

Phospholipase D1 Attenuation Therapeutics Promotes Resilience against Synaptotoxicity in 12-Month-Old 3xTg-AD Mouse Model of Progressive Neurodegeneration


Abstract

1. Introduction
2. Results
2.1. Increased PLD1 Levels and Increased Co-Localization with Amyloidogenic Proteins in Post-Mortem Human AD Hippocampal Slices Compared to Age-Matched Control
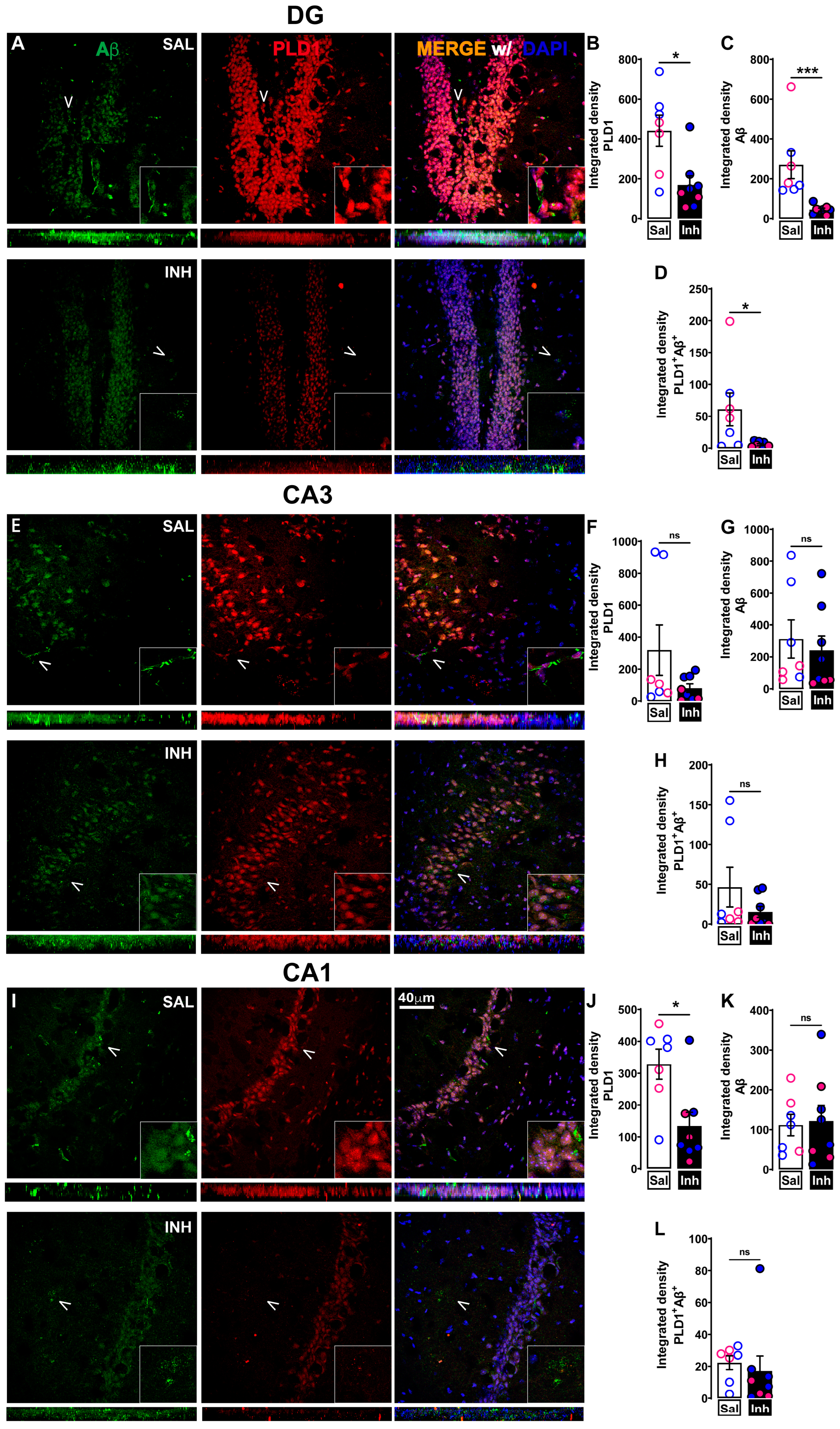
2.2. Chronic One-Month Treatment with PLD1 Inhibitor (VU0155069 or VU01 at 1 mg/kg/2 Days i.p.) in 12-Month-Old 3xTg-AD Mice Attenuates PLD1, PLD1-Aβ and PLD1-tau Co-Staining Expression Differentially in Hippocampal Subregions
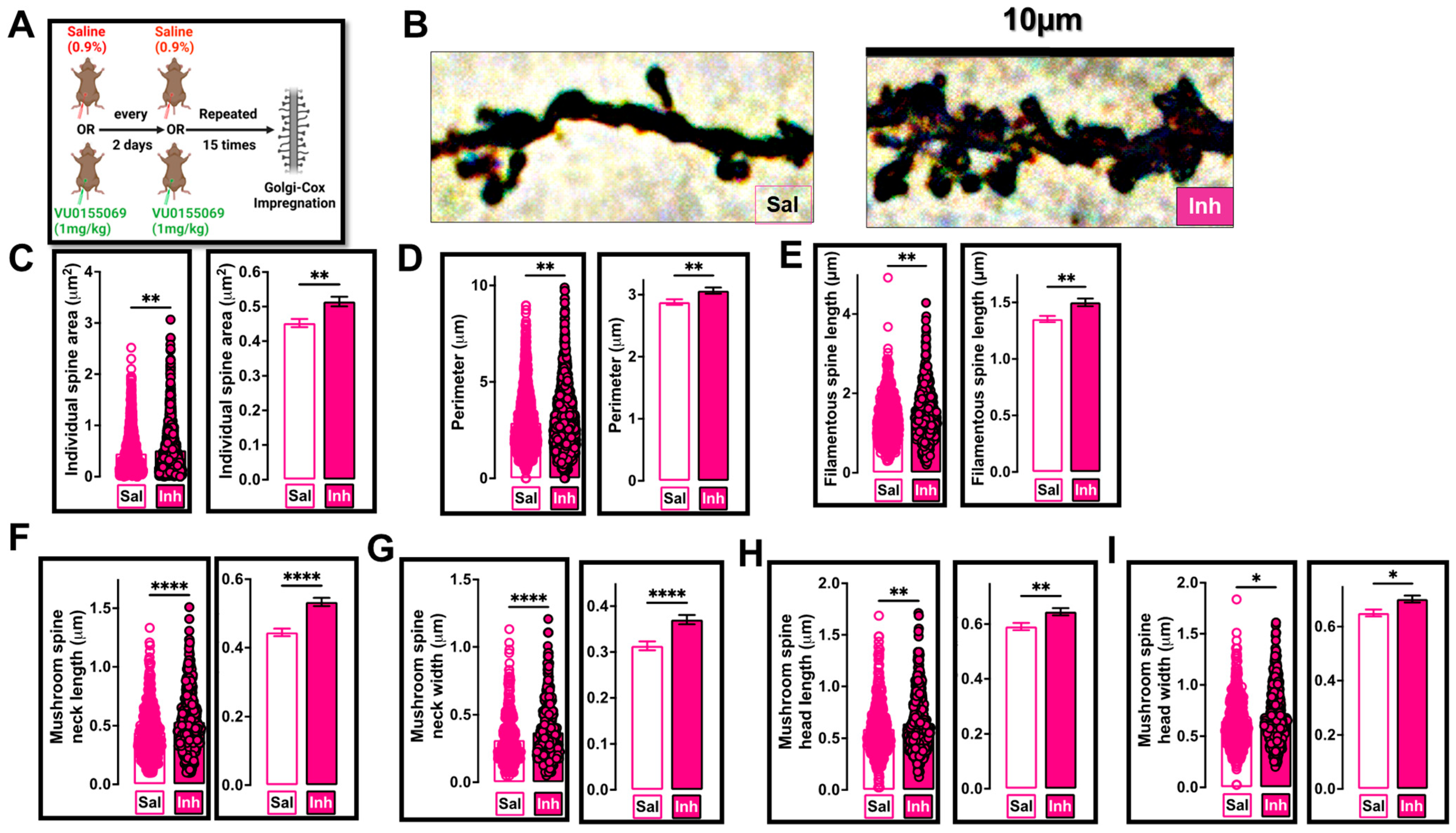
2.3. VU01 Dependent Chronic Inhibition of PLD1 Preserves Specific Dendritic Spine Morphologies in 12-Month-Old 3xTg-AD Mice
2.4. Restoration of Hippocampal Synaptic Function by Chronic PLD1 Inhibition Regimen in 12-Month-Old 3xTg-AD Mice
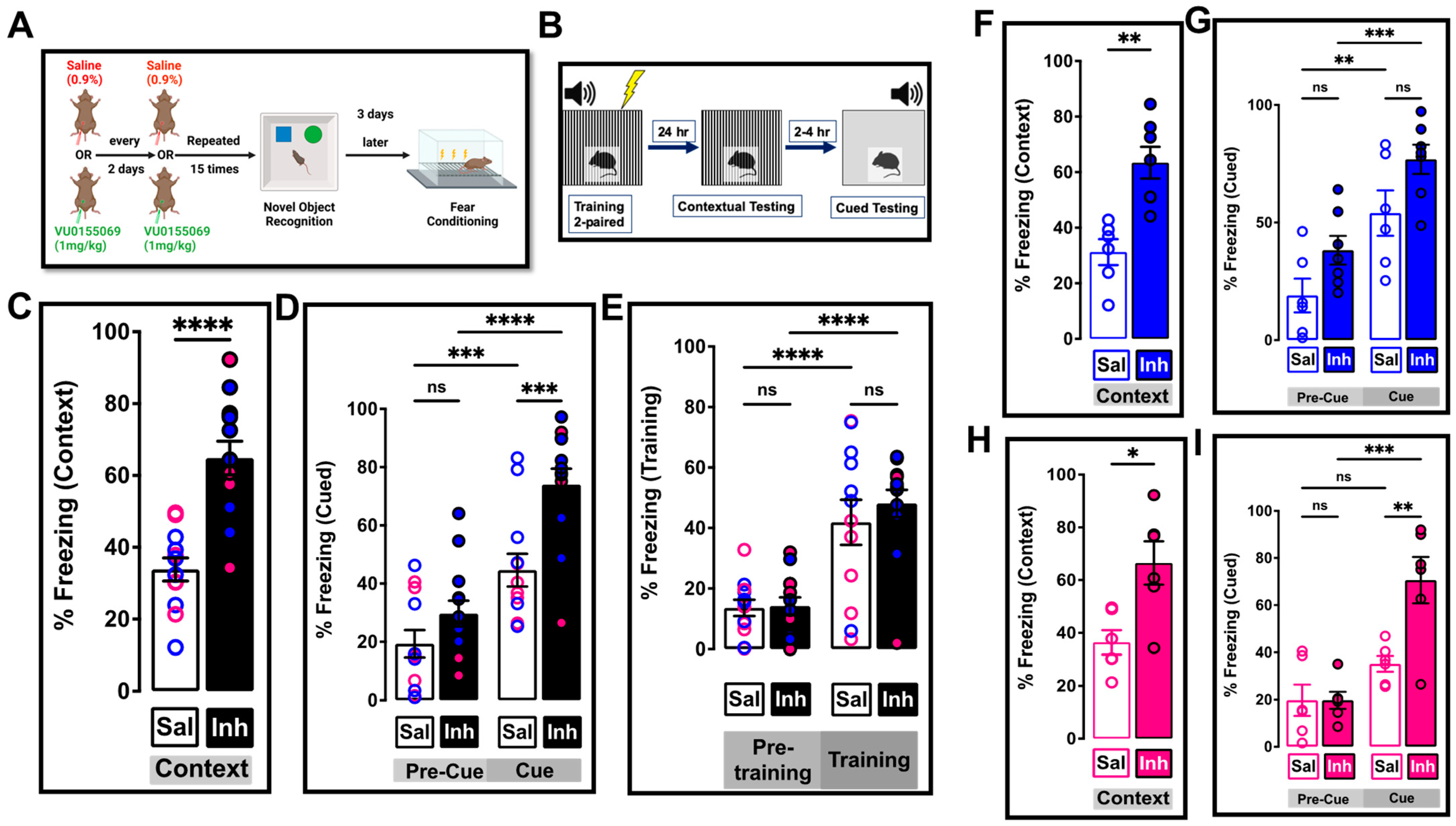
2.5. Chronic One-Month Treatment with PLD1 Inhibitor Ameliorates Memory Deficits in 12-Month-Old 3xTg-AD Mice
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Human Samples
4.3. Animals
4.4. Field Electrophysiological Recordings
4.5. Behaviors
4.5.1. Novel Object Recognition
4.5.2. Fear Conditioned Response
4.6. Tissue Processing and Golgi–Cox Staining
4.7. Dendrite Imaging
4.8. Immunofluorescence
4.9. Quantitative Microscopy
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gaugler, J.; James, B.; Johnson, T.; Reimer, J.; Solis, M.; Weuve, J.; Buckley, R.F.; Hohman, T.J. 2022 Alzheimer’s Disease Facts and Figures; Alzheimer’s Association: Chicago, IL, USA, 2022; pp. 1–122. [Google Scholar]
- Vina, J.; Lloret, A. Why women have more Alzheimer’s disease than men: Gender and mitochondrial toxicity of amyloid-beta peptide. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S527–S533. [Google Scholar] [CrossRef] [PubMed]
- Rocca, W.A.; Mielke, M.M.; Vemuri, P.; Miller, V.M. Sex and gender differences in the causes of dementia: A narrative review. Maturitas 2014, 79, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Dubal, D.B. Sex difference in Alzheimer’s disease: An updated, balanced and emerging perspective on differing vulnerabilities. Handb. Clin. Neurol 2020, 175, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Dominguez, S.; Fisher, D.W.; Dong, H. Sex differences in chronic stress responses and Alzheimer’s disease. Neurobiol. Stress 2018, 8, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.W.; Bennett, D.A.; Dong, H. Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol. Aging 2018, 70, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Reese, L.C.; Zhang, W.; Dineley, K.T.; Kayed, R.; Taglialatela, G. Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell 2008, 7, 824–835. [Google Scholar] [CrossRef]
- Liao, D.; Miller, E.C.; Teravskis, P.J. Tau acts as a mediator for Alzheimer’s disease-related synaptic deficits. Eur. J. Neurosci. 2014, 39, 1202–1213. [Google Scholar] [CrossRef]
- Miller, E.C.; Teravskis, P.J.; Dummer, B.W.; Zhao, X.; Huganir, R.L.; Liao, D. Tau phosphorylation and tau mislocalization mediate soluble Abeta oligomer-induced AMPA glutamate receptor signaling deficits. Eur. J. Neurosci. 2014, 39, 1214–1224. [Google Scholar] [CrossRef]
- Bilousova, T.; Miller, C.A.; Poon, W.W.; Vinters, H.V.; Corrada, M.; Kawas, C.; Hayden, E.Y.; Teplow, D.B.; Glabe, C.; Albay, R., 3rd; et al. Synaptic Amyloid-beta Oligomers Precede p-Tau and Differentiate High Pathology Control Cases. Am. J. Pathol. 2016, 186, 185–198. [Google Scholar] [CrossRef]
- Fa, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6, 19393. [Google Scholar] [CrossRef] [PubMed]
- Comerota, M.M.; Krishnan, B.; Taglialatela, G. Near infrared light decreases synaptic vulnerability to amyloid beta oligomers. Sci. Rep. 2017, 7, 15012. [Google Scholar] [CrossRef] [PubMed]
- Comerota, M.M.; Tumurbaatar, B.; Krishnan, B.; Kayed, R.; Taglialatela, G. Near Infrared Light Treatment Reduces Synaptic Levels of Toxic Tau Oligomers in Two Transgenic Mouse Models of Human Tauopathies. Mol. Neurobiol. Mol. Neurobiol. 2018, 56, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, B.; Kayed, R.; Taglialatela, G. Elevated phospholipase D isoform 1 in Alzheimer’s disease patients’ hippocampus: Relevance to synaptic dysfunction and memory deficits. Alzheimers Dement. 2018, 4, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Kanfer, J.N.; McCartney, D.G. Phosphatase and phospholipase activity in Alzheimer brain tissues. J. Neural Transmission. Suppl. 1987, 24, 183–188. [Google Scholar]
