Abstract
T-type calcium (CaV3) channels are involved in cardiac automaticity, development, and excitation–contraction coupling in normal cardiac myocytes. Their functional role becomes more pronounced in the process of pathological cardiac hypertrophy and heart failure. Currently, no CaV3 channel inhibitors are used in clinical settings. To identify novel T-type calcium channel ligands, purpurealidin analogs were electrophysiologically investigated. These compounds are alkaloids produced as secondary metabolites by marine sponges, and they exhibit a broad range of biological activities. In this study, we identified the inhibitory effect of purpurealidin I (1) on the rat CaV3.1 channel and conducted structure–activity relationship studies by characterizing the interaction of 119 purpurealidin analogs. Next, the mechanism of action of the four most potent analogs was investigated. Analogs 74, 76, 79, and 99 showed a potent inhibition on the CaV3.1 channel with IC50’s at approximately 3 μM. No shift of the activation curve could be observed, suggesting that these compounds act like a pore blocker obstructing the ion flow by binding in the pore region of the CaV3.1 channel. A selectivity screening showed that these analogs are also active on hERG channels. Collectively, a new class of CaV3 channel inhibitors has been discovered and the structure–function studies provide new insights into the synthetic design of drugs and the mechanism of interaction with T-type CaV channels.
1. Introduction
T-type calcium channels are members of the superfamily of voltage-gated calcium (CaV) channels. These channels are represented by three genes that encode three different CaV3 α1-subunits [1,2]. The CaV3.1 (α1G) and CaV3.2 (α1H) T-type CaV channel isoforms are present mainly in the heart [3]. However, it has been shown that CaV3.3 (α1I) channels are also expressed in Purkinje fibers [4]. The CaV3 channels are involved in cardiac automaticity, development, and excitation–contraction coupling in normal cardiac myocytes. Their functional role becomes more pronounced in the process of pathological cardiac hypertrophy and heart failure [5,6,7]. Studies have shown that CaV3.1 knockout mice present with bradycardia, confirming the role of these channels in pacemaking activity [8,9]. Mice lacking CaV3.2, unlike mice lacking CaV3.1, showed severely suppressed pressure overload-induced hypertrophy. Furthermore, angiotensin II-induced cardiac hypertrophy was suppressed in mice deficient for CaV3.2 [6,10]. CaV3.3 null mice appeared rather normal [11].
The family of CaV3 channels mediates a transient (or T-type) current. These channels open near-resting membrane potential due to their low activation threshold. Hence, they are classified as low-voltage-activated (LVA) channels. Moreover, they are characterized by fast inactivation and slow deactivation kinetics [2]. Given their unique biophysical properties, T-type CaV channels are ideally suited to regulating pacemaker activity, cellular excitability, and low-threshold firing [1,2]. High-voltage-activated (HVA) calcium channels include the CaV1 and CaV2 families, which conduct L-, P/Q-, N-, and R-type currents. In contrast to the HVA channels, co-assembly with auxiliary subunits, such as α2/δ-, β-, and γ-subunits, is not required for CaV3 channels [1].
Currently, no CaV3 channel inhibitors are used in clinical settings. Mibefradil is a potent T-type calcium channel blocker and is 10- to 15-fold more selective for T-type than L-type calcium channels [12,13,14]. This non-dihydropyridine molecule, which is sold under the name Posicor®, was previously used in the treatment of hypertension. In 1998, it had to be withdrawn from the market due to severe drug–drug interactions. Mibefradil has been repurposed as a therapeutical agent to treat solid tumors [15]. Currently, two compounds, Z944 and ACT-709478, are in phase II clinical trials for the treatment of pain and generalized epilepsy, respectively [11,16,17].
To identify novel T-type calcium channel ligands, purpurealidin analogs were electrophysiologically investigated on CaV3-expressing oocytes. Purpurealidins are alkaloid-type secondary metabolites produced by marine sponges. More specifically, the synthetic compounds investigated in this study are derived from sponges from the family Aplysinellidae (Order Verongiida) [18]. These sponges are widespread and can be found mostly in tropical and temperate regions of the world. Their secondary metabolites are reported to exhibit a broad range of biological activities, such as their antiproliferative, antiangiogenic, antibiotic, cytotoxic, and hemolytic properties [19]. In the past years, the U.S. Food and Drug Administration (FDA) has already approved several sponge-derived compounds for clinical use. For example, cytarabine is on the market as an anticancer agent, and vidarabine as an antiviral agent [20,21]. When they are converted into the triphosphate form, they are reported to inhibit DNA polymerase during the S-phase of DNA replication. This caused great interest in marine sponges as a source of compounds with pharmaceutical potential. Importantly, we noticed that the structure of purpurealidin I (1) resembles the structure of Z944, which was mentioned previously.
Following the observation of structural homology between Z944 and bromotyrosine purpurealidin I (1), we started by investigating the ex vivo activity of purpurealidin I (1). This bioactive compound can be found in the marine sponge Psammaplysilla purpurea [22]. We used the two-electrode voltage clamp technique to investigate its activity on CaV3.1 channels. We have previously reported syntheses of sets of compounds with bromotyrosine, bromotyramine, or diarylamine scaffold based on the purpurealidin I (1) structure [18,23,24,25]. These synthetic compounds are further referred to as purpurealidin analogs. In total, 119 purpurealidin analogs of this marine metabolite were screened, allowing us to investigate structure–activity relationships (SAR). Several synthetic analogs exhibited an effect on the CaV3.1 channel. The purpurealidin analogs 74, 76, 79, and 99 were found to potently inhibit CaV3.1 current, and their activity on CaV3.1-expressing oocytes was electrophysiologically characterized.
2. Results
2.1. Structure–Activity Relationship of Sponge-Derived Compounds
Secondary metabolites of marine sponges are known to possess a wide array of interesting bioactivities and, therefore, their effect on the cardiac T-type CaV channel CaV3.1 was evaluated. A first screening of purpurealidin I (1) (Figure 1) showed that this alkaloid was able to potently inhibit the rat CaV3.1 channel (Figure 2). In previous work, a library of 119 simplified analogs was synthesized with the aim of targeting KV10.1 [18,23,24,25]. Due to the interesting activity of purpurealidin I (1), we decided to repurpose this existing library to investigate the structure–activity relationship with respect to the CaV3.1 channels. All compounds were first dissolved in DMSO and then diluted in a 10 mM Ba2+ solution. Compounds were tested at a final concentration of 10 μM.
