells. Mol. Carcinog. 2014, 53 (Suppl. 1), E119–E129. [Google Scholar] [CrossRef] [PubMed]
- Kopke, R.D.; Jackson, R.L.; Coleman, J.K.; Liu, J.; Bielefeld, E.C.; Balough, B.J. NAC for noise: From the bench top to the clinic. Heart Res. 2007, 226, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Childs, A.; Jacobs, C.; Kaminski, T.; Halliwell, B.; Leeuwenburgh, C. Supplementation with vitamin C and N-acetyl-cysteine increases oxidative stress in humans after an acute muscle injury induced by eccentric exercise. Free Radic. Biol. Med. 2001, 31, 745–753. [Google Scholar] [CrossRef]
- Palmer, L.A.; Doctor, A.; Chhabra, P.; Sheram, M.L.; Laubach, V.; Karlinsey, M.Z.; Forbes, M.S.; Macdonald, T.; Gaston, B. S-Nitrosothiols signal hypoxia-mimetic vascular pathology. J. Clin. Investig. 2007, 117, 2592–2601. [Google Scholar] [CrossRef]
- Sprong, R.C.; Winkelhuyzen-Janssen, A.M.; Aarsman, C.J.; van Oirschot, J.F.; van der Bruggen, T.; van Asbeck, B.S. Low-dose N-acetylcysteine protects rats against endotoxin-mediated oxidative stress, but high-dose increases mortality. Am. J. Respir. Crit. Care Med. 1998, 157 Pt 1, 1283–1293. [Google Scholar] [CrossRef]
- Farr, S.A.; Poon, H.F.; Dogrukol-Ak, D.; Drake, J.; Banks, W.A.; Eyerman, E.L.; Butterfield, D.A.; Morley, J.E. The antioxidants α-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neurochem. 2003, 84, 1173–1183. [Google Scholar] [CrossRef]
- Fiorentino, D.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; O’Garra, A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991, 147, 3815–3822. [Google Scholar] [CrossRef]
- Terneus, M.V.; Brown, J.M.; Carpenter, A.B.; Valentovic, M.A. Comparison of S-adenosyl-l-methionine (SAMe) and N-acetylcysteine (NAC) protective effects on hepatic damage when administered after acetaminophen overdose. Toxicology 2008, 244, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008, 7, 156–167. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, Y.R.; Jeong, W.Y.; Lee, S.; Shin, J.H.; Lee, G.-J. Potential Protective Effect of Nitric Oxide-Releasing Nanofibers in Hypoxia/Reoxygenation-Induced Cardiomyocyte Injury. J. Nanosci. Nanotechnol. 2019, 19, 6539–6545. [Google Scholar] [CrossRef]
- Flanagan, R.J.; Meredith, T. Use of N-acetylcysteine in clinical toxicology. Am. J. Med. 1991, 91, S131–S139. [Google Scholar] [CrossRef]
- Guerini, M.; Condrò, G.; Friuli, V.; Maggi, L.; Perugini, P. N-acetylcysteine (NAC) and Its Role in Clinical Practice Management of Cystic Fibrosis (CF): A Review. Pharmaceuticals 2022, 15, 217. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Malhi, G.S.; Gray, L.J.; Dean, O.M. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol. Sci. 2013, 34, 167–177. [Google Scholar] [CrossRef]
- Branco, V.; Coppo, L.; Aschner, M.; Carvalho, C. N-Acetylcysteine or Sodium Selenite Prevent the p38-Mediated Production of Proinflammatory Cytokines by Microglia during Exposure to Mercury (II). Toxics 2022, 10, 433. [Google Scholar] [CrossRef]
- Moschou, M.; Kosmidis, E.K.; Kaloyianni, M.; Geronikaki, A.; Dabarakis, N.; Theophilidis, G. In vitro assessment of the neurotoxic and neuroprotective effects of N-acetyl-l-cysteine (NAC) on the rat sciatic nerve fibers. Toxicol. Vitr. 2008, 22, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.W.; Dewhurst, S.; Bellizzi, M.J.; Gelbard, H.A. Tumor necrosis factor-alpha in normal and diseased brain: Conflicting effects via intraneuronal receptor crosstalk? J. Neurovirol. 2002, 8, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef]
- Park, S.H.; Park-Min, K.-H.; Chen, J.; Hu, X.; Ivashkiv, L.B. Tumor necrosis factor induces GSK3 kinase–mediated cross-tolerance to endotoxin in macrophages. Nat. Immunol. 2011, 12, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef]
