
Functional Characterization of p.(Arg160Gln) PCSK9 Variant Accidentally Found in a Hypercholesterolemic Subject

 , , and
, , and
Abstract
:1. Introduction
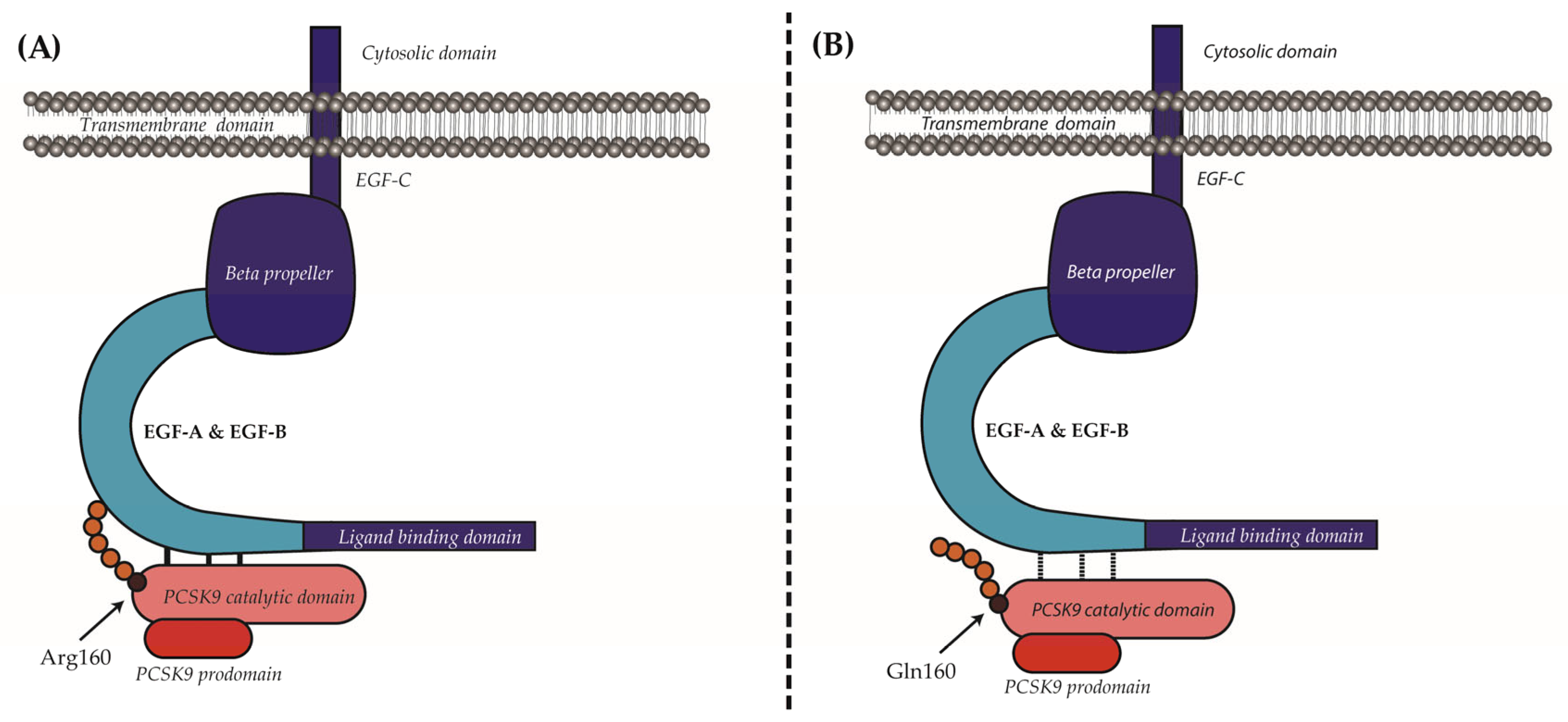
2. Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
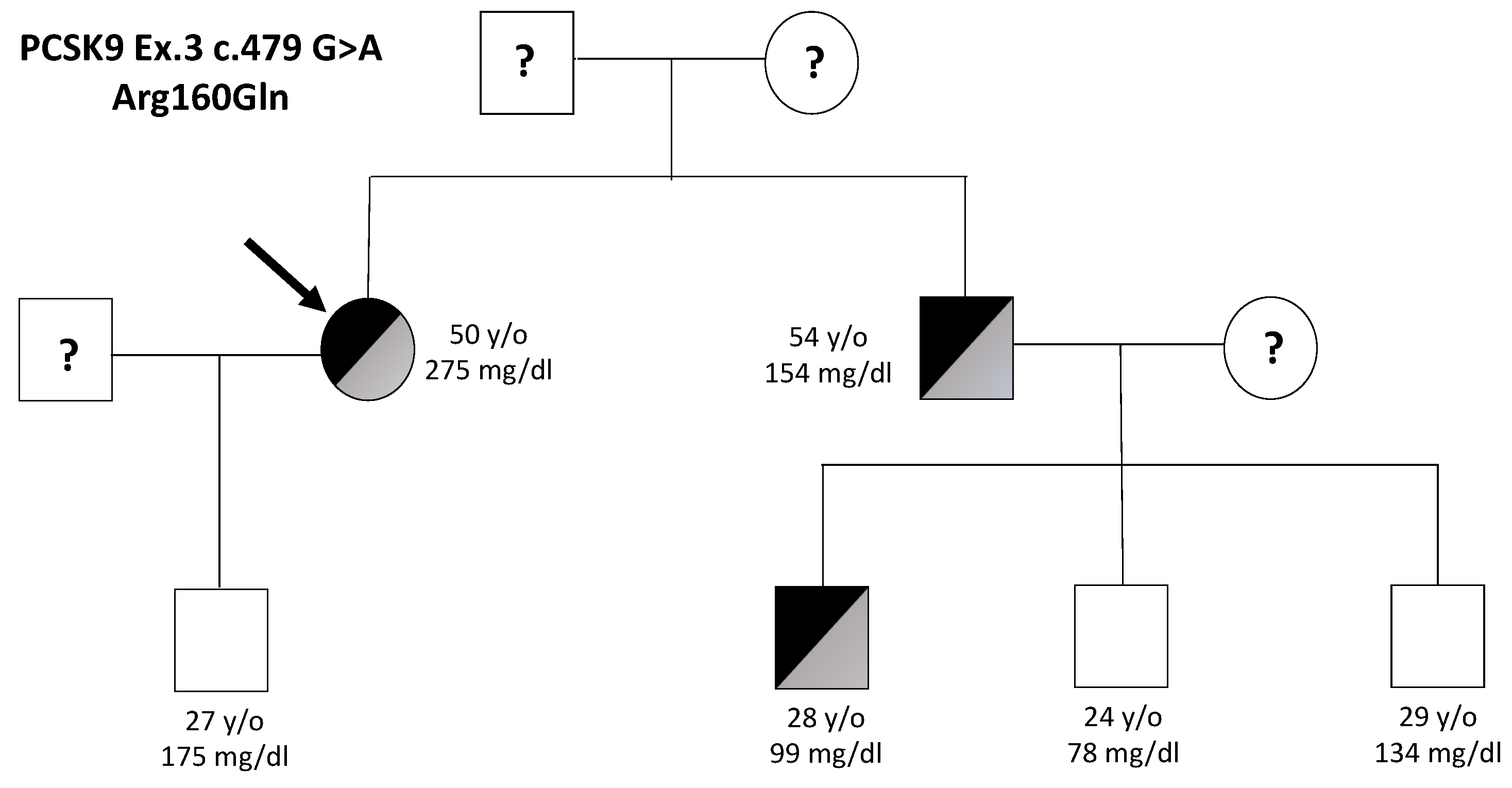
| Subject | Sex | Age | TC | LDL-C | HDL-C | TG | p.(Arg160Gln) |
|---|---|---|---|---|---|---|---|
| Proband | F | 50 | 341 | 275 | 59 | 139 | YES |
| Son | M | 27 | 282 | 175 | 76 | 199 | NO |
| Brother | M | 54 | 200 | 154 | 39 | 98 | YES |
| Nephew 1 | M | 28 | 169 | 99 | 53 | 88 | YES |
| Nephew 2 | M | 24 | 136 | 78 | 51 | 51 | NO |
| Nephew 3 | M | 29 | 187 | 134 | 44 | 132 | NO |
| Mutation Taster | PolyPhen-2 | PROVEAN | SIFT | Mutation Assessor | FATHMM | PANTHER | |
|---|---|---|---|---|---|---|---|
| p.(Arg160Gln) | Disease Causing | Probably damaging | Deleterious | Damaging | High Impact | Damaging | Probably Damaging |
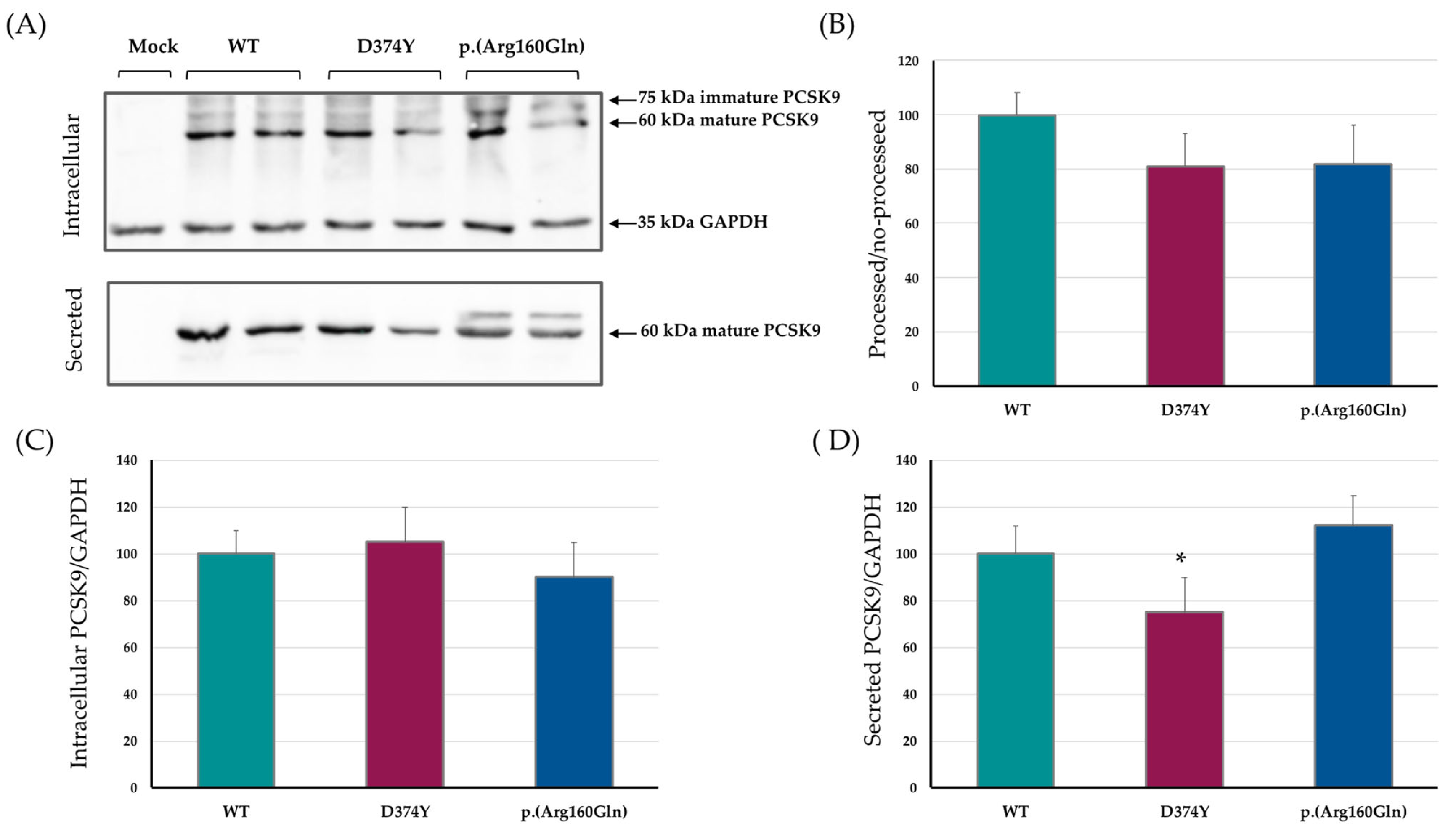
2.1. PCSK9 Expression, Maturation and Secretion
2.2. Effect of the p.(Arg160Gln) PCSK9 Variant on LDL Uptake
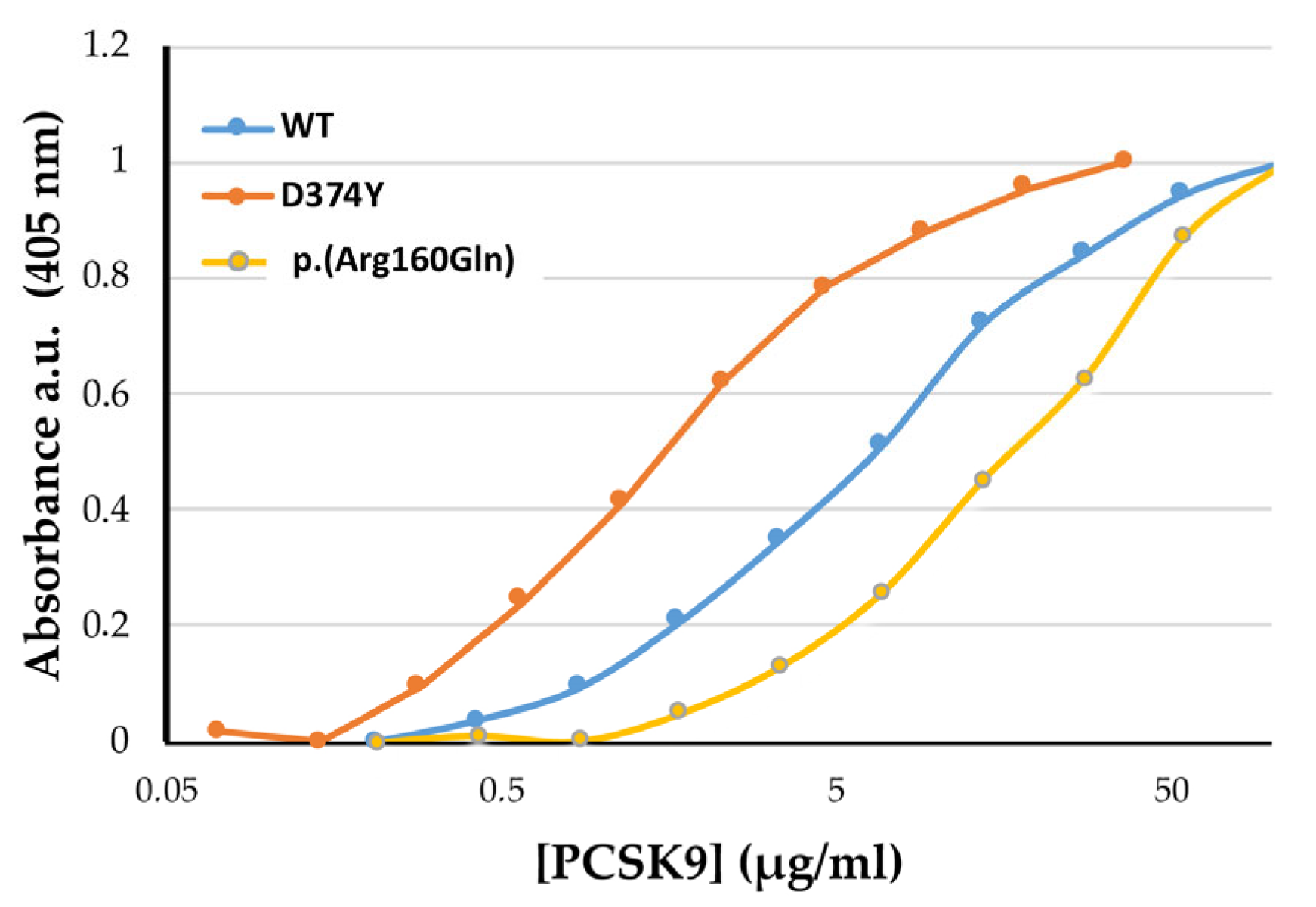
2.3. p.(Arg160Gln) PCSK9 Affinity (EC50) for the LDLr
| WT | D374Y | p.(Arg160Gln) | |
|---|---|---|---|