- Kanfer, J.N.; Pettegrew, J.W.; Moossy, J.; McCartney, D.G.; Bin, Q.; Strychor, S.; McKeag, D.W. Possible enzymatic causes for elevated phosphomonoesters in Alzheimer’s disease brain tissue. Ann. N. Y. Acad. Sci. 1991, 640, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Kanfer, J.N.; Pettegrew, J.W.; Moossy, J.; McCartney, D.G. Alterations of selected enzymes of phospholipid metabolism in Alzheimer’s disease brain tissue as compared to non-Alzheimer’s demented controls. Neurochem. Res. 1993, 18, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.P.T.; Flicker, L. Lecithin for dementia and cognitive impairment. Cochrane Database Syst. Rev. 2000. [Google Scholar] [CrossRef]
- Vitale, N.; Caumont, A.S.; Chasserot-Golaz, S.; Du, G.; Wu, S.; Sciorra, V.A.; Morris, A.J.; Frohman, M.A.; Bader, M.F. Phospholipase D1: A key factor for the exocytotic machinery in neuroendocrine cells. EMBO J. 2001, 20, 2424–2434. [Google Scholar] [CrossRef]
- Humeau, Y.; Vitale, N.; Chasserot-Golaz, S.; Dupont, J.L.; Du, G.; Frohman, M.A.; Bader, M.F.; Poulain, B. A role for phospholipase D1 in neurotransmitter release. Proc. Natl. Acad. Sci. USA 2001, 98, 15300–15305. [Google Scholar] [CrossRef]
- Ammar, M.R.; Thahouly, T.; Hanauer, A.; Stegner, D.; Nieswandt, B.; Vitale, N. PLD1 participates in BDNF-induced signalling in cortical neurons. Sci. Rep. 2015, 5, 14778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kanaho, Y.; Frohman, M.A.; Tsirka, S.E. Phospholipase D1-promoted release of tissue plasminogen activator facilitates neurite outgrowth. J. Neurosci. 2005, 25, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.C.; Zheng, J.Y.; Tang, L.J.; Huang, B.S.; Li, K.; Tao, Y.; Yu, W.; Zhu, R.L.; Li, S.; Li, L.X. MiR-133b Promotes neurite outgrowth by targeting RhoA expression. Cell Physiol. Biochem. 2015, 35, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Koshimizu, H.; Kiyosue, K.; Hara, T.; Hazama, S.; Suzuki, S.; Uegaki, K.; Nagappan, G.; Zaitsev, E.; Hirokawa, T.; Tatsu, Y.; et al. Multiple functions of precursor BDNF to CNS neurons: Negative regulation of neurite growth, spine formation and cell survival. Mol. Brain 2009, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Kanaho, Y.; Funakoshi, Y.; Hasegawa, H. Phospholipase D signalling and its involvement in neurite outgrowth. Biochim. Biophys. Acta 2009, 1791, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Zhong, M.; Wang, R.; Netzer, W.J.; Shields, D.; Zheng, H.; Sisodia, S.S.; Foster, D.A.; Gorelick, F.S.; Xu, H.; et al. Phospholipase D1 corrects impaired betaAPP trafficking and neurite outgrowth in familial Alzheimer’s disease-linked presenilin-1 mutant neurons. Proc. Natl. Acad. Sci. USA 2006, 103, 1936–1940. [Google Scholar] [CrossRef]
- Burkhardt, U.; Stegner, D.; Hattingen, E.; Beyer, S.; Nieswandt, B.; Klein, J. Impaired brain development and reduced cognitive function in phospholipase D-deficient mice. Neurosci. Lett. 2014, 572, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Dall’Armi, C.; Hurtado-Lorenzo, A.; Tian, H.; Morel, E.; Nezu, A.; Chan, R.B.; Yu, W.H.; Robinson, K.S.; Yeku, O.; Small, S.A.; et al. The phospholipase D1 pathway modulates macroautophagy. Nat. Commun. 2010, 1, 142. [Google Scholar] [CrossRef]
- Thakur, R.; Panda, A.; Coessens, E.; Raj, N.; Yadav, S.; Balakrishnan, S.; Zhang, Q.; Georgiev, P.; Basak, B.; Pasricha, R.; et al. Phospholipase D activity couples plasma membrane endocytosis with retromer dependent recycling. eLife 2016, 5, e18515. [Google Scholar] [CrossRef]
- Peng, X.; Frohman, M.A. Mammalian phospholipase D physiological and pathological roles. Acta Physiol. 2012, 204, 219–226. [Google Scholar] [CrossRef]
- Nelson, R.K.; Frohman, M.A. Physiological and pathophysiological roles for phospholipase D. J. Lipid Res. 2015, 56, 2229–2237. [Google Scholar] [CrossRef] [PubMed]
