

Molecular Mechanisms of Oxidative Stress Relief by CAPE in ARPE−19 Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
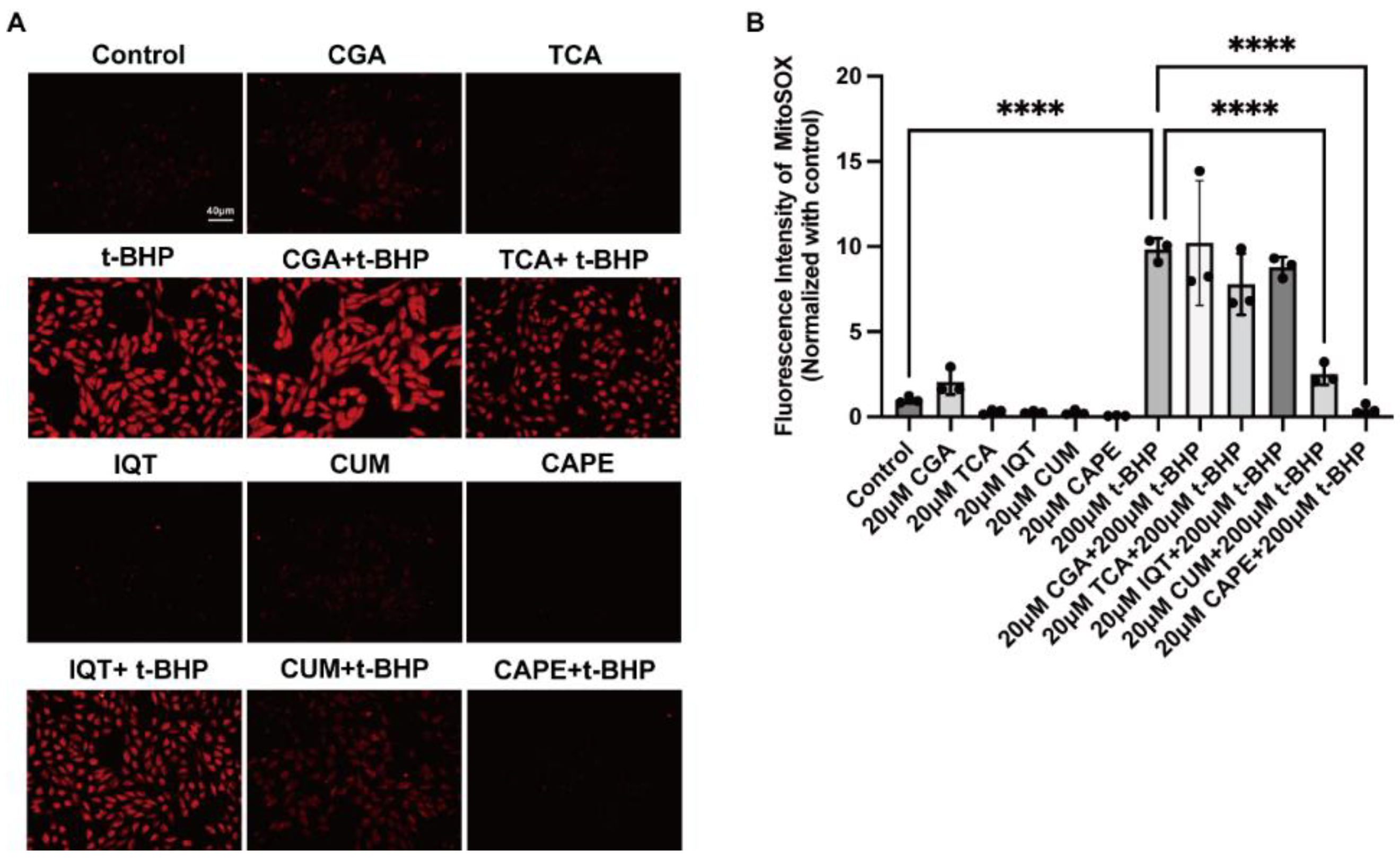
2.1. Natural Compounds with Antioxidant Properties Inhibited t-BHP Induced ROS Production in ARPE−19 Cells
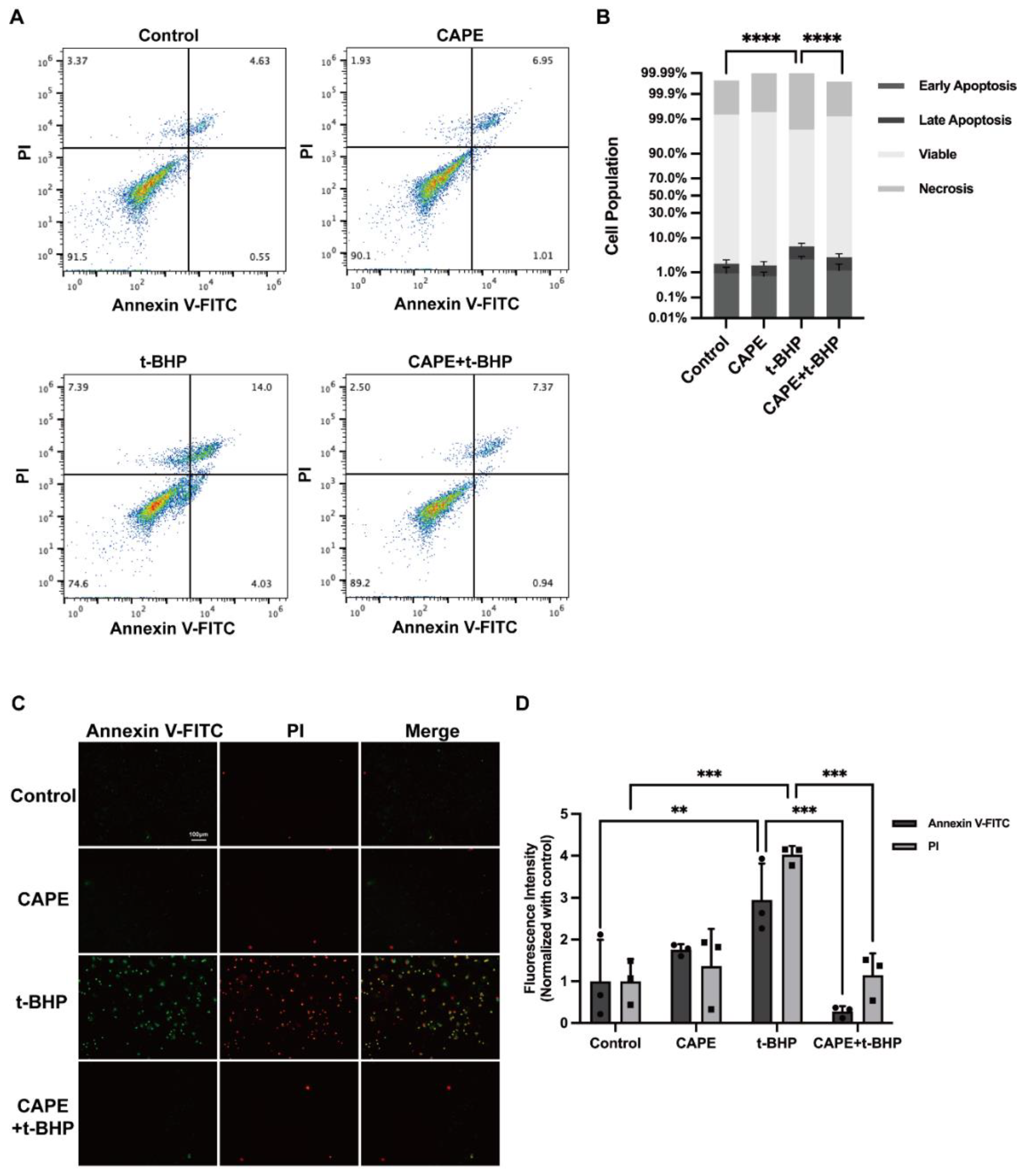
2.2. CAPE Prevents t-BHP-Induced Apoptosis in ARPE−19 Cells
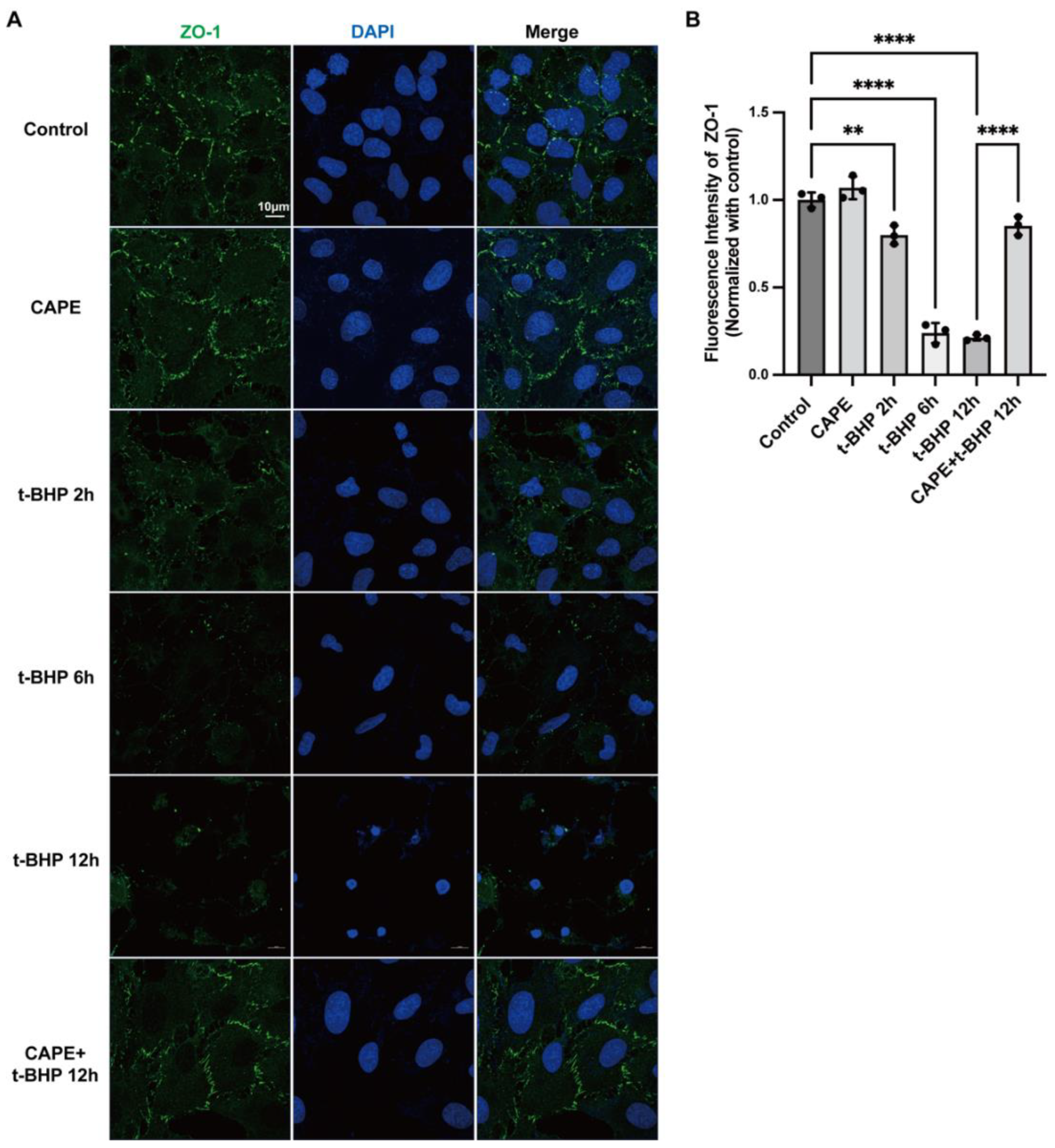
2.3. CAPE Inhibits t-BHP Induced Disruption of Tight Junction Protein ZO−1 in ARPE−19 Cells
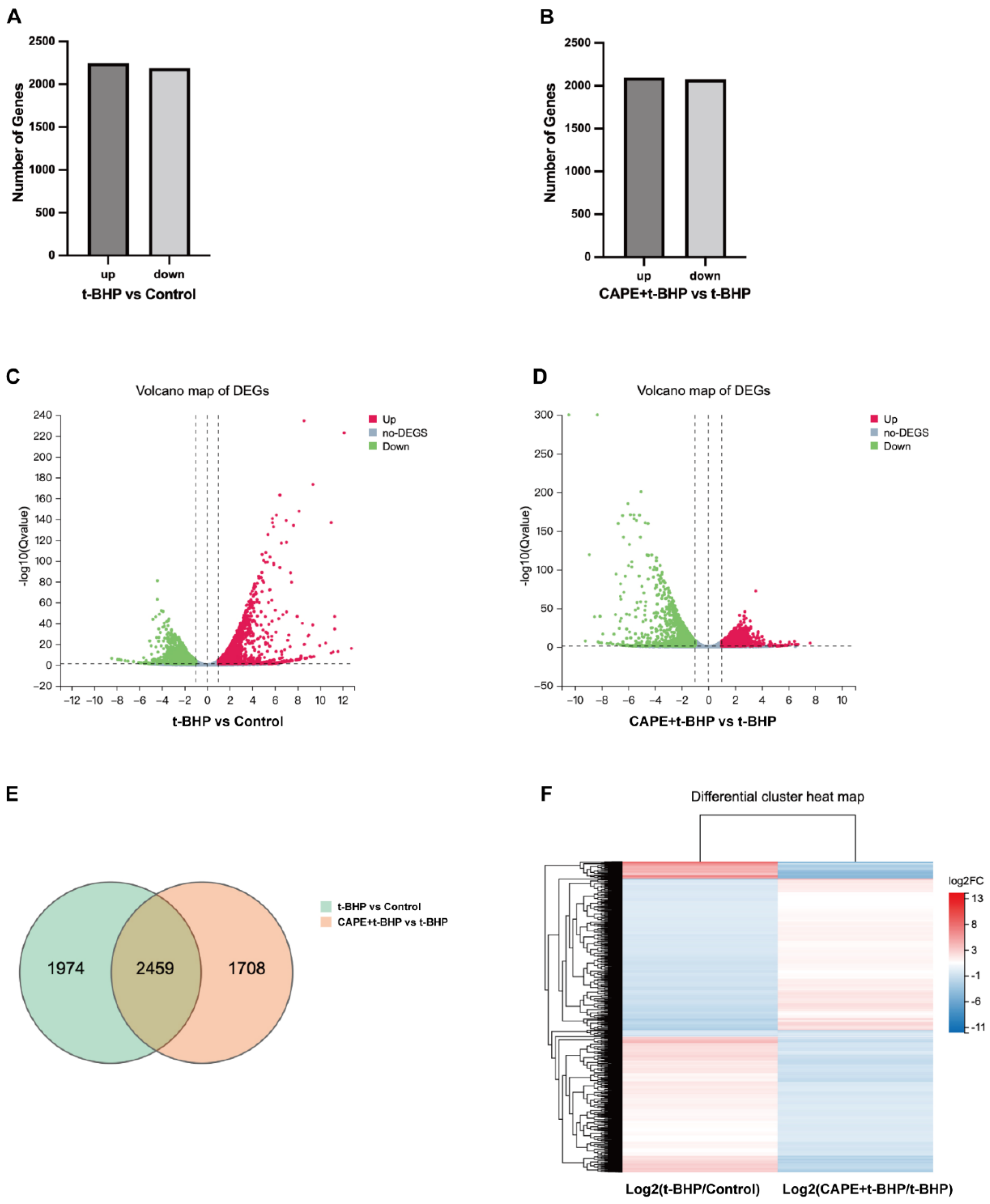
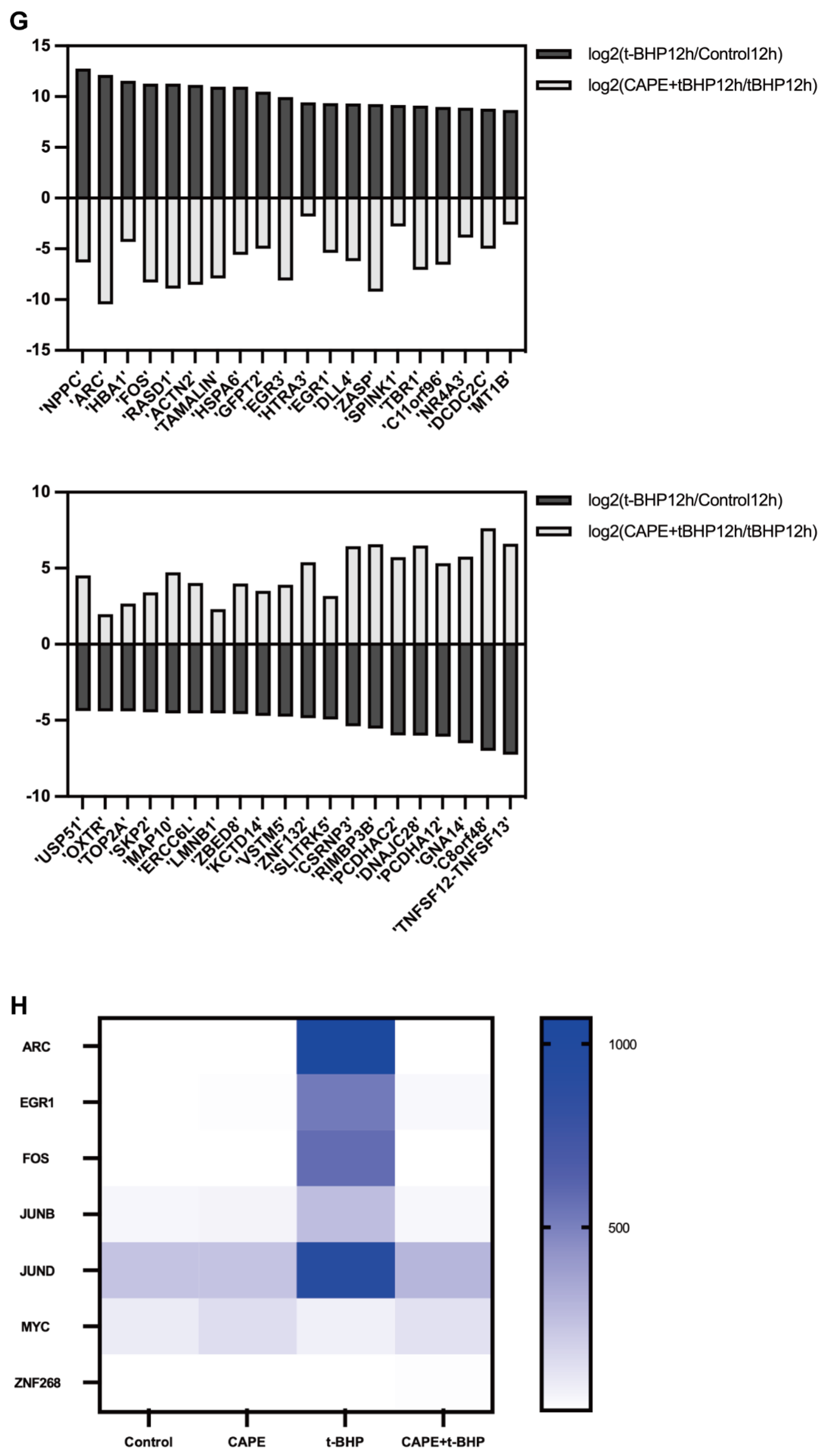
2.4. CAPE Modulates Oxidative Injury induced Gene Expression Profile Changes in ARPE−19 Cells
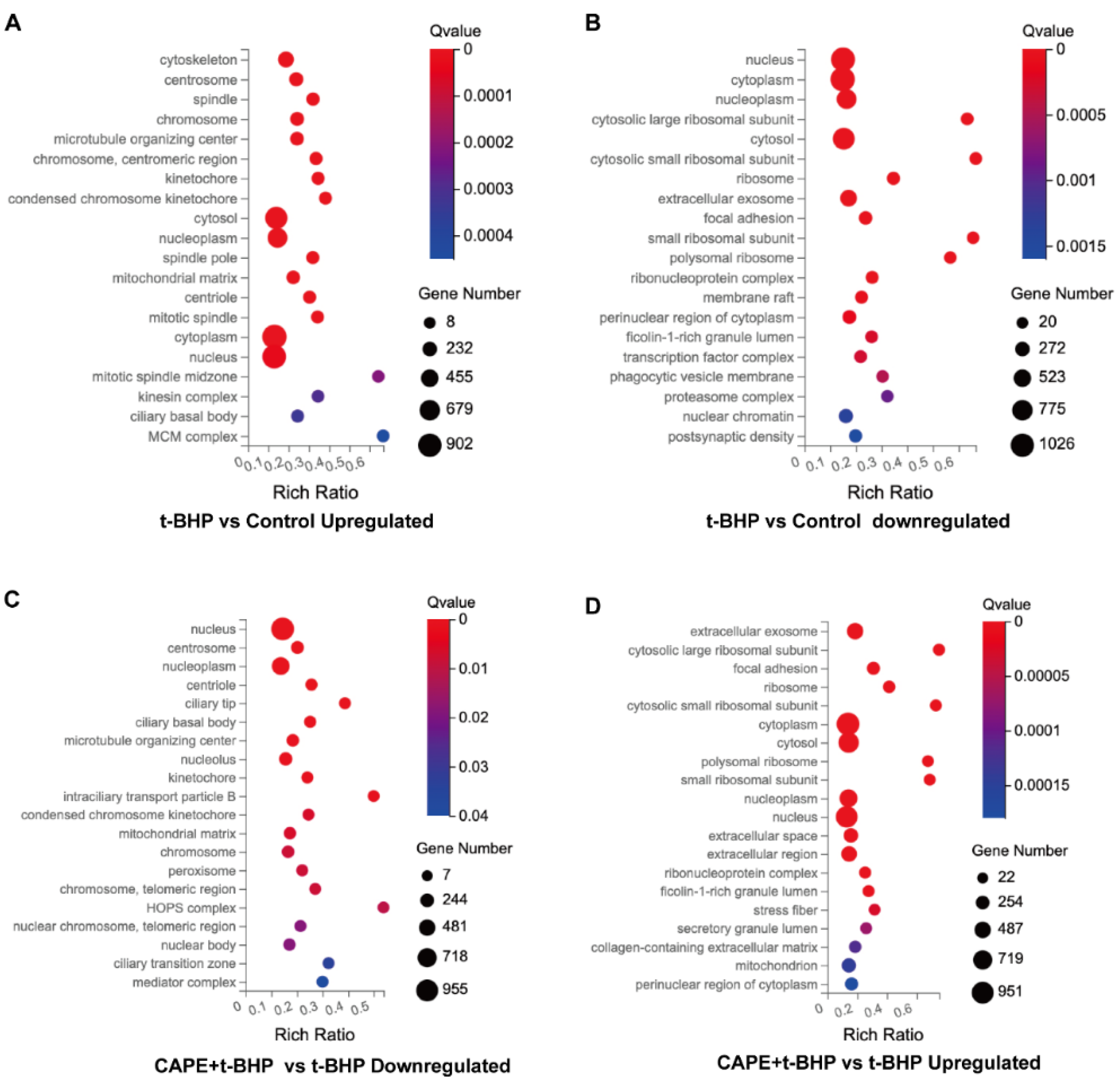
2.5. GO Enrichment Analyses of DEGs
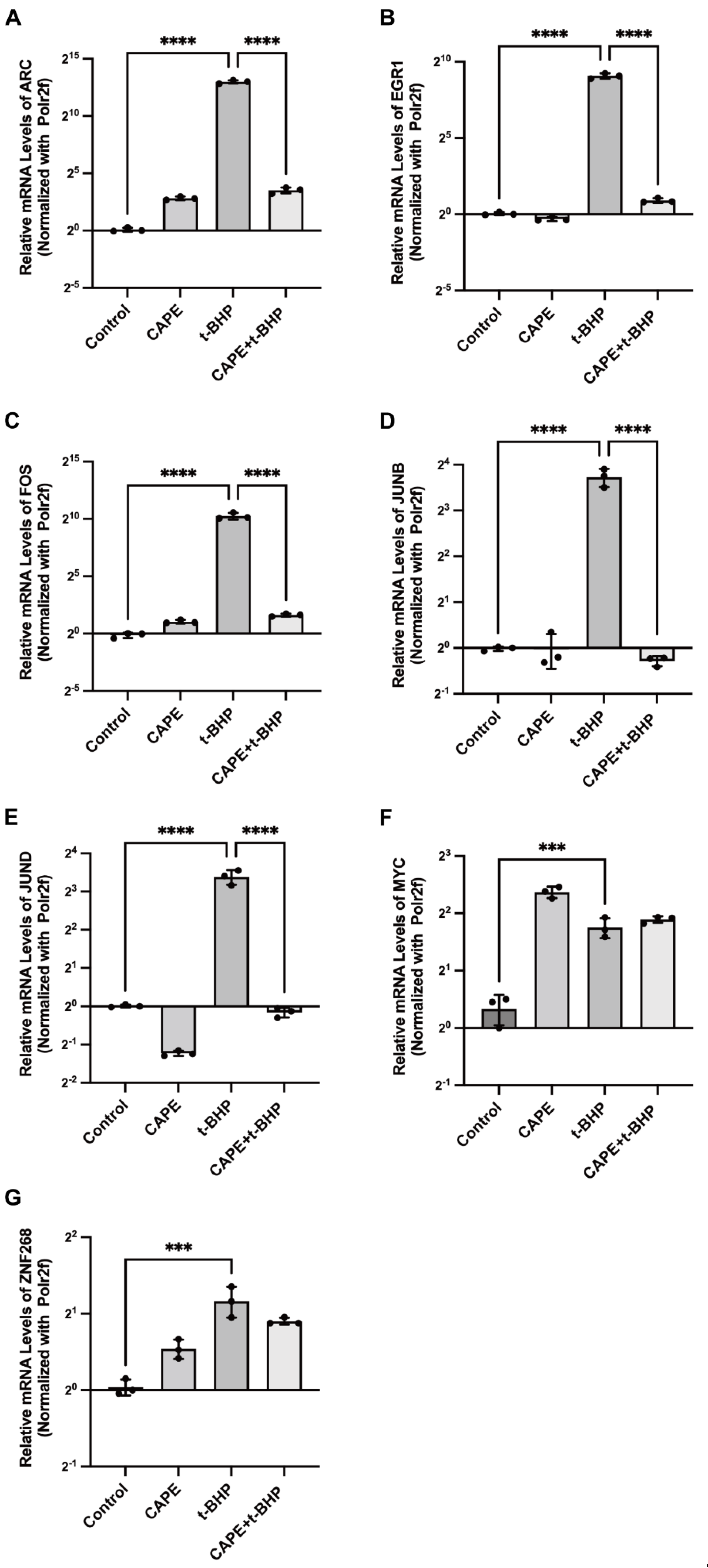
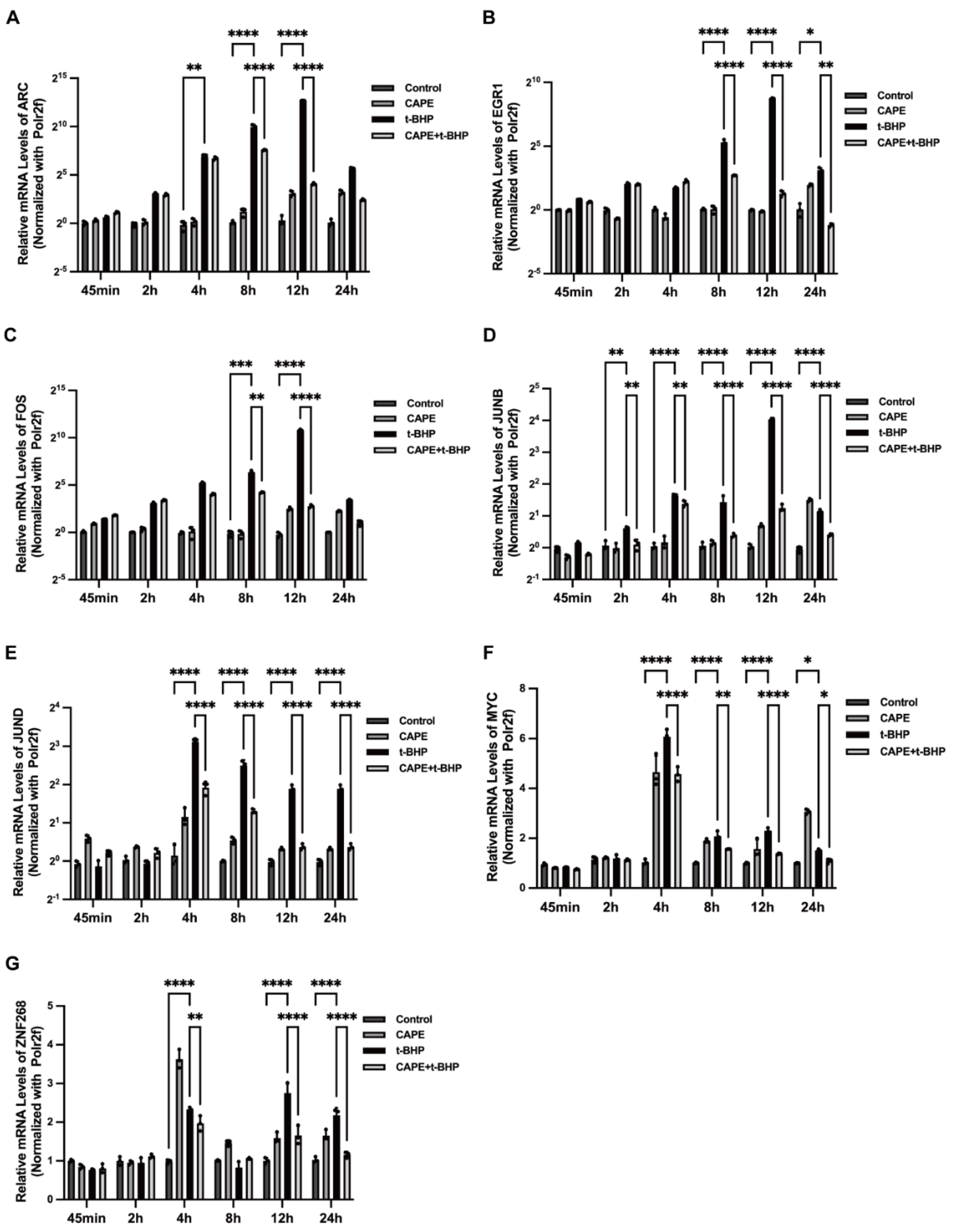
2.6. qPCR Validation of Changes in the Immediate Early Genes (IEGs) in the RNA-seq Data
2.7. Effects of CAPE on t-BHP-Induced Expression of IEGs
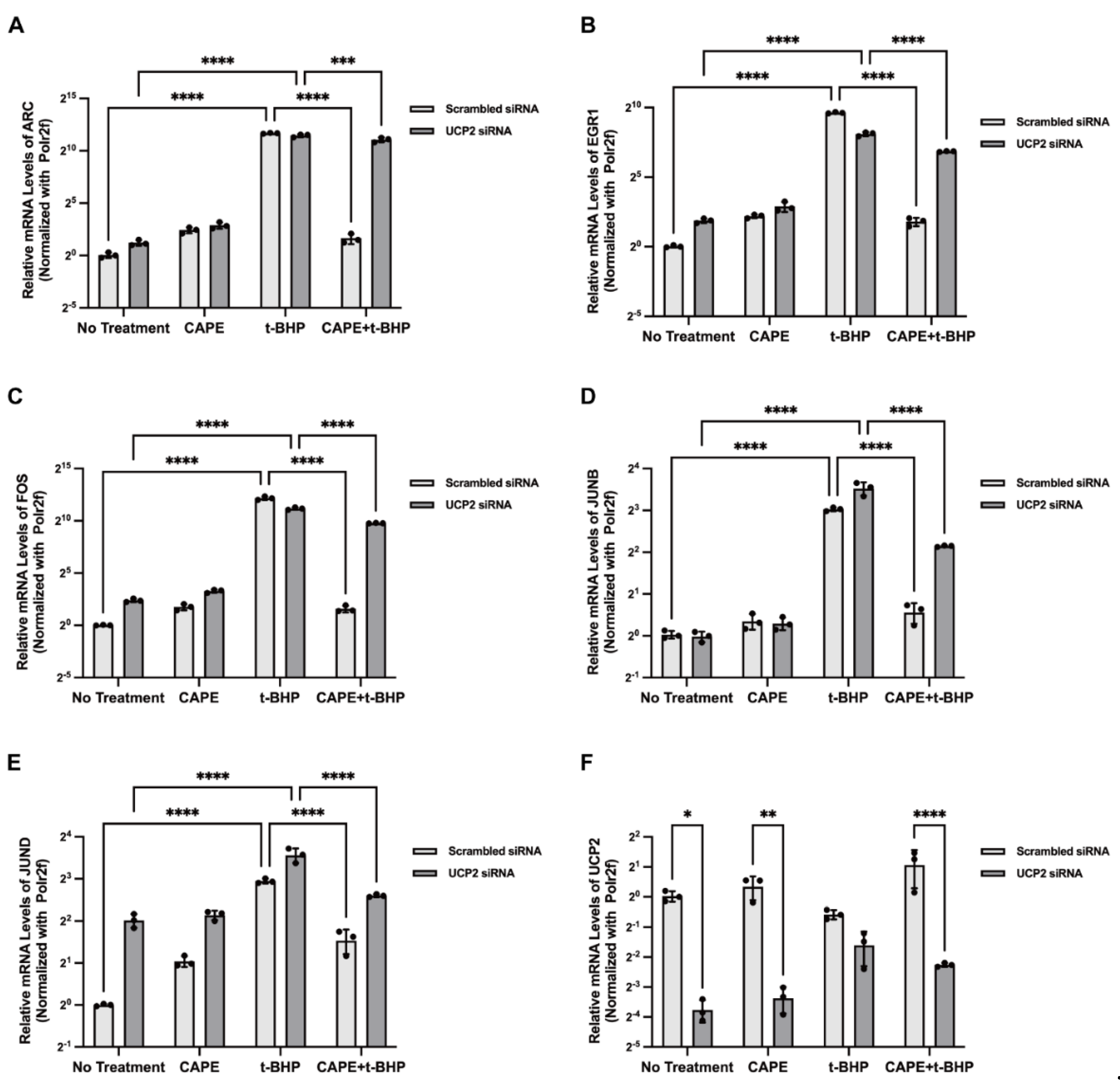
2.8. UCP2 Mediates CAPE Regulation of IEGs
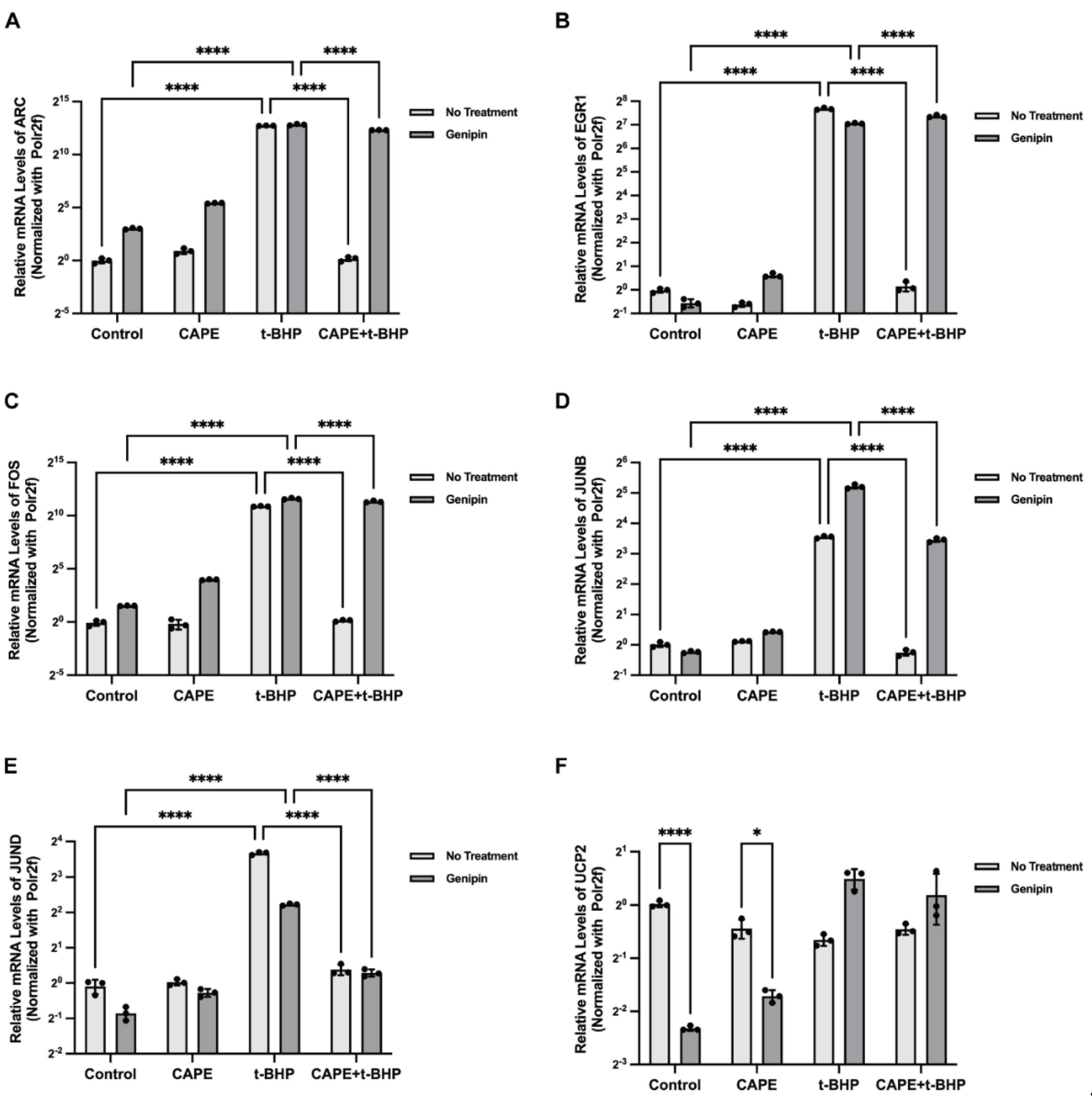
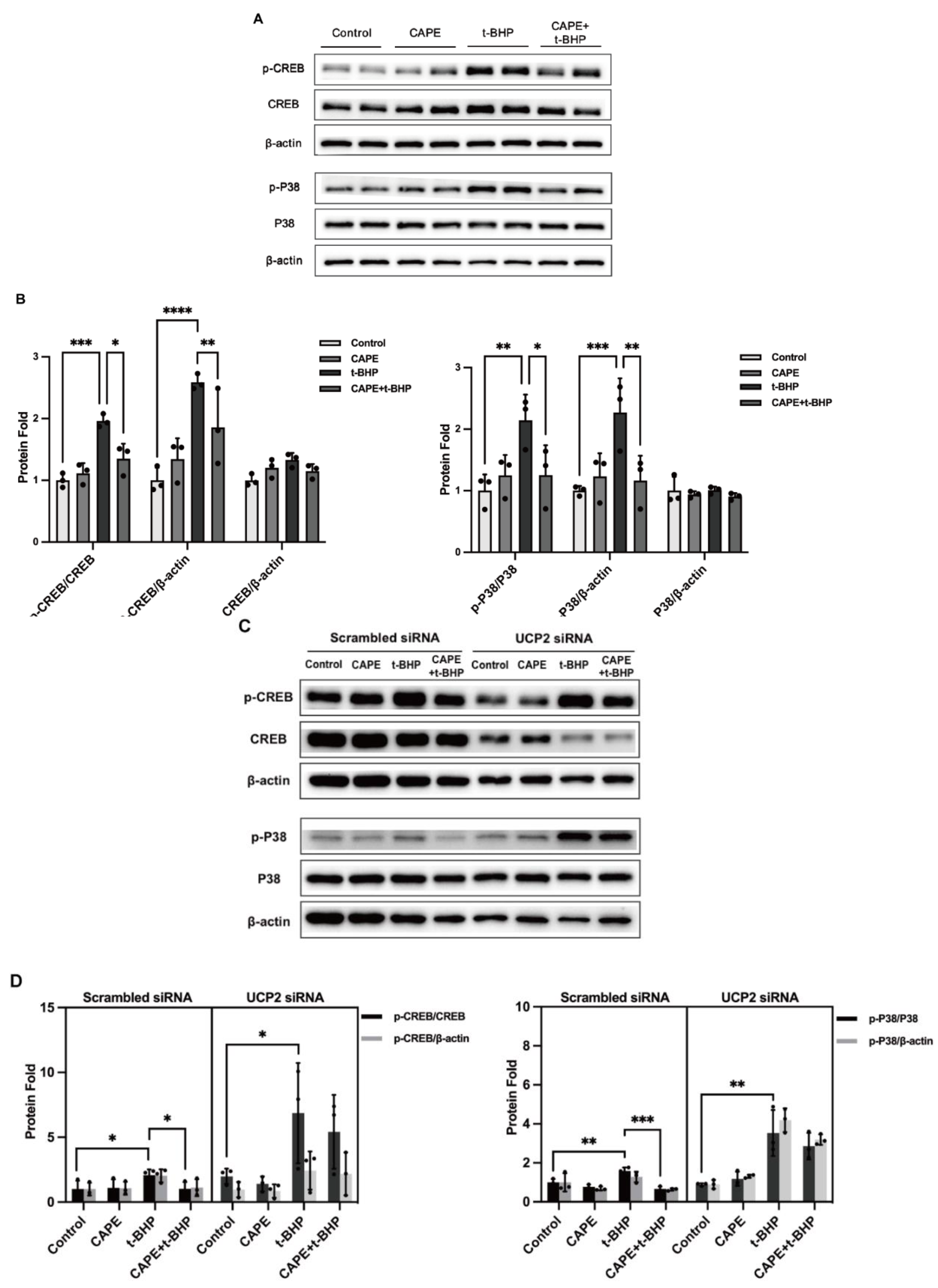
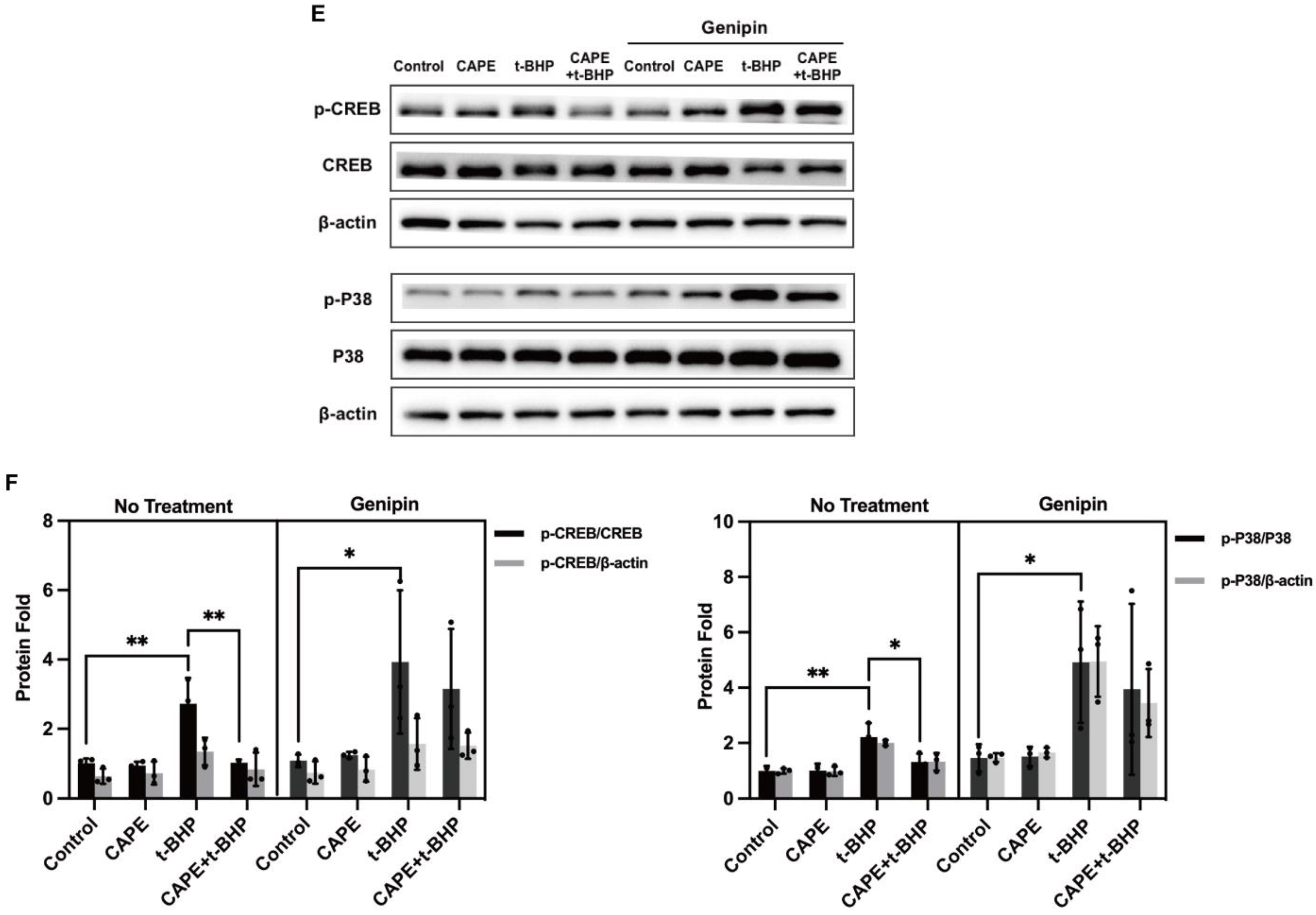
2.9. UCP2 Deficiency Interrupts the Inhibitory Actions of CAPE on p38 MAPK/CREB Activation during Oxidative Stress
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Human Retinal Pigment Epithelial (ARPE−19) Cells Culture and Treatment
4.3. RNA-Sequencing
4.4. Quantitative Real-Time PCR (qPCR)
4.5. Western Blot
4.6. Transient Transfection
4.7. Immunolabeling
4.8. Reactive Oxygen Species (ROS) Determination