| EC50 | 8.57 ± 0.76 | 1.54 ± 0.38 * | 25.96 ± 0.72 * |
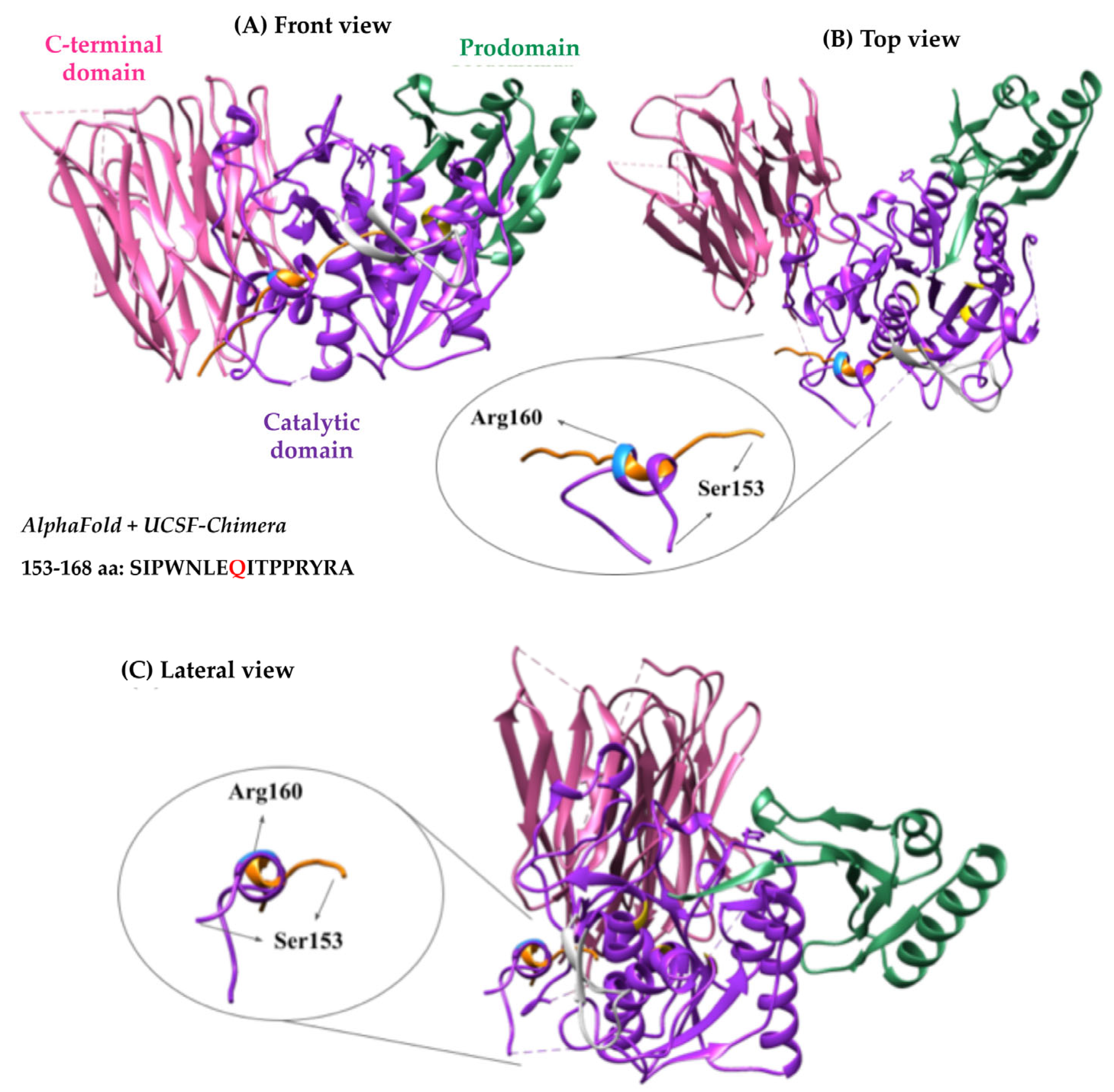
2.4. Bioinformatic Analysis of p.(Arg160Gln) PCSK9 3D Structure
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Sample Analysis
4.3. Site-Directed Mutagenesis and Cloning
4.4. PCSK9 Expression on HEK293 Cells
4.5. PCSK9 Secretion and Expression Analysis by Western Blot
4.6. Activity of PCKS9 Variants by Flow Cytometry
4.6.1. LDL Labelling with Fluorescein Isothiocyanate (FITC)
4.6.2. Flow Cytometry
4.6.3. PCSK9 Quantification
4.6.4. Solid-Phase Immunoassay for PCSK9-LDLr Ectodomain Affinity
- PCSK9 purification from stably transfected HEK293
- LDLr-ectodomain production and purification
- ELISA assay
4.7. PCSK9 3D Structure Bioinformatics Analysis
4.8. Statistical Analysis
4.9. Study Limitation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. Int. J. Mol. Sci. 2018, 19, 3426. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Breslow, J.L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 7100–7105. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Prat, A. The proprotein convertases are potential targets in the treatment of dyslipidemia. J. Mol. Med. 2007, 85, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Lee, H.S.; Kim, K.S.; Kim, Y.K.; Yoon, D.; Park, S.W. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J. Lipid Res. 2008, 49, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Awan, Z.; Chretien, M.; Mbikay, M. PCSK9: A key modulator of cardiovascular health. Circ. Res. 2014, 114, 1022–1036. [Google Scholar] [CrossRef]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Prat, A.; Chretien, M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [CrossRef]
- Piper, D.E.; Jackson, S.; Liu, Q.; Romanow, W.G.; Shetterly, S.; Thibault, S.T.; Shan, B.; Walker, N.P. The crystal structure of PCSK9: A regulator of plasma LDL-cholesterol. Structure 2007, 15, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Uribe, K.B.; Chemello, K.; Larrea-Sebal, A.; Benito-Vicente, A.; Galicia-Garcia, U.; Bourane, S.; Jaafar, A.K.; Lambert, G.; Martin, C. A Systematic Approach to Assess the Activity and Classification of PCSK9 Variants. Int. J. Mol. Sci. 2021, 22, 3602. [Google Scholar] [CrossRef]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H., Jr.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Lange, L.A.; Hu, Y.; Zhang, H.; Xue, C.; Schmidt, E.M.; Tang, Z.Z.; Bizon, C.; Lange, E.M.; Smith, J.D.; Turner, E.H.; et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am. J. Hum. Genet. 2014, 94, 233–245. [Google Scholar] [CrossRef]