- Frohman, M.A. The phospholipase D superfamily as therapeutic targets. Trends Pharmacol. Sci. 2015, 36, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Chen, Q.; Frohman, M.A. Targeting phospholipase D with small-molecule inhibitors as a potential therapeutic approach for cancer metastasis. Future Oncol. 2009, 5, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A.; Thomas, P.G.; Lindsley, C.W. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat. Rev. Drug Discov. 2017, 16, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Lavieri, R.R.; Scott, S.A.; Selvy, P.E.; Kim, K.; Jadhav, S.; Morrison, R.D.; Daniels, J.S.; Brown, H.A.; Lindsley, C.W. Design, synthesis, and biological evaluation of halogenated N-(2-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)benzamides: Discovery of an isoform-selective small molecule phospholipase D2 inhibitor. J. Med. Chem. 2010, 53, 6706–6719. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.A.; Scott, S.A.; Lavieri, R.; Buck, J.R.; Selvy, P.E.; Stoops, S.L.; Armstrong, M.D.; Brown, H.A.; Lindsley, C.W. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures for PLD1 specificity. Bioorganic Med. Chem. Lett. 2009, 19, 1916–1920. [Google Scholar] [CrossRef]
- Waterson, A.G.; Scott, S.A.; Kett, N.R.; Blobaum, A.L.; Alex Brown, H.; Lindsley, C.W. Isoform selective PLD inhibition by novel, chiral 2,8-diazaspirodecan-1-one derivatives. Bioorganic Med. Chem. Lett. 2018, 28, 3670–3673. [Google Scholar] [CrossRef]
- Jin, J.K.; Ahn, B.H.; Na, Y.J.; Kim, J.I.; Kim, Y.S.; Choi, E.K.; Ko, Y.G.; Chung, K.C.; Kozlowski, P.B.; Min do, S. Phospholipase D1 is associated with amyloid precursor protein in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1015–1027. [Google Scholar] [CrossRef]
- Jin, J.K.; Kim, N.H.; Min, D.S.; Kim, J.I.; Choi, J.K.; Jeong, B.H.; Choi, S.I.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Increased expression of phospholipase D1 in the brains of scrapie-infected mice. J. Neurochem. 2005, 92, 452–461. [Google Scholar] [CrossRef]
- Jin, J.K.; Kim, N.H.; Lee, Y.J.; Kim, Y.S.; Choi, E.K.; Kozlowski, P.B.; Park, M.H.; Kim, H.S.; Min do, S. Phospholipase D1 is up-regulated in the mitochondrial fraction from the brains of Alzheimer’s disease patients. Neurosci. Lett. 2006, 407, 263–267. [Google Scholar] [CrossRef]
- Bourne, K.Z.; Natarajan, C.; Perez, C.X.M.; Tumurbaatar, B.; Taglialatela, G.; Krishnan, B. Suppressing aberrant phospholipase D1 signaling in 3xTg Alzheimer’s disease mouse model promotes synaptic resilience. Sci. Rep. 2019, 9, 18342. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Clinton, L.K.; Billings, L.M.; Green, K.N.; Caccamo, A.; Ngo, J.; Oddo, S.; McGaugh, J.L.; LaFerla, F.M. Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol. Dis. 2007, 28, 76–82. [Google Scholar] [CrossRef]
- Stover, K.R.; Campbell, M.A.; Van Winssen, C.M.; Brown, R.E. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer’s disease. Behav. Brain Res. 2015, 289, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, R.; Rodin, A.; Ferreira, E.; Velazquez, R.; Branca, C.; Caccamo, A.; Oddo, S. Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging Cell 2019, 18, e12873. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Llort, L.; García, Y.; Buccieri, K.; Revilla, S.; Suñol, C.; Cristofol, R.; Sanfeliu, C. Gender-Specific Neuroimmunoendocrine Response to Treadmill Exercise in 3xTg-AD Mice. Int. J. Alzheimers Dis. 2010, 2010, 128354. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dennison, J.L.; Ricciardi, N.R.; Lohse, I.; Volmar, C.H.; Wahlestedt, C. Sexual Dimorphism in the 3xTg-AD Mouse Model and Its Impact on Pre-Clinical Research. J. Alzheimers Dis. 2021, 80, 41–52. [Google Scholar] [CrossRef]
- Fertan, E.; Wong, A.A.; Vienneau, N.A.; Brown, R.E. Age and sex differences in motivation and spatial working memory in 3xTg-AD mice in the Hebb-Williams maze. Behav. Brain Res. 2019, 370, 111937. [Google Scholar] [CrossRef]
- Mastrangelo, M.A.; Bowers, W.J. Detailed immunohistochemical characterization of temporal and spatial progression of Alzheimer’s disease-related pathologies in male triple-transgenic mice. BMC Neurosci. 2008, 9, 81. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.J.; Guerriero, L.E.; Kohler, K.; Beechem, L.E.; Gillis, B.D.; Salisbury, F.; Wessel, C.; Wang, J.; Sunderam, S.; Bachstetter, A.D.; et al. Chronic Fragmentation of the Daily Sleep-Wake Rhythm Increases Amyloid-beta Levels and Neuroinflammation in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Neuroscience 2022, 481, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef] [PubMed]