4.9. Analysis of Apoptosis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nishimura, Y.; Hara, H.; Kondo, M.; Hong, S.; Matsugi, T. Oxidative Stress in Retinal Diseases. Oxid. Med. Cell Longev. 2017, 2017, 4076518. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Gu, L.; Kwong, J.M.; Caprioli, J.; Piri, N. DNA and RNA oxidative damage in the retina is associated with ganglion cell mitochondria. Sci. Rep. 2022, 12, 8705. [Google Scholar] [CrossRef] [PubMed]
- Abokyi, S.; Shan, S.-W.; Lam, C.H.-I.; Catral, K.P.; Pan, F.; Chan, H.H.-L.; To, C.-H.; Tse, D.Y.-Y. Targeting Lysosomes to Reverse Hydroquinone-Induced Autophagy Defects and Oxidative Damage in Human Retinal Pigment Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 9042. [Google Scholar] [CrossRef]
- Lakkaraju, A.; Umapathy, A.; Tan, L.X.; Daniele, L.; Philp, N.J.; Boesze-Battaglia, K.; Williams, D.S. The cell biology of the retinal pigment epithelium. Prog. Retin. Eye Res. 2020, 78, 100846. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ge, J.; Burke, J.M.; Myers, R.L.; Dong, Z.Z.; Tombran-Tink, J. Mitochondria impairment correlates with increased sensitivity of aging RPE cells to oxidative stress. J. Ocul. Biol. Dis. Inform. 2010, 3, 92–108. [Google Scholar] [CrossRef]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef]
- Barnstable, C.J.; Zhang, M.; Tombran-Tink, J. Uncoupling Proteins as Therapeutic Targets for Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5672. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, C.; Feng, C.; Yan, C.; Yu, Y.; Chen, Z.; Guo, C.; Wang, X. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Rep. 2022, 27, 45–52. [Google Scholar] [CrossRef]