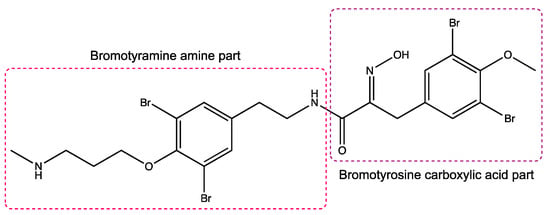
Figure 1.
Structure of purpurealidin I (compound 1) [22].
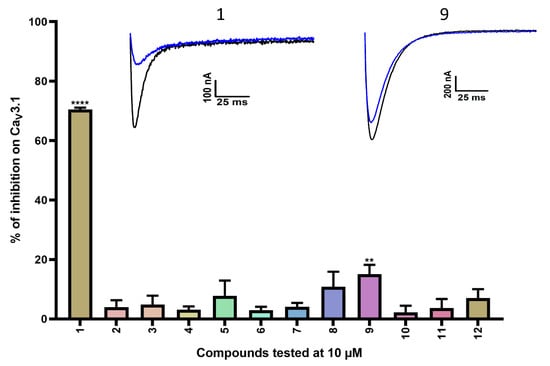
Figure 2.
Inhibition of CaV3.1 after application of purpurealidin analogs (compounds 1–12). Compounds were tested at 10 μM. Data are presented as means ± SEM (n ≥ 3). Statistical significance was determined using one-way ANOVA with Dunnett’s post-test; ** p < 0.01; **** p < 0.0001. The inset shows representative CaV3.1 current traces in control (black) and after application of 10 μM compound 1 or compound 9 (blue).
A first series of compounds is shown in Table S1. Compounds 2–12 are simplified analogs that show modifications to the bromotyramine part of the structure of purpurealidin I (Figure 1 and Table S1). The synthesis of these bromotyrosine analogs is described in more detail by Bhat et al. [24]. The average current inhibition (%) is shown in Figure 2.
A first screening showed potent inhibition of CaV3.1 currents by purpurealidin I (compound 1, 70.5 ± 0.6%). For the simplified analogs, only compound 9 (15.1 ± 3.1%) showed a low but significant inhibition of the CaV3.1 channels. Purpurealidin I (1) showed the strongest inhibition of CaV3.1 currents at 10 μM, but unfortunately, not enough material was available to determine a concentration–response curve for this compound.
Next, compounds 13–40 (Table S2) are simplified analogs that show modifications to the bromotyrosine part of the purpurealidin I (1) structure (Figure 1). More specifically, these compounds are simpler amide analogs containing the tyramine fragment in combination with substituted phenyl rings (Ar in Table S2). The synthesis of these bromotyramine analogs is described in more detail by Moreels et al. [18]. The average current inhibition (%) is shown in Figure 3.
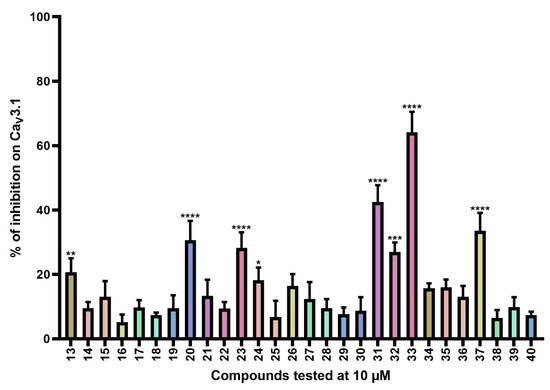
Figure 3.
Inhibition of CaV3.1 after application of purpurealidin analogs (compounds 13–40). Compounds were tested at 10 μM. Data are presented as means ± SEM (n ≥ 3). Statistical significance was determined using one-way ANOVA with Dunnett’s post-test; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
CaV3.1 currents were significantly inhibited by compounds 13 (20.7 ± 4.3%), 20 (30.6 ± 6.0%), 23 (28.2 ± 4.9%), 24 (18.2 ± 3.9), 31 (42.4 ± 5.3%), 32 (26.9 ± 3.0%), 33 (64.1 ± 6.4%), and 37 (33.5 ± 5.6%).
Compounds 41–72 (Table S3) consist of synthesized marine bromotyrosine clavatadine C 41 and its spiro-structured analogs, which here are called spirocyclic bromotyrosines. Structurally, these analogs are more rigid and occupy the chemical space better than the open-chain bromotyrosine. The synthesis of these compounds is described in more detail by Patel et al. [23]. Synthesis of chloro spiro compounds 65–72 is described in the supporting material (Supplemental Scheme S1).
The average current inhibition (%) is shown in Figure 4.
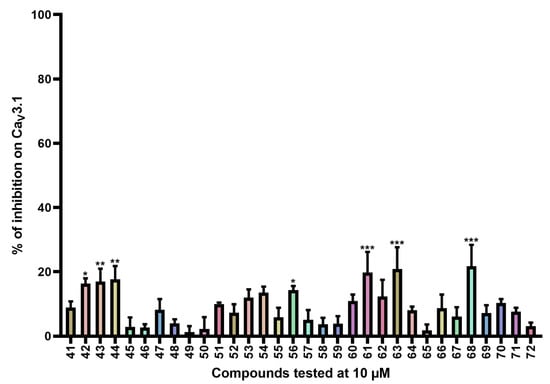
Figure 4.
Inhibition of CaV3.1 after application of purpurealidin analogs (compounds 41–72). Compounds were tested at 10 μM. Data are presented as means ± SEM (n ≥ 3). Statistical significance was determined using one-way ANOVA with Dunnett’s post-test; * p < 0.05; ** p < 0.01; *** p < 0.001.
CaV3.1 currents were significantly inhibited by compounds 42 (16.3 ± 1.7%), 43 (17.0 ± 4.0%), 44 (17.7 ± 4.1%), 61 (19.8 ± 6.4%), 63 (20.9 ± 6.7%), and 68 (21.7 ± 6.7%).
For the last series of compounds, another strategy has been applied. Based on the structure of the purpurealidin analogs, a pharmacophore model was utilized for the virtual screening of commercially available compounds. One of the hit compounds was compound 76 (Table S4). This compound has a unique diarylamine structure and, therefore, represents a new structural class of CaV3 channel inhibitors. Compounds 73–75 and 77–119 (Tables S4 and S5) are analogous structures to compound 76. The synthesis of these compounds is described in more detail by Toplak et al. [25] and Gubič et al. [26]. The average current inhibition (%) is shown in Figure 5 and Figure 6.