- Shekari, A.; Fahnestock, M. Cholinergic neurodegeneration in Alzheimer disease mouse models. Handb. Clin. Neurol. 2021, 182, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Angulo, S.L.; Orman, R.; Neymotin, S.A.; Liu, L.; Buitrago, L.; Cepeda-Prado, E.; Stefanov, D.; Lytton, W.W.; Stewart, M.; Small, S.A.; et al. Tau and amyloid-related pathologies in the entorhinal cortex have divergent effects in the hippocampal circuit. Neurobiol. Dis. 2017, 108, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Wegmann, S.; Dujardin, S.; Commins, C.; Schiantarelli, J.; Klickstein, N.; Kamath, T.V.; Carlson, G.A.; Nelken, I.; Hyman, B.T. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nature Neurosci. 2019, 22, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing Endogenous Tau Ameliorates Amyloid ß-Induced Deficits in an Alzheimer’s Disease Mouse Model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense Reduction of Tau in Adult Mice Protects against Seizures. J. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef]
- Cai, D.; Netzer, W.J.; Zhong, M.; Lin, Y.; Du, G.; Frohman, M.; Foster, D.A.; Sisodia, S.S.; Xu, H.; Gorelick, F.S.; et al. Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc. Natl. Acad. Sci. USA 2006, 103, 1941–1946. [Google Scholar] [CrossRef]
- Winner, B.; Winkler, J. Adult neurogenesis in neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2015, 7, a021287. [Google Scholar] [CrossRef] [PubMed]
- Hollands, C.; Bartolotti, N.; Lazarov, O. Alzheimer’s Disease and Hippocampal Adult Neurogenesis; Exploring Shared Mechanisms. Front. Neurosci. 2016, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Kim, Y.S.; Bae, G.H.; Lee, H.Y.; Bae, Y.-S. VU0155069 inhibits inflammasome activation independent of phospholipase D1 activity. Sci. Rep. 2019, 9, 14349. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.L. Synaptic targeting by A beta oligomers (ADDLS) as a basis for memory loss in early Alzheimer’s disease. Alzheimers Dement. 2006, 2, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Spires, T.L.; Meyer-Luehmann, M.; Stern, E.A.; McLean, P.J.; Skoch, J.; Nguyen, P.T.; Bacskai, B.J.; Hyman, B.T. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 2005, 25, 7278–7287. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Meyer-Luehmann, M.; Osetek, J.D.; Jones, P.B.; Stern, E.A.; Bacskai, B.J.; Hyman, B.T. Impaired spine stability underlies plaque-related spine loss in an Alzheimer’s disease mouse model. Am. J. Pathol. 2007, 171, 1304–1311. [Google Scholar] [CrossRef]
- Yu, W.; Polepalli, J.; Wagh, D.; Rajadas, J.; Malenka, R.; Lu, B. A critical role for the PAR-1/MARK-tau axis in mediating the toxic effects of Abeta on synapses and dendritic spines. Hum. Mol. Genet. 2012, 21, 1384–1390. [Google Scholar] [CrossRef]
- Dickstein, D.L.; Brautigam, H.; Stockton, S.D., Jr.; Schmeidler, J.; Hof, P.R. Changes in dendritic complexity and spine morphology in transgenic mice expressing human wild-type tau. Brain Struct. Funct. 2010, 214, 161–179. [Google Scholar] [CrossRef]
- Sydow, A.; Van der Jeugd, A.; Zheng, F.; Ahmed, T.; Balschun, D.; Petrova, O.; Drexler, D.; Zhou, L.; Rune, G.; Mandelkow, E.; et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J. Neurosci. 2011, 31, 2511–2525. [Google Scholar] [CrossRef]
- Dubal, D.B. The Way of Tau: Secretion and Synaptic Dysfunction. Trends Mol. Med. 2018, 24, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wei, Q.; Liu, F.F.; Hu, F.; Xie, A.J.; Zhu, L.Q.; Liu, D. Synaptic Dysfunction in Alzheimer’s Disease: Abeta, Tau, and Epigenetic Alterations. Mol. Neurobiol. 2018, 55, 3021–3032. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.E.; Gan, L. Tau-mediated synaptic and neuronal dysfunction in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 51, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Micci, M.A.; Krishnan, B.; Bishop, E.; Zhang, W.R.; Guptarak, J.; Grant, A.; Zolochevska, O.; Tumurbaatar, B.; Franklin, W.; Marino, C.; et al. Hippocampal stem cells promotes synaptic resistance to the dysfunctional impact of amyloid beta oligomers via secreted exosomes. Mol. Neurodegener 2019, 14, 25. [Google Scholar] [CrossRef]
- Marino, C.; Krishnan, B.; Cappello, F.; Taglialatela, G. Hsp60 Protects against Amyloid beta Oligomer Synaptic Toxicity via Modification of Toxic Oligomer Conformation. ACS Chem. Neurosci. 2019, 10, 2858–2867. [Google Scholar] [CrossRef]
- Franklin, W.; Krishnan, B.; Taglialatela, G. Chronic synaptic insulin resistance after traumatic brain injury abolishes insulin protection from amyloid beta and tau oligomer-induced synaptic dysfunction. Sci. Rep. 2019, 9, 8228. [Google Scholar] [CrossRef]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. Biol. Sci. 2014, 369, 20130144. [Google Scholar] [CrossRef]