- Hass, D.T.; Barnstable, C.J. Uncoupling proteins in the mitochondrial defense against oxidative stress. Prog. Retin. Eye Res. 2021, 83, 100941. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Locy, M.L.; Chanda, D.; Kurundkar, A.; Kurundkar, D.; Larson-Casey, J.L.; Londono, P.; Bagchi, R.A.; Deskin, B.; Elajaili, H.; et al. Mitochondrial uncoupling protein-2 reprograms metabolism to induce oxidative stress and myofibroblast senescence in age-associated lung fibrosis. Aging Cell 2022, 21, e13674. [Google Scholar] [CrossRef]
- Pan, J.A.; Zhang, H.; Lin, H.; Gao, L.; Zhang, H.L.; Zhang, J.F.; Wang, C.Q.; Gu, J. Irisin ameliorates doxorubicin-induced cardiac perivascular fibrosis through inhibiting endothelial-to-mesenchymal transition by regulating ROS accumulation and autophagy disorder in endothelial cells. Redox Biol. 2021, 46, 102120. [Google Scholar] [CrossRef]
- Hanus, J.; Kolkin, A.; Chimienti, J.; Botsay, S.; Wang, S. 4-Acetoxyphenol Prevents RPE Oxidative Stress-Induced Necrosis by Functioning as an NRF2 Stabilizer. Invest. Ophthalmol. Vis. Sci. 2015, 56, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.A.; Wong, H.-S.; Brand, M.D. Use of S1QELs and S3QELs to link mitochondrial sites of superoxide and hydrogen peroxide generation to physiological and pathological outcomes. Biochem. Soc. Trans. 2019, 47, 1461–1469. [Google Scholar] [CrossRef]
- He, Y.; Leung, K.W.; Ren, Y.; Pei, J.; Ge, J.; Tombran-Tink, J. PEDF improves mitochondrial function in RPE cells during oxidative stress. Invest. Ophthalmol. Vis. Sci. 2014, 55, 6742–6755. [Google Scholar] [CrossRef]
- He, Y.; Wang, X.; Liu, X.; Ji, Z.; Ren, Y. Decreased uncoupling protein 2 expression in aging retinal pigment epithelial cells. Int. J. Ophthalmol. 2019, 12, 375–380. [Google Scholar] [CrossRef]
- Szliszka, E.; Czuba, Z.P.; Domino, M.; Mazur, B.; Zydowicz, G.; Krol, W. Ethanolic extract of propolis (EEP) enhances the apoptosis- inducing potential of TRAIL in cancer cells. Molecules 2009, 14, 738–754. [Google Scholar] [CrossRef]