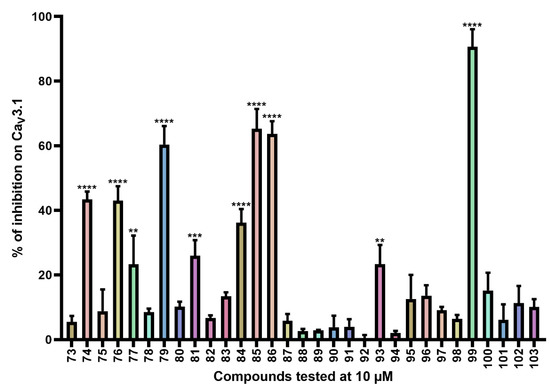
Figure 5.
Inhibition of CaV3.1 after application of purpurealidin analogs (compounds 73–103). Compounds were tested at 10 μM. Data are presented as means ± SEM (n ≥ 3). Statistical significance was determined using one-way ANOVA with Dunnett’s post-test; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
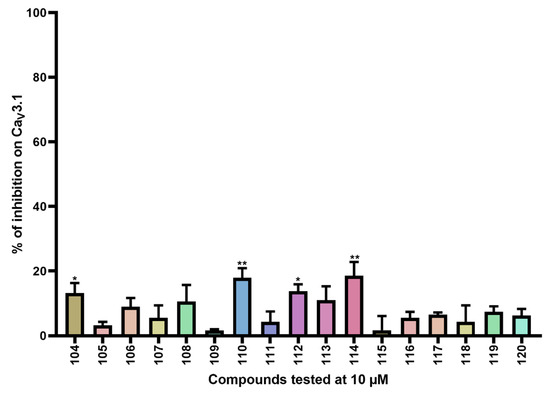
Figure 6.
Inhibition of CaV3.1 after application of purpurealidin analogs (compounds 104–120). Compounds were tested at 10 μM. Data are presented as means ± SEM (n ≥3). Statistical significance was determined using one-way ANOVA with Dunnett’s post-test; * p < 0.05; ** p < 0.01.
CaV3.1 currents were significantly inhibited by compounds 74 (43.4 ± 2.4%), 76 (43.0 ± 4.5%), 77 (23.3 ± 8.9%), 79 (60.4 ± 5.7%), 81 (26.0 ± 4.8%), 84 (36.2 ± 4.2%), 85 (65.3 ± 6.1%), 86 (63.7 ± 3.9%), 93 (23.4 ± 5.9%), and 99 (90.7 ± 5.4%). An electrophysiological characterization is conducted for compounds 74, 76, 79, and 99 and is reported below.
Furthermore, CaV3.1 currents were significantly inhibited by compounds 104 (13.2 ± 3.1%), 110 (17.9 ± 3.0%), 112 (13.8 ± 2.1%), and 114 (18.6 ± 4.2%).
2.2. Electrophysiological Characterization of Compounds 74, 76, 79, and 99 on the T-Type CaV Channels
The activity of the purpurealidin compounds 74, 76, 79 and 99 (Figure 7) was investigated using electrophysiology on oocytes that expressed the CaV3.1 channel.
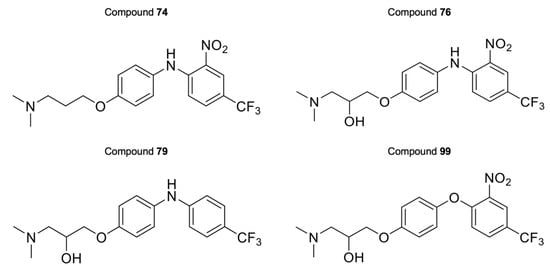
Figure 7.
Structures of compounds 74, 76, 79, and 99.
In a first stage, an electrophysiological characterization was conducted for compounds 74, 76, and 79. The concentration-dependency of the inhibition was evaluated using increasing compound concentrations on all three CaV3 isoforms (Figure 8). The calculated IC50 values and Hill coefficients are shown in Table 1.
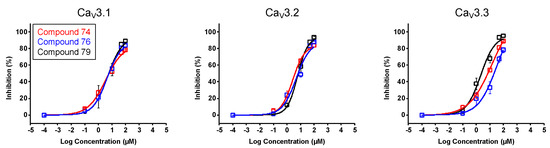
Figure 8.
Concentration–response determination for compounds 74, 76, and 79. The concentration dependency was assessed by measuring the current inhibition in the presence of increasing compound concentrations (0.1 μM, 1 μM, 10 μM, 50 μM, and 100 μM). Red curve shows the concentration–response for compound 74. Blue curve shows the concentration–response for compound 76. Black curve shows the concentration–response for compound 79. Data are presented as means ± SEM (n ≥ 3).

Table 1.
IC50 values and Hill coefficients obtained for compounds 74, 76, and 79, on CaV3 channel isoforms.
Further electrophysiological characterization was performed with a compound concentration of 10 μM. Current traces of the CaV3 channel isoforms before (control, black) and after the application of 10 μM compound (blue) are shown in the top row of Figure 9A for compound 74, the top row of Figure 9B for compound 76, and the top row of Figure 9C for compound 79. In the middle row of each panel, the normalized current amplitude (I/Imax,control) in control conditions (black circles) and compound conditions (blue circles) is plotted against the pulse potential. To evaluate the effect of the compounds on the activation and inactivation of CaV3 channels, the normalized current amplitude (I/Imax) is plotted against the corresponding pulse potentials and fitted with the Boltzmann equation (Figure 9, bottom row of each panel). No significant shift in the voltage dependency of the CaV3 channels was observed upon compound binding (Figure 9, middle and bottom row of each panel). Wash-in and wash-out studies showed that the binding of these compounds to the CaV3 channels is reversible (Supplemental Figure S1).
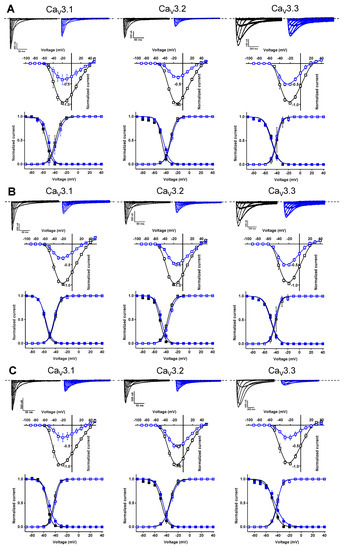
Figure 9.
Electrophysiological characterization of compounds 74 (A), 76 (B), and 79 (C) on all three CaV3 channel isoforms. (A–C) Top row: current traces before (black) and after (blue) application of 10 μM compound. In 10 mM Ba2+, currents were elicited by depolarizing pulses from −90 to +50 mV for CaV3.1 and CaV3.2, and from −90 to +40 mV for CaV3.3. Holding potential was −90 mV. First 300 ms of the depolarizing pulses are shown for CaV3.1 and CaV3.2, and 600 ms for CaV3.3. Middle row: Normalized voltage–current relationship in control (black symbols) and compound (10 μM, blue symbols) conditions. Bottom row: steady-state activation (open symbols) and inactivation (closed symbols) curves in control (black symbols) and compound (10 μM, blue symbols) conditions. Data are presented as means ± SEM (n ≥ 3).