- Huijgen, R.; Blom, D.J.; Hartgers, M.L.; Chemello, K.; Benito-Vicente, A.; Uribe, K.B.; Behardien, Z.; Blackhurst, D.M.; Brice, B.C.; Defesche, J.C.; et al. Novel PCSK9 (Proprotein Convertase Subtilisin Kexin Type 9) Variants in Patients with Familial Hypercholesterolemia from Cape Town. Atheroscler. Thromb. Vasc. Biol. 2021, 41, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Hernandez, R.M.; Di Taranto, M.D.; Benito-Vicente, A.; Uribe, K.B.; Lamiquiz-Moneo, I.; Larrea-Sebal, A.; Jebari, S.; Galicia-Garcia, U.; Novoa, F.J.; Boronat, M.; et al. The Arg499His gain-of-function mutation in the C-terminal domain of PCSK9. Atherosclerosis 2019, 289, 162–172. [Google Scholar] [CrossRef]
- Saavedra, Y.G.; Day, R.; Seidah, N.G. The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 protein is needed for the extracellular low-density lipoprotein receptor (LDLR) degradation pathway. J. Biol. Chem. 2012, 287, 43492–43501. [Google Scholar] [CrossRef] [PubMed]
- Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Validation of LDLr Activity as a Tool to Improve Genetic Diagnosis of Familial Hypercholesterolemia: A Retrospective on Functional Characterization of LDLr Variants. Int. J. Mol. Sci. 2018, 19, 1676. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wu, W.F.; Sun, L.Y.; Chen, P.P.; Wang, W.; Benito-Vicente, A.; Zhang, F.; Pan, X.D.; Cui, W.; Yang, S.W.; et al. The use of targeted exome sequencing in genetic diagnosis of young patients with severe hypercholesterolemia. Sci. Rep. 2016, 6, 36823. [Google Scholar] [CrossRef]
- Alves, A.C.; Azevedo, S.; Benito-Vicente, A.; Graca, R.; Galicia-Garcia, U.; Barros, P.; Jordan, P.; Martin, C.; Bourbon, M. LDLR variants functional characterization: Contribution to variant classification. Atherosclerosis 2021, 329, 14–21. [Google Scholar] [CrossRef]
- Zhang, Y.; Ultsch, M.; Skelton, N.J.; Burdick, D.J.; Beresini, M.H.; Li, W.; Kong-Beltran, M.; Peterson, A.; Quinn, J.; Chiu, C.; et al. Discovery of a cryptic peptide-binding site on PCSK9 and design of antagonists. Nat. Struct. Mol. Biol. 2017, 24, 848–856. [Google Scholar] [CrossRef]
- Kwon, H.J.; Lagace, T.A.; McNutt, M.C.; Horton, J.D.; Deisenhofer, J. Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. USA 2008, 105, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, M.J.; Cirillo, A.; Orsatti, L.; Ruggeri, L.; Fisher, T.S.; Santoro, J.C.; Cummings, R.T.; Cubbon, R.M.; Lo Surdo, P.; Calzetta, A.; et al. Structural and biochemical characterization of the wild type PCSK9-EGF(AB) complex and natural familial hypercholesterolemia mutants. J. Biol. Chem. 2009, 284, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Fasano, T.; Trenti, C.; Negri, E.A.; Guiducci, V.; Foracchia, M.; Bonelli, E.; Canovi, S.; Besutti, G.; Bertolini, S.; Calandra, S. Search for familial hypercholesterolemia patients in an Italian community: A real-life retrospective study. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Etxebarria, A.; Benito-Vicente, A.; Stef, M.; Ostolaza, H.; Palacios, L.; Martin, C. Activity-associated effect of LDL receptor missense variants located in the cysteine-rich repeats. Atherosclerosis 2015, 238, 304–312. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larrea-Sebal, A.; Trenti, C.; Jebari-Benslaiman, S.; Bertolini, S.; Calandra, S.; Negri, E.A.; Bonelli, E.; Benito-Vicente, A.; Uraga-Gracianteparaluceta, L.; Martín, C.; et al. Functional Characterization of p.(Arg160Gln) PCSK9 Variant Accidentally Found in a Hypercholesterolemic Subject. Int. J. Mol. Sci. 2023, 24, 3330. https://doi.org/10.3390/ijms24043330