- Jia, Y.; Jiang, S.; Chen, C.; Lu, G.; Xie, Y.; Sun, X.; Huang, L. Caffeic acid phenethyl ester attenuates nuclear factor-κB-mediated inflammatory responses in Müller cells and protects against retinal ganglion cell death. Mol. Med. Rep. 2019, 19, 4863–4871. [Google Scholar] [CrossRef] [PubMed]
- Şahin, A.; Kaya, S.; Baylan, M. The effects of caffeic acid phenethyl ester on retina in a diabetic rat model. Cutan. Ocul. Toxicol. 2021, 40, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, L.; Wen, D.; Ren, C.; Chen, S.; Zhang, Z.; Hu, L.; Yu, Z.; Tombran-Tink, J.; Zhang, X.; et al. Neuroprotection of retinal cells by Caffeic Acid Phenylethyl Ester (CAPE) is mediated by mitochondrial uncoupling protein UCP2. Neurochem. Int. 2021, 151, 105214. [Google Scholar] [CrossRef]
- Liang, N.; Kitts, D.D. Role of Chlorogenic Acids in Controlling Oxidative and Inflammatory Stress Conditions. Nutrients 2015, 8, 16. [Google Scholar] [CrossRef]
- Lee, B.-H.; Choi, H.-S.; Hong, J. Roles of anti- and pro-oxidant potential of cinnamic acid and phenylpropanoid derivatives in modulating growth of cultured cells. Food Sci. Biotechnol. 2022, 31, 463–473. [Google Scholar] [CrossRef]
- Chagas, M.D.S.S.; Behrens, M.D.; Moragas-Tellis, C.J.; Penedo, G.X.M.; Silva, A.R.; Gonçalves-De-Albuquerque, C.F. Flavonols and Flavones as Potential Anti-Inflammatory, Antioxidant, and Antibacterial Compounds. Oxidative Med. Cell. Longev. 2022, 2022, 9966750. [Google Scholar] [CrossRef]
- Sathyabhama, M.; Dharshini, L.C.P.; Karthikeyan, A.; Kalaiselvi, S.; Min, T. The Credible Role of Curcumin in Oxidative Stress-Mediated Mitochondrial Dysfunction in Mammals. Biomolecules 2022, 12, 1405. [Google Scholar] [CrossRef]