In the next stage of the structure–activity relationship studies, we discovered that compound 99 could potently inhibit the CaV3.1 channels. Moreover, for this compound, an electrophysiological characterization was conducted.
First, the concentration-dependent inhibitory effect of compound 99 was determined by measuring the current inhibition in the presence of increasing compound concentrations. The calculated IC50 value and the Hill coefficient of compound 99 on CaV3.1 are 4.1 ± 0.1 μM and 1.6 ± 0.1, respectively. To investigate isoform selectivity between the two cardiac CaV3 channels, the IC50 value and Hill coefficient were also determined for CaV3.2. The IC50 value is 2.3 ± 0.5 μM with a Hill coefficient of 1.4 ± 0.3. Further electrophysiological characterization was performed with 5 μM of compound 99 on the CaV3.1 channel.
Representative current traces of CaV3.1 are shown in control conditions (black line) and after application of 5 μM compound 99 (blue line, Figure 10A). At a concentration of 5 μM, CaV3.1 currents are inhibited by 57.0 ± 5.8%. The normalized current amplitude (I/Imax,control) in control conditions (black circles) and compound conditions (blue circles) is plotted against the pulse potential (Figure 10B). Next, the effect of the compounds on the activation and inactivation of the CaV3.1 channels is investigated by plotting the normalized current amplitude (I/Imax) against the corresponding pulse potentials and by fitting this with the Boltzmann equation. Steady-state activation and inactivation curves showed no significant shift in the voltage dependency of CaV3.1 channels upon compound binding (Figure 10B,C). For the midpoint of activation, V1/2 values yielded −39.9 ± 0.1 mV in control conditions and −37.4 ± 0.1 mV in the presence of 5 μM compound 99. For the inactivation curves, the V1/2 shifted from −56.0 ± 0.1 mV to −53.0 ± 0.2 mV in control and compound situations, respectively. In Figure 10D, a representative normalized time-dependent profile of the CaV3.1 currents during wash-in and wash-out studies of 5 μM compound 99 is shown for one experiment. The effect of the compound was not completely reversible upon wash-out with compound-free bath solution (Figure 10D). Unfortunately, due to the limited amount available, compound 99 could not be tested on CaV3.3 channels.
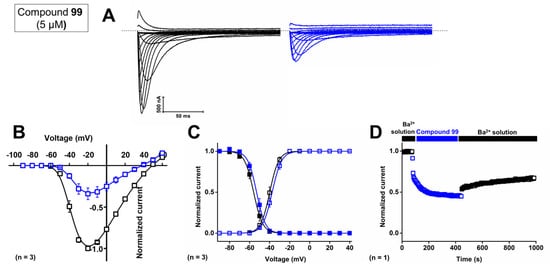
Figure 10.
Electrophysiological characterization of activity of compound 99 on the CaV3.1 channel. (A) Current traces before (black) and after (blue) application of 5 μM compound 99. In 10 mM Ba2+, currents were elicited by depolarizing pulses from −90 to +60 mV. Holding potential was −90 mV. First 300 ms of the depolarizing pulses are shown. (B) Normalized voltage–current relationship in control (black symbols) and compound 99 (5 μM, blue symbols) conditions. (C) Steady-state activation (open symbols) and inactivation (closed symbols) curves in control (black symbols) and compound 99 (5 μM, blue symbols) conditions. Data are presented as means ± SEM (n = 3). (D) Normalized CaV3.1 channel current during wash-in and wash-out of compound 99 over time.
2.3. Selectivity Screening
As these purpurealidin analogs represent a new class of potent CaV3.1 inhibitors, they could potentially be used as pharmacological tools to investigate the role of CaV3 channels in various disease models. Therefore, compounds 74, 76, 79, and 99 were further evaluated at a concentration of 10 μM for their selectivity against cardiac voltage-gated potassium and sodium channels. No significant inhibition was observed for compounds 74, 76, 79, and 99 against KV1.4, KV4.2 and NaV1.5 channels. Nevertheless, the affinity for the KV11.1 channel is an important issue for all four compounds (Table 2).
Table 2.
Percentages of inhibition of the selectivity screening of the selected compounds against cardiac voltage-gated potassium and sodium channels at a concentration of 10 μM. Data are presented as means ± SEM (n ≥ 3). NS—not significant.
3. Discussion
3.1. Structure–Activity Relationship
In this study, we report the identification and characterization of a new group of CaV3 channel blockers. These compounds are analogs based on the structure of purpurealidin I (1). Both the bromotyrosine and bromotyramine parts of the structure have been investigated.
In the first series of compounds (Table S1), the bromotyramine part of the purpurealidin I structure was modified. Only compound 9 showed a low but significant inhibition of 15.1 ± 3.1%. An interesting comparison in structure can be made between purpurealidin I (1) and compound 4. Although their bromotyrosine part is identical, the bromotyramine part of these compounds shows a few differences. The aryl group of compound 1 contains two bromine atoms in the meta position, whereas compound 4 contains only one. Furthermore, compound 4 lacks an ethylmethylamine group that is present in compound 1. Finally, the linker between hydroxyamino amide and the aryl group of compound 1 contains two carbon atoms, whereas this linker is missing in compound 4. These modifications cause a difference in inhibition of approximately 70%. When the structure of compound 4 is compared to the structure of compound 9, only a small difference in structure can be noticed. Compound 4 is halogenated with a bromine atom, whereas compound 9 is halogenated with a chlorine atom, causing a shift in effect from not significant (compound 4) to significant (compound 9) inhibition of the CaV3.1 currents.