- Regan, P.; Piers, T.; Yi, J.-H.; Kim, D.-H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 4804–4812. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006, 443, 768–773. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Synaptic depression and aberrant excitatory network activity in Alzheimer’s disease: Two faces of the same coin? Neuromol. Med. 2010, 12, 48–55. [Google Scholar] [CrossRef]
- Falke, E.; Nissanov, J.; Mitchell, T.W.; Bennett, D.A.; Trojanowski, J.Q.; Arnold, S.E. Subicular dendritic arborization in Alzheimer’s disease correlates with neurofibrillary tangle density. Am. J. Pathol. 2003, 163, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Mijalkov, M.; Volpe, G.; Fernaud-Espinosa, I.; DeFelipe, J.; Pereira, J.B.; Merino-Serrais, P. Dendritic spines are lost in clusters in Alzheimer’s disease. Sci. Rep. 2021, 11, 12350. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, I.; Furini, C.R.; Myskiw, J.C. Fear Memory. Physiol. Rev. 2016, 96, 695–750. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Martinez-Coria, H.; Wu, J.W.; LaFerla, F.; Glabe, C.G. Systemic vaccination with anti-oligomeric monoclonal antibodies improves cognitive function by reducing Aβ deposition and tau pathology in 3xTg-AD mice. J. Neurochem. 2013, 126, 473–482. [Google Scholar] [CrossRef]
- Montoliu-Gaya, L.; Esquerda-Canals, G.; Bronsoms, S.; Villegas, S. Production of an anti-Aβ antibody fragment in Pichia pastoris and in vitro and in vivo validation of its therapeutic effect. PLoS ONE 2017, 12, e0181480. [Google Scholar] [CrossRef]
- Herline, K.; Prelli, F.; Mehta, P.; MacMurray, C.; Goñi, F.; Wisniewski, T. Immunotherapy to improve cognition and reduce pathological species in an Alzheimer’s disease mouse model. Alzheimers Res. Ther. 2018, 10, 54. [Google Scholar] [CrossRef]
- Montoliu-Gaya, L.; Güell-Bosch, J.; Esquerda-Canals, G.; Roda, A.R.; Serra-Mir, G.; Lope-Piedrafita, S.; Sánchez-Quesada, J.L.; Villegas, S. Differential effects of apoE and apoJ mimetic peptides on the action of an anti-Aβ scFv in 3xTg-AD mice. Biochem. Pharmacol. 2018, 155, 380–392. [Google Scholar] [CrossRef]
- Rosenberg, R.N.; Fu, M.; Lambracht-Washington, D. Active full-length DNA Aβ(42) immunization in 3xTg-AD mice reduces not only amyloid deposition but also tau pathology. Alzheimers Res. Ther. 2018, 10, 115. [Google Scholar] [CrossRef]
- Acero, G.; Garay, C.; Venegas, D.; Ortega, E.; Gevorkian, G. Novel monoclonal antibody 3B8 specifically recognizes pyroglutamate-modified amyloid β 3-42 peptide in brain of AD patients and 3xTg-AD transgenic mice. Neurosci. Lett. 2020, 724, 134876. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.K.; Jang, B.; Jin, H.T.; Choi, E.K.; Jung, C.G.; Akatsu, H.; Kim, J.I.; Carp, R.I.; Kim, Y.S. Phosphatidylinositol-glycan-phospholipase D is involved in neurodegeneration in prion disease. PLoS ONE 2015, 10, e0122120. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Lee, H.J.; Jang, Y.H.; Michael, S.; Masliah, E.; Min, D.S.; Lee, S.J. Phospholipase D1 regulates autophagic flux and clearance of alpha-synuclein aggregates. Cell Death Differ. 2014, 21, 1132–1141. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hazama, K.; Ishida, K.; Komori, M.; Nishimura, K.; Matsuo, S. Inhibition of PLD1 activity causes ER stress via regulation of COPII vesicle formation. Biochem. Biophys. Res. Commun. 2017, 490, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Soreng, K.; Knaevelsrud, H.; Holland, P.; Simonsen, A. HS1BP3 inhibits autophagy by regulation of PLD1. Autophagy 2017, 13, 985–986. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, L.O.; Pufe, T.; Koch, T. Role of phospholipase d in g-protein coupled receptor function. Membranes 2014, 4, 302–318. [Google Scholar] [CrossRef] [PubMed]
- Gavin, A.L.; Huang, D.; Huber, C.; Mårtensson, A.; Tardif, V.; Skog, P.D.; Blane, T.R.; Thinnes, T.C.; Osborn, K.; Chong, H.S.; et al. PLD3 and PLD4 are single-stranded acid exonucleases that regulate endosomal nucleic-acid sensing. Nat. Immunol. 2018, 19, 942–953. [Google Scholar] [CrossRef]
- Hwang, W.C.; Seo, S.H.; Kang, M.; Kang, R.H.; Di Paolo, G.; Choi, K.Y.; Min, D.S. PLD1 and PLD2 differentially regulate the balance of macrophage polarization in inflammation and tissue injury. J. Cell. Physiol. 2021, 236, 5193–5211. [Google Scholar] [CrossRef]