- Shi, X.; Li, P.; Liu, H.; Prokosch, V. Oxidative Stress, Vascular Endothelium, and the Pathology of Neurodegeneration in Retina. Antioxidants 2022, 11, 543. [Google Scholar] [CrossRef]
- Subramaniam, M.D.; Iyer, M.; Nair, A.P.; Venkatesan, D.; Mathavan, S.; Eruppakotte, N.; Kizhakkillach, S.; Chandran, M.K.; Roy, A.; Gopalakrishnan, A.V.; et al. Oxidative stress and mitochondrial transfer: A new dimension towards ocular diseases. Genes Dis. 2020, 9, 610–637. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, P.R.; Madanagopalan, V.G. Role of Curcumin in Retinal Diseases—A review. Graefes Arch. Clin. Exp. Ophthalmol. 2022, 260, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Wang, Z.; Luo, Y.; Zhang, Y.; He, W.; Mei, Y.; Xue, J.; Li, M.; Pan, H.; Li, W.; et al. FA-97, a New Synthetic Caffeic Acid Phenethyl Ester Derivative, Protects against Oxidative Stress-Mediated Neuronal Cell Apoptosis and Scopolamine-Induced Cognitive Impairment by Activating Nrf2/HO-1 Signaling. Oxid. Med. Cell. Longev. 2019, 2019, 8239642. [Google Scholar] [CrossRef] [PubMed]
- Olgierd, B.; Kamila, Ż.; Anna, B.; Emilia, M. The Pluripotent Activities of Caffeic Acid Phenethyl Ester. Molecules 2021, 26, 1335. [Google Scholar] [CrossRef]
- Balaha, M.; De Filippis, B.; Cataldi, A.; di Giacomo, V. CAPE and Neuroprotection: A Review. Biomolecules 2021, 11, 176. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, X.; Huang, Q.; Li, S.; Zhou, Y.; Li, Z. Cardioprotection of CAPE-oNO2 against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF-κB pathway in vivo and in vitro. Redox Biol. 2017, 15, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Soares, V.E.M.; Carmo, T.I.T.D.; dos Anjos, F.; Wruck, J.; Maciel, S.F.V.D.O.; Bagatini, M.D.; Silva, D.T.D.R.E. Role of inflammation and oxidative stress in tissue damage associated with cystic fibrosis: CAPE as a future therapeutic strategy. Mol. Cell. Biochem. 