In the next series of compounds (Table S2), structural changes have been made to the bromotyrosine part of purpurealidin I (1). The three most potent compounds from this series are compounds 31, 33, and 37, as they produce 42.4 ± 5.3%, 64.1 ± 6.4%, and 33.5 ± 5.6% inhibition, respectively. Remarkably, all three analogs show a minor difference in the structure that sets them apart from the other compounds in this series. They have a monomethylamine at the end of the propoxy chain, which is identical to the purpurealidin I (1) structure, whereas this is replaced by an N,N-dimethylamino moiety in the other compounds, except for compound 26. In compounds 16 and 19, the monomethylamine is replaced by isopropyl, and no significant effect could be observed. Nonetheless, it is uncertain whether the loss of activity can be attributed to this or to the para-methoxy group that is missing in the aryl group (Ar). Between the three analogs 31, 33, and 37, the only difference is the halogenation of the meta-position, which influences the activity as follows: dihalogenation with chlorine atoms (compound 33) > monohalogenation with a chlorine atom (compound 31) > dihalogenation with fluorine atoms (compound 37). It appears that compounds dihalogenated with chlorine atoms show a greater effect. A similar effect is seen when two other compounds are compared: compound 30 is monohalogenated with a chlorine atom and caused 8.7 ± 4.2% inhibition; compound 32 is dihalogenated with chlorine atoms and inhibited the channels with 26.9 ± 3.0%. Although the tyrosine part of compound 1 is halogenated with two bromine atoms, halogenation with bromine or iodine atoms appears to cause a loss of activity for this series of analogs. Further investigation is required to understand the structurally important moieties in detail.
The third series of compounds (Table S3) include the spirocyclic bromotyrosines. For these analogs, the open chain of the bromotyrosine part is rendered spirocyclic, and the bromotyramine part is replaced by different side chains. Although seven analogs retained some significant activity, it appears that the spirocyclic structure causes a decrease in activity. This difference could be attributed to the more rigid structure of the spirocyclic bromotyrosine analogs. Because there are no large differences in activity between the analogs of this series, it is difficult to determinate any SAR. One thing we could confirm is the importance of halogenation. Non-halogenated compounds showed no significant effect. For the halogenated compounds, no significant difference was observed between bromine or chlorine dihalogenation for compounds with identical R-groups, for example, compounds 48 and 65, compounds 53 and 68, compounds 55 and 66, and compounds 56 and 67. However, a significant difference was observed between the activity of compound 68 (21.7 ± 6.7% inhibition) and the activity of compounds 58 (3.7 ± 2.0% inhibition) and 72 (3.1 ± 1.1% inhibition). Remarkably, there is only a minor structural difference. All three compounds contain the same pyridine ring, but the pyridine ring of compound 68 is immediately linked to the amide. However, compounds 58 and 72 contain a methylene unit between the pyridine ring and the amide, which completely abolishes the effect.
Finally, the last series comprises the diarylamine analogs (Table S4). Compound 99 is the most active compound of this series, with an inhibition of 90.7 ± 5.4%. This analog has a unique structure compared to other analogs of this series. Although the diarylamine group is a common feature among most compounds, it is replaced by a diarylether group in compound 99. Other features of this compound include an ortho-nitro and para-trifluoromethyl substituent on the phenyl ring (R1 and R2 in Table S4). Furthermore, at the end of the aliphatic chain is a basic dimethylamine group (R3 in Table S4), and finally, the aliphatic chain between the aromatic ring and the basic center contains a hydroxy group (R4 in Table S4). An interesting comparison can be made between compounds 99 and 76. The latter compound shares the same features as compound 99, with the only exception that it contains the diarylamine structure instead of the diarylether group. This structural difference reduced the activity of compound 76 by half compared to compound 99. Another noteworthy comparison can be made between compounds 76 and 79. Removal of the nitro group on the phenyl ring of compound 76 causes an increase in activity by approximately 20% for compound 79, although this difference was not shown to be significant. When the nitro group on the phenyl ring is replaced by a methyl ester (compound 81), no difference in activity could be observed. However, compound 77, in which the nitro group is replaced by an amino group, lost approximately 20% activity compared to compound 76. For two analogs, compounds 73 and 74, the hydroxy group was removed. Despite this structural modification, no difference in effect was observed between these compounds and their analogs that contain the hydroxy group (compounds 75 and 76, respectively). Furthermore, a significant increase in activity was observed for compounds that contain a para-trifluoromethyl group versus the ones without, for example, compounds 73 and 74, 75 and 76, 78 and 79, and 80 and 81. Replacement of the amine linker between the two aryl groups by an amide, resulting in an N-aryl-arylamide structure (such as compounds 102 and 103), abolished all activity. Finally, it appears that compounds with a large group at the end of the aliphatic chain are less potent. This is observed in compounds with a morpholino moiety (such as compounds 88–93) or an aniline moiety (compound 94), or even in larger groups such as compounds 95–98. Remarkably, a structural difference was noticed that is similar to what is earlier described for the bromotyramine analogs (second series, Table S2). Namely, at the end of the aliphatic chain, compound 85 contains a monomethylamine that is identical to the purpurealidin I (1) structure, causing a higher percentage of inhibition than its analogous compound with an N,N-dimethylamino moiety (compound 76).
The other diarylamine analogs (Table S5) show no big changes in effect, although a few differences may be worth mentioning. First, no significant difference in activity was observed upon non-halogenation, bromine dehalogenation, or chlorine dihalogenation of the aryl ring. Next, compound 104 shows a structure that is comparable with compound 74. They both contain an ortho-nitro and para-trifluoromethyl substituent on the phenyl ring, and they do not show a hydroxy group on their aliphatic chain. The only difference is that the aliphatic chain of compound 104 lacks one carbon atom compared to compound 74. This causes a reduction in activation for compound 104 by approximately 30%. Lastly, compounds 115–119 have an N-aryl-arylamide structure, contain a phenyl ring directly linked to the ether that connects it to the aryl ring, and show alterations in the positioning of the nitro groups. Those differences in structure caused a complete loss of activity. However, further structure–function studies are required to draw more definite conclusions.
3.2. Electrophysiological Characterization
After observing the interesting effect of compounds 74, 76, 79, and 99 on CaV3.1 channels, further characterization was conducted. The calculated IC50 values yielded 2.6 ± 0.6 μM, 3.7 ± 0.6 μM, 5.9 ± 1.9 μM, and 4.1 ± 0.1 μM, respectively. No significant difference between these values was observed, although, at a concentration of 10 μM, the effect of compound 99 was significantly higher. These variances may be explained by differences in the properties of the compounds. Namely, the effect of compounds 74, 76, and 79 on all three CaV3 channels were shown to be reversible, but this seemed not the case for compound 99. After up to ten minutes of washing out with a compound-free solution, the effect of the compound was still present. Furthermore, our electrophysiological studies suggested that these compounds act like a pore blocker obstructing the ion flow by binding in the pore region of the CaV3.1 channel rather than acting as a voltage-sensor modifier because no significant shift in the activation and steady-state inactivation curves could be observed.