- Lee, S.M.; Choi, H.J.; Oh, C.H.; Oh, J.W.; Han, J.S. Leptin increases TNF-alpha expression and production through phospholipase D1 in Raw 264.7 cells. PLoS ONE 2014, 9, e102373. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Lee, S.Y.; Park, S.-Y.; Choi, H.-J.; Kim, E.-G.; Han, J.-S. Therapeutic potential of a phospholipase D1 inhibitory peptide fused with a cell-penetrating peptide as a novel anti-asthmatic drug in a Der f 2-induced airway inflammation model. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Urbahn, M.A.; Kaup, S.C.; Reusswig, F.; Kruger, I.; Spelleken, M.; Jurk, K.; Klier, M.; Lang, P.A.; Elvers, M. Phospholipase D1 regulation of TNF-alpha protects against responses to LPS. Sci. Rep. 2018, 8, 10006. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Wang, Z.; Luu, T.T.T.; Zhang, D.; Finke, B.; He, J.; Tay, L.W.R.; Di Paolo, G.; Du, G. PLD1 promotes reactive oxygen species production in vascular smooth muscle cells and injury-induced neointima formation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159062. [Google Scholar] [CrossRef] [PubMed]
- Usatyuk, P.V.; Kotha, S.R.; Parinandi, N.L.; Natarajan, V. Phospholipase D signaling mediates reactive oxygen species-induced lung endothelial barrier dysfunction. Pulm Circ. 2013, 3, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Suryadevara, V.; Huang, L.; Kim, S.J.; Cheresh, P.; Shaaya, M.; Bandela, M.; Fu, P.; Feghali-Bostwick, C.; Di Paolo, G.; Kamp, D.W.; et al. Role of phospholipase D in bleomycin-induced mitochondrial reactive oxygen species generation, mitochondrial DNA damage, and pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L175–L187. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Min, G.; Bae, Y.S.; Min, D.S. Phospholipase D is involved in oxidative stress-induced migration of vascular smooth muscle cells via tyrosine phosphorylation and protein kinase C. Exp. Mol. Med. 2004, 36, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Oguin, T.H., 3rd; Sharma, S.; Stuart, A.D.; Duan, S.; Scott, S.A.; Jones, C.K.; Daniels, J.S.; Lindsley, C.W.; Thomas, P.G.; Brown, H.A. Phospholipase D facilitates efficient entry of influenza virus, allowing escape from innate immune inhibition. J. Biol. Chem. 2014, 289, 25405–25417. [Google Scholar] [CrossRef] [PubMed]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavik, J.; Machala, M.; Zimmermann, P. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef]
- Han, X.; Yu, R.; Ji, L.; Zhen, D.; Tao, S.; Li, S.; Sun, Y.; Huang, L.; Feng, Z.; Li, X.; et al. InlB-mediated Listeria monocytogenes internalization requires a balanced phospholipase D activity maintained through phospho-cofilin. Mol. Microbiol. 2011, 81, 860–880. [Google Scholar] [CrossRef]
- Koffie, R.M.; Meyer-Luehmann, M.; Hashimoto, T.; Adams, K.W.; Mielke, M.L.; Garcia-Alloza, M.; Micheva, K.D.; Smith, S.J.; Kim, M.L.; Lee, V.M.; et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. USA 2009, 106, 4012–4017. [Google Scholar] [CrossRef]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef]
- Wei, W.; Nguyen, L.N.; Kessels, H.W.; Hagiwara, H.; Sisodia, S.; Malinow, R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 2010, 13, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, T.; Lechat, B.; Demedts, D.; Gielis, L.; Devijver, H.; Borghgraef, P.; Duimel, H.; Verheyen, F.; Kügler, S.; Van Leuven, F. Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am. J. Pathol. 2011, 179, 2001–2015. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Cooke, S.F. Long-term potentiation and long-term depression: A clinical perspective. Clinics 2011, 66 (Suppl. S1), 3–17. [Google Scholar] [CrossRef] [PubMed]
- Vitolo, O.V.; Sant’Angelo, A.; Costanzo, V.; Battaglia, F.; Arancio, O.; Shelanski, M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13217–13221. [Google Scholar] [CrossRef]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fà, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar Amyloid-β Positively Modulates Synaptic Plasticity and Memory in Hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Fa, M.; Staniszewski, A.; Hashimoto, G.; Aziz, F.; Sakurai, M.; Ribe, E.M.; Troy, C.M.; Mercken, M.; et al. Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 2011, 69, 819–830. [Google Scholar] [CrossRef]
- McIntire, L.B.; Berman, D.E.; Myaeng, J.; Staniszewski, A.; Arancio, O.; Di Paolo, G.; Kim, T.W. Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer’s disease. J. Neurosci. 2012, 32, 15271–15276. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 2004, 44, 181–193. [Google Scholar] [CrossRef]