2021, 477, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Marin, E.H.; Paek, H.; Li, M.; Ban, Y.; Karaga, M.K.; Shashidharamurthy, R.; Wang, X. Caffeic acid phenethyl ester exerts apoptotic and oxidative stress on human multiple myeloma cells. Invest. New Drugs 2018, 37, 837–848. [Google Scholar] [CrossRef]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Chen, H.; Tran, J.-T.A.; Anderson, R.E.; Mandal, N.A. Caffeic acid phenethyl ester protects 661W cells from H2O2-mediated cell death and enhances electroretinography response in dim-reared albino rats. Mol. Vis. 2012, 18, 1325–1338. [Google Scholar]
- Zhang, C.; Xie, H.; Yang, Q.; Yang, Y.; Li, W.; Tian, H.; Lu, L.; Wang, F.; Xu, J.; Gao, F.; et al. Erythropoietin protects outer blood-retinal barrier in experimental diabetic retinopathy by up-regulating ZO-1 and occludin. Clin. Exp. Ophthalmol. 2019, 47, 1182–1197. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Fung, T.S.; He, J.; Chen, R.A.; Liu, D.X. Modulation of viral replication, apoptosis and antiviral response by induction and mutual regulation of EGR and AP-1 family genes during coronavirus infection. Emerg. Microbes Infect. 2022, 11, 1717–1729. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.; Um, J.W. Synapse development organized by neuronal activity-regulated immediate-early genes. Exp. Mol. Med. 2018, 50, 1–7. [Google Scholar] [CrossRef]