Zhao et al. observed a similar phenomenon, in that the Z944 compound acted as a pore blocker on the CaV3.1 channel [27]. After detailed investigations, they showed that the Z944 compound exhibited an arch-shaped conformation, reclining in the central cavity of the pore domain, with its wide end embedded in the II–III fenestration and its narrow end situated above the intracellular gate like a plug [27]. It may be noticed that the structure of purpurealidin I (1) and compound 99 resembles the Z944 structure (Figure 11). This could indicate that our compounds share the same binding location, which would be consistent with our findings that the compounds are pore blockers of the CaV3.1 channel.
Figure 11.
Structures of Z944, purpurealidin I (1), and compound 99.
3.3. Selectivity Screening
In the last part of this study, analogs 74, 76, 79, and 99 were evaluated for their selectivity against other cardiac ion channels, in particular the KV11.1 channel. The currents of potassium channels, KV1.4 and KV4.2, and the sodium channel, NaV1.5, play an important role in the generation of action potentials in the heart [28]. Therefore, the effect of the compounds was investigated on these channels, but no activity could be observed. What is of concern, however, is their activity on KV11.1 channels. All four compounds were shown to potently inhibit hERG channels.
Whereas compound 99 is equipotent across CaV3.1 and hERG channels, Z944 is 150-fold more potent against CaV3.1 compared to hERG channels. Remarkably, the hERG activity of compound 99 (69.1 ± 1.9% at a concentration of 10 μM) is comparable to the hERG activity of Z944 (IC50 = 7.8 μM).
This selectivity issue is of major concern during drug development projects. Inhibition of the hERG channel can cause prolongation of the AP and lead to an increase in the length of time between the start of the Q-wave and the end of the T-wave on an electrocardiogram (QT interval), resulting in lethal cardiac arrhythmia, e.g., Torsade de Pointes (TdP) [29]. Jamieson et al. summarized different in vitro, in vivo, and in silico approaches to determine and overcome hERG blockade. They suggest disrupting any putative π-stacking interactions with the channel, which, in the case of compound 99, could be related to its diarylether group. Furthermore, they propose attenuating hERG inhibition by creating subtle changes in the molecular architecture, such as by removing electron-donating groups, adding electron-withdrawing moieties, or modifying the template of the structure, such as by deleting distal aromatic groups or incorporating heterocyclic groups [29]. These techniques can be used in the future to further optimize our lead compounds. Moreover, the published cryo-EM structure of the hERG channel can help in predicting the hERG liability of compounds [30].
4. Materials and Methods
4.1. Compound Synthesis
For this study, we made use of an existing library of purpurealidin analogs. These compounds were chemically synthesized in previous studies, as described in detail by Moreels et al. [18], Patel et al. [23], Bhat et al. [24], Toplak et al. [25], and Gubič et al. [26]. Synthesis of compounds 65–72 and 115–120 is described in the supporting material.
4.2. Xenopus Laevis Surgery
Stage V–VI oocytes were isolated via partial ovariectomy from X. laevis frogs (African clawed frogs), as described previously [31]. Mature female frogs were purchased from CRB Xénopes (Rennes, France) and housed in the Aquatic Facility (KU Leuven) in compliance with the regulations of the European Union (EU) concerning the welfare of laboratory animals, as declared in Directive 2010/63/EU. After the frogs were anesthetized by a 15 min submersion in 0.1% tricaine methanesulfonate (pH 7.0), the oocytes were collected. The isolated oocytes were then washed with a 1.5 mg/mL collagenase solution to remove the follicle layer.
4.3. Expression of CaV3 Channels in Xenopus Laevis Oocytes
Rat CaV3.1, human CaV3.2, rat CaV3.3, human NaV1.5, rat KV1.4, rat KV4.2, and human KV11.1 were expressed in X. laevis oocytes by linearizing the plasmids and subsequent in vitro transcription using a commercial T7 mMESSAGE mMACHINE transcription kit (Ambion, Carlsbad, CA, USA). Defolliculated Xenopus oocytes were injected with 50 nL of cRNA at a concentration of 1 ng/nL by using a microinjector (Drummond Scientific Company, Broomall, PA, USA). The oocytes were incubated in a solution containing (in mM): NaCl, 96; KCl, 2; CaCl2, 1.8; MgCl2, 2; and HEPES, 5 (pH 7.5). This solution was supplemented with 50 mg/L gentamicin sulfate and 90 mg theophylline.
4.4. Electrophysiological Recordings
Two-electrode voltage clamp recordings were performed at room temperature (18–22 °C) by using a GeneClamp 500 amplifier (Molecular Devices, San Jose, CA, USA) controlled by a pClamp data acquisition system (Axon Instruments, Union City, CA, USA). Whole-cell currents from oocytes were recorded 5–10 days after mRNA injection. The bath solution composition was the following (in mM): BaCl2, 10; NaOH, 90; KOH, 1; EDTA, 0.1; and HEPES, 5 (pH 7.5). Voltage and current electrodes were filled with 3 M KCl. Resistances of both electrodes were kept between 0.8 and 1.5 MΩ. The elicited CaV3.1, CaV3.2, and CaV3.3 currents were filtered at 2 kHz and sampled at 4 kHz using a four-pole low-pass Bessel filter. Leak subtraction was performed by using a -P/4 protocol. Oocytes were placed in a measuring chamber filled with 200 μL ND96. The compounds were added directly to the measuring chamber under continuous application of the described voltage protocol. The percentage channel modulation was calculated when steady-state conditions were reached.
For the electrophysiological analysis of peptides, a number of protocols were applied from a holding potential of −90 mV. Currents for CaV3.1 and CaV3.2, which were carried by Ba2+, were evoked by 300 ms depolarizing pulses to −25 mV or Vmax (the voltage corresponding to maximal Ba2+ current in control conditions). Currents for CaV3.3, which were carried by Ba2+, were evoked by 1 s depolarizing pulses to −25 mV or Vmax. The current–voltage relationships were determined by 600 ms step depolarizations between −90 and +60 mV, using 10 mV increments. The values of IPBa were normalized to the maximal Ba2+ current amplitude and plotted as a function of voltage. To investigate the effect of the peptide toxins on the activation and steady-state inactivation, a standard 2-step protocol was used. In this protocol, 600 ms conditioning 10 mV step prepulses, ranging from −90 to +60 mV, were followed by a 400 ms test pulse to −25 mV. Data were normalized to the maximal Ba2+ current amplitude, plotted against prepulse potential, and fitted by using the Boltzmann function: ICa/Imax = {(1 − C)/(1 + exp[(V − Vh)/k)]} + C, where Imax is the maximal IBa, Vh is the voltage corresponding to half-maximal inactivation, V is the test voltage, k is the slope factor, and C is a constant representing a non-inactivating persistent fraction (close to 0 in control). The concentration–response relationship was determined by fitting the data with the Hill equation: y = 100/{1 + [IC50/(toxin)]h}, where y is the amplitude of the toxin-induced effect, IC50 is the toxin concentration at half-maximal efficacy, toxin is the toxin concentration, and h is the Hill coefficient.