Larrea-Sebal A, Trenti C, Jebari-Benslaiman S, Bertolini S, Calandra S, Negri EA, Bonelli E, Benito-Vicente A, Uraga-Gracianteparaluceta L, Martín C, et al. Functional Characterization of p.(Arg160Gln) PCSK9 Variant Accidentally Found in a Hypercholesterolemic Subject. International Journal of Molecular Sciences. 2023; 24(4):3330. https://doi.org/10.3390/ijms24043330
Chicago/Turabian StyleLarrea-Sebal, Asier, Chiara Trenti, Shifa Jebari-Benslaiman, Stefano Bertolini, Sebastiano Calandra, Emanuele A. Negri, Efrem Bonelli, Asier Benito-Vicente, Leire Uraga-Gracianteparaluceta, César Martín, and et al. 2023. "Functional Characterization of p.(Arg160Gln) PCSK9 Variant Accidentally Found in a Hypercholesterolemic Subject" International Journal of Molecular Sciences 24, no. 4: 3330. https://doi.org/10.3390/ijms24043330
APA StyleLarrea-Sebal, A., Trenti, C., Jebari-Benslaiman, S., Bertolini, S., Calandra, S., Negri, E. A., Bonelli, E., Benito-Vicente, A., Uraga-Gracianteparaluceta, L., Martín, C., & Fasano, T. (2023). Functional Characterization of p.(Arg160Gln) PCSK9 Variant Accidentally Found in a Hypercholesterolemic Subject. International Journal of Molecular Sciences, 24(4), 3330. https://doi.org/10.3390/ijms24043330