- Karthikkeyan, G.; Nareshkumar, R.N.; Aberami, S.; Sulochana, K.N.; Vedantham, S.; Coral, K. Hyperglycemia induced early growth response-1 regulates vascular dysfunction in human retinal endothelial cells. Microvasc. Res. 2018, 117, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, R.; Hu, G.; Liu, H.; Lv, K.; Duan, Y.; Shen, N.; Wu, J.; Hu, J.; Liu, Y.; et al. Single cell RNA sequencing (scRNA-Seq) deciphering pathological alterations in streptozotocin-induced diabetic retinas. Exp. Eye Res. 2021, 210, 108718. [Google Scholar] [CrossRef] [PubMed]
- Rickhag, M.; Teilum, M.; Wieloch, T. Rapid and long-term induction of effector immediate early genes (BDNF, Neuritin and Arc) in peri-infarct cortex and dentate gyrus after ischemic injury in rat brain. Brain Res. 2007, 1151, 203–210. [Google Scholar] [CrossRef]
- Guo, H.; Golczer, G.; Wittner, B.S.; Langenbucher, A.; Zachariah, M.; Dubash, T.D.; Hong, X.; Comaills, V.; Burr, R.; Ebright, R.Y.; et al. NR4A1 regulates expression of immediate early genes, suppressing replication stress in cancer. Mol. Cell 2021, 81, 4041–4058.e15. [Google Scholar] [CrossRef] [PubMed]
- Didier, S.; Sauvé, F.; Domise, M.; Buée, L.; Marinangeli, C.; Vingtdeux, V. AMP-activated Protein Kinase Controls Immediate Early Genes Expression Following Synaptic Activation Through the PKA/CREB Pathway. Int. J. Mol. Sci. 2018, 19, 3716. [Google Scholar] [CrossRef]
- Chan, C.-M.; Huang, D.-Y.; Sekar, P.; Hsu, S.-H.; Lin, W.-W. Reactive oxygen species-dependent mitochondrial dynamics and autophagy confer protective effects in retinal pigment epithelial cells against sodium iodate-induced cell death. J. Biomed. Sci. 2019, 26, 40. [Google Scholar] [CrossRef]
- Gupta, A.K.; Roy, S.; Das, P.K. Antileishmanial effect of the natural immunomodulator genipin through suppression of host negative regulatory protein UCP2. J. Antimicrob. Chemother. 2020, 76, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Kundumani-Sridharan, V.; Kumar, S.; White, C.W.; Das, K.A.-O. Thioredoxin Prevents Loss of UCP2 in Hyperoxia via MKK4-p38 MAPK-PGC1α Signaling and Limits Oxygen Toxicity. Am. J. Respir. Cell Mol. Biol. 2022, 66, 323–336. [Google Scholar] [CrossRef]