- Lei, M.; Xu, H.; Li, Z.; Wang, Z.; O’Malley, T.T.; Zhang, D.; Walsh, D.M.; Xu, P.; Selkoe, D.J.; Li, S. Soluble Abeta oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol. Dis. 2016, 85, 111–121. [Google Scholar] [CrossRef]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s disease: Synaptic dysfunction and Abeta. Mol. Neurodegener. 2009, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Pickett, E.; Spires-Jones, T.L. Synaptic pathology: A shared mechanism in neurological disease. Ageing Res. Rev. 2016, 28, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Ramakers, G.M.; Urban, I.J.; Hens, J.J.; Oestreicher, A.B.; de Graan, P.N.; Gispen, W.H. Long-term potentiation and synaptic protein phosphorylation. Behav. Brain Res. 1995, 66, 53–59. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patel, H.; Zamani, R. The role of PKMζ in the maintenance of long-term memory: A review. Rev. Neurosci. 2021, 32, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Sanhueza, M.; Lisman, J. The CaMKII/NMDAR complex as a molecular memory. Mol. Brain 2013, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Norris, C.M.; Blalock, E.M.; Chen, K.C.; Porter, N.M.; Landfield, P.W. Calcineurin enhances L-type Ca(2+) channel activity in hippocampal neurons: Increased effect with age in culture. Neuroscience 2002, 110, 213–225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martin, Z.S.; Neugebauer, V.; Dineley, K.T.; Kayed, R.; Zhang, W.; Reese, L.C.; Taglialatela, G. alpha-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: Relevance to human synucleopathic diseases. J. Neurochem. 2012, 120, 440–452. [Google Scholar] [CrossRef]
- Yamin, G. NMDA receptor-dependent signaling pathways that underlie amyloid beta-protein disruption of LTP in the hippocampus. J. Neurosci. Res. 2009, 87, 1729–1736. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Peineau, S.; Howland, J.G.; Wang, Y.T. Long-term depression in the CNS. Nat. Rev. Neurosci. 2010, 11, 459–473. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Javonillo, D.I.; Tran, K.M.; Phan, J.; Hingco, E.; Kramár, E.A.; da Cunha, C.; Forner, S.; Kawauchi, S.; Milinkeviciute, G.; Gomez-Arboledas, A.; et al. Systematic Phenotyping and Characterization of the 3xTg-AD Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2022, 15, 785276. [Google Scholar] [CrossRef] [PubMed]
- Oblak, A.L.; Forner, S.; Territo, P.R.; Sasner, M.; Carter, G.W.; Howell, G.R.; Sukoff-Rizzo, S.J.; Logsdon, B.A.; Mangravite, L.M.; Mortazavi, A.; et al. Model organism development and evaluation for late-onset Alzheimer’s disease: MODEL-AD. Alzheimers Dement. 2020, 6, e12110. [Google Scholar] [CrossRef]
- Parhizkar, S.; Holtzman, D.M. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin. Immunol. 2022, 59, 101594. [Google Scholar] [CrossRef] [PubMed]
- Sienski, G.; Narayan, P.; Bonner, J.M.; Kory, N.; Boland, S.; Arczewska, A.A.; Ralvenius, W.T.; Akay, L.; Lockshin, E.; He, L.; et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. 2021, 13, aaz4564. [Google Scholar] [CrossRef] [PubMed]
- Grone, B.; Zalocusky, K.; Hao, Y.; Yoon, S.Y.; Arriola, P.; Huang, Y. Early and lifelong effects of APOE4 on neuronal gene expression networks relevant to Alzheimer’s disease. bioRxiv 2022. [CrossRef]
- Hirata-Fukae, C.; Li, H.F.; Hoe, H.S.; Gray, A.J.; Minami, S.S.; Hamada, K.; Niikura, T.; Hua, F.; Tsukagoshi-Nagai, H.; Horikoshi-Sakuraba, Y.; et al. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008, 1216, 92–103. [Google Scholar] [CrossRef]
- Stimmell, A.C.; Baglietto-Vargas, D.; Moseley, S.C.; Lapointe, V.; Thompson, L.M.; LaFerla, F.M.; McNaughton, B.L.; Wilber, A.A. Impaired Spatial Reorientation in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Sci. Rep. 2019, 9, 1311. [Google Scholar] [CrossRef]
- Creighton, S.D.; Mendell, A.L.; Palmer, D.; Kalisch, B.E.; MacLusky, N.J.; Prado, V.F.; Prado, M.A.M.; Winters, B.D. Dissociable cognitive impairments in two strains of transgenic Alzheimer’s disease mice revealed by a battery of object-based tests. Sci. Rep. 2019, 9, 57. [Google Scholar] [CrossRef]
- Omori, C.; Motodate, R.; Shiraki, Y.; Chiba, K.; Sobu, Y.; Kimura, A.; Nakaya, T.; Kondo, H.; Kurumiya, S.; Tanaka, T.; et al. Facilitation of brain mitochondrial activity by 5-aminolevulinic acid in a mouse model of Alzheimer’s disease. Nutr. Neurosci. 2017, 20, 538–546. [Google Scholar] [CrossRef]