- Golan, H.; Levav, T.; Mendelsohn, A.; Huleihel, M. Involvement of Tumor Necrosis Factor Alpha in Hippocampal Development and Function. Cereb. Cortex 2004, 14, 97–105. [Google Scholar] [CrossRef]
- Aloe, L.; Properzi, F.; Probert, L.; Akassoglou, K.; Kassiotis, G.; Micera, A.; Fiore, M. Learning abilities, NGF and BDNF brain levels in two lines of TNF-alpha transgenic mice, one characterized by neurological disorders, the other phenotypically normal. Brain Res. 1999, 840, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Lewitus, G.M.; Konefal, S.C.; Greenhalgh, A.D.; Pribiag, H.; Augereau, K.; Stellwagen, D. Microglial TNF-α Suppresses Cocaine-Induced Plasticity and Behavioral Sensitization. Neuron 2016, 90, 483–491. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Favuzzi, E.; Huang, S.; Saldi, G.A.; Binan, L.; Ibrahim, L.A.; Fernández-Otero, M.; Cao, Y.; Zeine, A.; Sefah, A.; Zheng, K.; et al. GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell 2021, 184, 4048–4063.e32. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.; Littlewood, T.; Soucek, L. Tamoxifen Administration to Mice. Cold Spring Harb. Protoc. 2015, 2015, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Kitaoka, S.; Tanaka, K.; Segi-Nishida, E.; Imoto, Y.; Ogawa, A.; Nakano, F.; Tomohiro, A.; Nakayama, K.; Taniguchi, M.; et al. The Innate Immune Receptors TLR2/4 Mediate Repeated Social Defeat Stress-Induced Social Avoidance through Prefrontal Microglial Activation. Neuron 2018, 99, 464–479.e7. [Google Scholar] [CrossRef] [PubMed]
- Kuprash, D.V.; Tumanov, A.V.; Liepinsh, D.J.; Koroleva, E.P.; Drutskaya, M.S.; Kruglov, A.A.; Shakhov, A.N.; Southon, E.; Murphy, W.J.; Tessarollo, L.; et al. Novel tumor necrosis factor-knockout mice that lack Peyer’s patches. Eur. J. Immunol. 2005, 35, 1592–1600. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., III; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Golden, S.A.; Covington, H.E.; Berton, O.; Russo, S.J. A standardized protocol for repeated social defeat stress in mice. Nat. Protoc. 2011, 6, 1183–1191. [Google Scholar] [CrossRef]
- Can, A.; Dao, D.T.; Arad, M.; Terrillion, C.E.; Piantadosi, S.C.; Gould, T.D. The mouse forced swim test. J. Vis. Exp. 2012, 59, e3638. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakai, M.; Yu, Z.; Taniguchi, M.; Picotin, R.; Oyama, N.; Stellwagen, D.; Ono, C.; Kikuchi, Y.; Matsui, K.; Nakanishi, M.; et al. N-Acetylcysteine Suppresses Microglial Inflammation and Induces Mortality Dose-Dependently via Tumor Necrosis Factor-α Signaling. Int. J. Mol. Sci. 2023, 24, 3798. https://doi.org/10.3390/ijms24043798
Sakai M, Yu Z, Taniguchi M, Picotin R, Oyama N, Stellwagen D, Ono C, Kikuchi Y, Matsui K, Nakanishi M, et al. N-Acetylcysteine Suppresses Microglial Inflammation and Induces Mortality Dose-Dependently via Tumor Necrosis Factor-α Signaling. International Journal of Molecular Sciences. 2023; 24(4):3798. https://doi.org/10.3390/ijms24043798
Chicago/Turabian StyleSakai, Mai, Zhiqian Yu, Masayuki Taniguchi, Rosanne Picotin, Nanami Oyama, David Stellwagen, Chiaki Ono, Yoshie Kikuchi, Ko Matsui, Miharu Nakanishi, and et al. 2023. "N-Acetylcysteine Suppresses Microglial Inflammation and Induces Mortality Dose-Dependently via Tumor Necrosis Factor-α Signaling" International Journal of Molecular Sciences 24, no. 4: 3798. https://doi.org/10.3390/ijms24043798
APA StyleSakai, M., Yu, Z., Taniguchi, M., Picotin, R., Oyama, N., Stellwagen, D., Ono, C., Kikuchi, Y., Matsui, K., Nakanishi, M., Yoshii, H., Furuyashiki, T., Abe, T., & Tomita, H. (2023). N-Acetylcysteine Suppresses Microglial Inflammation and Induces Mortality Dose-Dependently via Tumor Necrosis Factor-α Signaling. International Journal of Molecular Sciences, 24(4), 3798. https://doi.org/10.3390/ijms24043798