- Healy, S.; Khan, P.; Davie, J.R. Immediate early response genes and cell transformation. Pharmacol. Ther. 2013, 137, 64–77. [Google Scholar] [CrossRef]
- Gao, P.; You, M.; Li, L.; Zhang, Q.; Fang, X.; Wei, X.; Zhou, Q.; Zhang, H.; Wang, M.; Lu, Z.; et al. Salt-Induced Hepatic Inflammatory Memory Contributes to Cardiovascular Damage Through Epigenetic Modulation of SIRT3. Circulation 2022, 145, 375–391. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, C.; Zhou, P.; Zhang, M.; Yu, Z.; Zhang, X.; Tombran-Tink, J.; Barnstable, C.J.; Li, X. Molecular Mechanisms of Oxidative Stress Relief by CAPE in ARPE−19 Cells. Int. J. Mol. Sci. 2023, 24, 3565. https://doi.org/10.3390/ijms24043565
Ren C, Zhou P, Zhang M, Yu Z, Zhang X, Tombran-Tink J, Barnstable CJ, Li X. Molecular Mechanisms of Oxidative Stress Relief by CAPE in ARPE−19 Cells. International Journal of Molecular Sciences. 2023; 24(4):3565. https://doi.org/10.3390/ijms24043565
Chicago/Turabian StyleRen, Changjie, Peiran Zhou, Mingliang Zhang, Zihao Yu, Xiaomin Zhang, Joyce Tombran-Tink, Colin J. Barnstable, and Xiaorong Li. 2023. "Molecular Mechanisms of Oxidative Stress Relief by CAPE in ARPE−19 Cells" International Journal of Molecular Sciences 24, no. 4: 3565. https://doi.org/10.3390/ijms24043565
APA StyleRen, C., Zhou, P., Zhang, M., Yu, Z., Zhang, X., Tombran-Tink, J., Barnstable, C. J., & Li, X. (2023). Molecular Mechanisms of Oxidative Stress Relief by CAPE in ARPE−19 Cells. International Journal of Molecular Sciences, 24(4), 3565. https://doi.org/10.3390/ijms24043565