The selectivity screening was conducted with the following protocols: the elicited KV1.4, KV4.2, and KV11.1 currents were filtered at 0.5 kHz and sampled at 2 kHz; NaV1.5 currents were filtered at 2 kHz and sampled at 20 kHz using a four-pole low-pass Bessel filter. Leak subtraction was performed using a -P/4 protocol. The oocytes were measured in a bath solution composition of ND96 (in mM): NaCl, 96; KCl, 2; CaCl2, 1.8; MgCl2, 2; and HEPES, 5 (pH 7.5). For the electrophysiological analysis of the compounds, a number of protocols were applied from a holding potential of −90 mV. Currents for KV1.4 and KV4.2 were evoked by 0.5 s depolarizing pulses to 0 mV, followed by 0.5 s repolarizing pulses to −50 mV. Currents for KV11.1 were evoked by 2.5 s depolarizing prepulses to +40 mV, followed by hyperpolarizing pulses to −120 mV for 2.5 s. Currents for NaV1.5 were evoked by 100 ms depolarizing pulses to 0 mV.
4.5. Data Analysis
All electrophysiological data are presented as means ± SEM of at least three independent experiments (n ≥ 3), unless otherwise indicated. All data were analyzed using pClamp Clampfit 10.4 (Molecular Devices, Downingtown, PA, USA) and OriginPro 9 (Originlab, Northampton, MA, USA) or GraphPad Prism 8 software (GraphPad Software, Inc., San Diego, CA, USA). Statistical significance was determined using one-way ANOVA with Dunnett’s post-test or one-way ANOVA with Tukey’s post-test; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
5. Conclusions
In this study, we discovered a new class of CaV3 inhibitors. The effect of the sponge-derived purpurealidin I (1) compound and 119 bromotyrosine analogs were examined using two-electrode voltage clamp electrophysiology on oocytes expressing the CaV3.1 channel. Although some conclusions could be drawn, additional studies are still required to further analyze and correlate differences in structure with differences in activity on the CaV3.1 channel. Moreover, molecular modeling can be used to determine the binding location of these compounds in the CaV3.1 channel. Further electrophysiological characterization was conducted with potent inhibitor compounds 74, 76, 79, and 99. Our data suggest that these compounds act on the channel as pore blockers.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24043429/s1.
Author Contributions
Conceptualization, S.P. and J.T.; methodology, A.-S.D. and S.P.; validation, A.-S.D.; formal analysis, A.-S.D.; investigation, A.-S.D., E.L.P.-J. and L.H.; synthesis, P.A.P., C.B., M.V., I.E., F.V., T.B., A.L., N.H., E.M.-L., Ž.T., T.T. and L.P.M.; resources, J.Y.-K. and J.T.; data curation, A.-S.D., E.L.P.-J. and L.H.; writing—original draft preparation, A.-S.D.; writing—review and editing, A.-S.D., P.K., J.Y.-K., C.B., S.P. and J.T.; visualization, A.-S.D.; supervision, P.K., S.P. and J.T.; project administration, S.P. and J.T.; funding acquisition, J.Y.-K., P.K., S.P. and J.T. All authors have read and agreed to the published version of the manuscript.
Funding
J.T. was funded by F.W.O.-Vlaanderen (grants GOC2319N, GOA4919N, and G0E7120N). S.P. was supported by KU Leuven funding (PDM/19/164) and by F.W.O.-Vlaanderen (grant 12W7822N). University of Helsinki research was supported by the Academy of Finland (Grant Nos. 307464 J.Y.-K. and 315937 P.K.).
Institutional Review Board Statement
The use of the frogs was in accordance with license number LA1210239 of the Laboratory of Toxicology & Pharmacology, University of Leuven. All animal care and experimental procedures complied with the guidelines of the ‘European Convention for the protection of vertebrate animals used for experimental and other scientific purposes’ (Strasbourg, 18.III.1986).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank J.-H. Lee (Sogang University, Seoul, Republic of Korea) for sharing the hCaV3.2 clone. The rCaV3.3 cDNA clone was kindly provided by E. Perez-Reyes (University of Virginia, Charlottesville, VA, USA). We thank Nina Sipari from the Viikki Metabolomics Unit (Helsinki Institute of Life Science, University of Helsinki; Biocenter Finland) for her expertise with the LC-MS analyses.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed]
- Perez-Reyes, E. Molecular Physiology of Low-Voltage-Activated T-type Calcium Channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Iijima, T. Cardiac T-type Ca2+ channels in the heart. J. Mol. Cell. Cardiol. 2010, 48, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Bao, W.; Wang, Z.; Nattel, S. Comparison of ion-channel subunit expression in canine cardiac purkinje fibers and ventricular muscle. Circ. Res. 2002, 91, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Visa, A.; Shaikh, S.; Alza, L.; Herreros, J.; Canti, C. The Hard-To-Close Window of T-Type Calcium Channels. Trends Mol. Med. 2019, 25, 571–584. [Google Scholar] [CrossRef]
- Chiang, C.-S.; Huang, C.-H.; Chieng, H.; Chang, Y.-T.; Chang, D.; Chen, J.-J.; Chen, Y.-C.; Chen, Y.-H.; Shin, H.-S.; Campbell, K.P.; et al. The CaV3.2 T-type Ca2+ channel is required for pressure overload–induced cardiac hypertrophy in mice. Circ. Res. 2009, 104, 522–530. [Google Scholar] [CrossRef]
- Cribbs, L. T-type calcium channel expression and function in the diseased heart. Channels 2010, 4, 447–452. [Google Scholar] [CrossRef]
- Mangoni, M.E.; Traboulsie, A.; Leoni, A.L.; Couette, B.; Marger, L.; Le Quang, K.; Kupfer, E.; Cohen-Solal, A.; Vilar, J.; Shin, H.S.; et al. Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/alpha1G T-type calcium channels. Circ. Res. 2006, 98, 1422–1430. [Google Scholar] [CrossRef]