- Blázquez, G.; Cañete, T.; Tobeña, A.; Giménez-Llort, L.; Fernández-Teruel, A. Cognitive and emotional profiles of aged Alzheimer’s disease (3×TgAD) mice: Effects of environmental enrichment and sexual dimorphism. Behav. Brain Res. 2014, 268, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.M.; Brown, R.E. Reference and working memory deficits in the 3xTg-AD mouse between 2 and 15-months of age: A cross-sectional study. Behav. Brain Res. 2015, 278, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Juottonen, K.; Laakso, M.P.; Insausti, R.; Lehtovirta, M.; Pitkänen, A.; Partanen, K.; Soininen, H. Volumes of the entorhinal and perirhinal cortices in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.J.; Stackman, R.W., Jr. Assessing rodent hippocampal involvement in the novel object recognition task. A review. Behav. Brain Res. 2015, 285, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Garber, J.C. Guide for the Care and Use of Laboratory Animals, 8th ed.; The National Academies Press: Washington, DC, USA, 2011. [Google Scholar] [CrossRef]
- Krishnan, B.; Sallam, H.S.; Tumurbataar, B.; Saieva, S.; Baymon, D.M.; Tuvdendorj, D.; Micci, M.A.; Abate, N.; Taglialatela, G. Amelioration of hippocampal dysfunction by adipose tissue-targeted stem cell transplantation in a mouse model of type 2 diabetes. J. Neurochem. 2020, 153, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, B.; Natarajan, C.; Bourne, K.Z.; Alikhani, L.; Wang, J.; Sowa, A.; Groen, K.; Perry, B.; Dickstein, D.L.; Baulch, J.E.; et al. Chronic Low Dose Neutron Exposure Results in Altered Neurotransmission Properties of the Hippocampus-Prefrontal Cortex Axis in Both Mice and Rats. Int. J. Mol. Sci. 2021, 22, 3668. [Google Scholar]
- Mathis, D.M.; Furman, J.L.; Norris, C.M. Preparation of acute hippocampal slices from rats and transgenic mice for the study of synaptic alterations during aging and amyloid pathology. J. Vis. Exp. 2011, 46, e2330. [Google Scholar] [CrossRef]
- Dineley, K.T.; Kayed, R.; Neugebauer, V.; Fu, Y.; Zhang, W.; Reese, L.C.; Taglialatela, G. Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J. Neurosci. Res. 2010, 88, 2923–2932. [Google Scholar] [CrossRef]
- Smith, D.L.; Pozueta, J.; Gong, B.; Arancio, O.; Shelanski, M. Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc. Natl. Acad. Sci. USA 2009, 106, 16877–16882. [Google Scholar] [CrossRef]
- Boros, B.D.; Greathouse, K.M.; Gentry, E.G.; Curtis, K.A.; Birchall, E.L.; Gearing, M.; Herskowitz, J.H. Dendritic spines provide cognitive resilience against Alzheimer’s disease. Ann. Neurol. 2017, 82, 602–614. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case No. | Diagnosis | Age (Years) | Gender | Braak Stage | MMSE | PMI (h) |
|---|---|---|---|---|---|---|
| 1220 | CTRL | >89 | F | 2 | 30 | 12 |
| 2467 | CTRL | 99 | F | 3 | 28 | 4.5 |
| 2553 | CTRL | 100 | M | 2 | 28 | 4 |
| 2682 | CTRL | 90 | F | 2 | 29 | 9 |
| 2755 | CTRL | 95 | F | 2 | 29 | 18 |
| 2953 | CTRL | 100 | F | 3 | 27 | 2.5 |
| 1678 | AD | 76 | F | 6 | 1 | 25 |
| 2312 | AD | 87 | F | 6 | - | 2.5 |
| 2316 | AD | 83 | M | 5 | - | 13 |
| 2317 | AD | 88 | M | 6 | - | 4 |
| 2374 | AD | 91 | M | 6 | - | 24 |
| 2543 | AD | 95 | M | 6 | 21 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natarajan, C.; Cook, C.; Ramaswamy, K.; Krishnan, B. Phospholipase D1 Attenuation Therapeutics Promotes Resilience against Synaptotoxicity in 12-Month-Old 3xTg-AD Mouse Model of Progressive Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 3372. https://doi.org/10.3390/ijms24043372
Natarajan C, Cook C, Ramaswamy K, Krishnan B. Phospholipase D1 Attenuation Therapeutics Promotes Resilience against Synaptotoxicity in 12-Month-Old 3xTg-AD Mouse Model of Progressive Neurodegeneration. International Journal of Molecular Sciences. 2023; 24(4):3372. https://doi.org/10.3390/ijms24043372
Chicago/Turabian StyleNatarajan, Chandramouli, Charles Cook, Karthik Ramaswamy, and Balaji Krishnan. 2023. "Phospholipase D1 Attenuation Therapeutics Promotes Resilience against Synaptotoxicity in 12-Month-Old 3xTg-AD Mouse Model of Progressive Neurodegeneration" International Journal of Molecular Sciences 24, no. 4: 3372. https://doi.org/10.3390/ijms24043372
APA StyleNatarajan, C., Cook, C., Ramaswamy, K., & Krishnan, B. (2023). Phospholipase D1 Attenuation Therapeutics Promotes Resilience against Synaptotoxicity in 12-Month-Old 3xTg-AD Mouse Model of Progressive Neurodegeneration. International Journal of Molecular Sciences, 24(4), 3372. https://doi.org/10.3390/ijms24043372