- Baudot, M.; Torre, E.; Bidaud, I.; Louradour, J.; Torrente, A.G.; Fossier, L.; Talssi, L.; Nargeot, J.; Barrere-Lemaire, S.; Mesirca, P.; et al. Concomitant genetic ablation of L-type CaV1.3 (alpha1D) and T-type CaV3.1 (alpha1G) Ca2+ channels disrupts heart automaticity. Sci. Rep. 2020, 10, 18906. [Google Scholar] [CrossRef]
- Chen, C.C.; Lamping, K.G.; Nuno, D.W.; Barresi, R.; Prouty, S.J.; Lavoie, J.L.; Cribbs, L.L.; England, S.K.; Sigmund, C.D.; Weiss, R.M.; et al. Abnormal coronary function in mice deficient in alpha1H T-type Ca2+ channels. Science 2003, 302, 1416–1418. [Google Scholar] [CrossRef]
- Lee, M. Z944: A first in class T-type calcium channel modulator for the treatment of pain. J. Peripher. Nerv. Syst. 2014, 19, S11–S12. [Google Scholar] [CrossRef] [PubMed]
- Mehrke, G.; Zong, X.G.; Flockerzi, V.; Hofmann, F. The Ca2+-channel blocker Ro 40-5967 blocks differently T-type and L-type Ca2+ channels. J. Pharmacol. Exp. Ther. 1994, 271, 1483–1488. [Google Scholar]
- Mádle, A.; Linhartová, K.; Koza, J. Effects of the T-type calcium channel blockade with oral mibefradil on the electrophysiologic properties of the human heart. Med. Sci. Monit. 2001, 7, 74–77. [Google Scholar] [PubMed]
- Ertel, S.I.; Clozel, J.P. Mibefradil (Ro 40-5967): The first selective T-type Ca2+ channel blocker. Expert Opin. Investig. Drugs 1997, 6, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Rubaiy, H.N.; Chen, G.L.; Hallett, T.; Zaibi, N.; Zeng, B.; Saurabh, R.; Xu, S.Z. Mibefradil, a T-type Ca2+ channel blocker also blocks Orai channels by action at the extracellular surface. Br. J. Pharmacol. 2019, 176, 3845–3856. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Zamponi, G.W. T-Type channel druggability at a crossroads. ACS Chem. Neurosci. 2019, 10, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.; Kaufmann, P.; Ort, M.; Kornberger, R.; Dingemanse, J. Multiple-ascending dose study in healthy subjects to assess the pharmacokinetics, tolerability, and CYP3A4 interaction potential of the T-type calcium channel blocker ACT-709478, a potential new antiepileptic drug. CNS Drugs 2020, 34, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Moreels, L.; Bhat, C.; Voráčová, M.; Peigneur, S.; Goovaerts, H.; Mäki-Lohiluoma, E.; Zahed, F.; Pardo, L.A.; Yli-Kauhaluoma, J.; Kiuru, P.; et al. Synthesis of novel purpurealidin analogs and evaluation of their effect on the cancer-relevant potassium channel KV10.1. PLoS ONE 2017, 12, e0188811. [Google Scholar] [CrossRef]
- Peng, J.; Li, J.; Hamann, M.T. The marine bromotyrosine derivatives. Alkaloids Chem. Biol. 2005, 61, 59–262. [Google Scholar]
- Barreca, M.; Spanò, V.; Montalbano, A.; Cueto, M.; Díaz Marrero, A.R.; Deniz, I.; Erdoğan, A.; Lukić Bilela, L.; Moulin, C.; Taffin-De-Givenchy, E.; et al. Marine anticancer agents: An overview with a particular focus on their chemical classes. Mar. Drugs 2020, 18, 619. [Google Scholar] [CrossRef]
- Huyck, T.K.; Gradishar, W.; Manuguid, F.; Kirkpatrick, P. Eribulin mesylate. Nat. Rev. Drug Discov. 2011, 10, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Tilvi, S.; D’Souza, L. Identifying the related compounds using electrospray ionization tandem mass spectrometry: Bromotyrosine alkaloids from marine sponge Psammaplysilla purpurea. Eur. J. Mass Spectrom. 2012, 18, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.A.; Bruun, T.; Ilina, P.; Mäkkylä, H.; Lempinen, A.; Yli-Kauhaluoma, J.; Tammela, P.; Kiuru, P.S. Synthesis and cytotoxicity evaluation of spirocyclic bromotyrosine clavatadine C analogs. Mar. Drugs 2021, 19, 400. [Google Scholar] [CrossRef]
- Bhat, C.; Ilina, P.; Tilli, I.; Voráčová, M.; Bruun, T.; Barba, V.; Hribernik, N.; Lillsunde, K.-E.; Mäki-Lohiluoma, E.; Rüffer, T.; et al. Synthesis and antiproliferative activity of marine bromotyrosine purpurealidin I and its derivatives. Mar. Drugs 2018, 16, 481. [Google Scholar] [CrossRef] [PubMed]
- Toplak, Ž.; Hendrickx, L.A.; Gubič, Š.; Možina, Š.; Žegura, B.; Štern, A.; Novak, M.; Shi, X.; Peigneur, S.; Tytgat, J.; et al. 3D pharmacophore-based discovery of novel KV10.1 inhibitors with antiproliferative activity. Cancers 2021, 13, 1244. [Google Scholar] [CrossRef]
- Gubič, Š.; Toplak, Ž.; Shi, X.; Dernovšek, J.; Hendrickx, L.A.; Pinheiro-Junior, E.L.; Peigneur, S.; Tytgat, J.; Pardo, L.A.; Peterlin Mašič, L.; et al. New Diarylamine KV10.1 Inhibitors and Their Anticancer Potential. Pharmaceutics 2022, 14, 1963. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, G.; Wu, Q.; Wu, K.; Li, R.; Lei, J.; Pan, X.; Yan, N. Cryo-EM structures of apo and antagonist-bound human CaV3.1. Nature 2019, 576, 492–497. [Google Scholar] [CrossRef]
- Grant, A.O. Cardiac ion channels. Circ. Arrhythmia Electrophysiol. 2009, 2, 185–194. [Google Scholar] [CrossRef]
- Jamieson, C.; Moir, E.M.; Rankovic, Z.; Wishart, G. Medicinal chemistry of hERG optimizations: Highlights and hang-ups. J. Med. Chem. 2006, 49, 5029–5046. [Google Scholar] [CrossRef]
- Wang, W.; MacKinnon, R. Cryo-EM structure of the open human ether-à-go-go-related K+ channel hERG. Cell 2017, 169, 422–430. [Google Scholar] [CrossRef]
- Kalina, R.S.; Peigneur, S.; Zelepuga, E.A.; Dmitrenok, P.S.; Kvetkina, A.N.; Kim, N.Y.; Leychenko, E.V.; Tytgat, J.; Kozlovskaya, E.P.; Monastyrnaya, M.M.; et al. New insights into the type II toxins from the sea anemone Heteractis crispa. Toxins 2020, 12, 44. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).