
Type I Diabetes Pathoetiology and Pathophysiology: Roles of the Gut Microbiome, Pancreatic Cellular Interactions, and the ‘Bystander’ Activation of Memory CD8+ T Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Classical Type 1 Diabetes Mellitus Pathoetiology
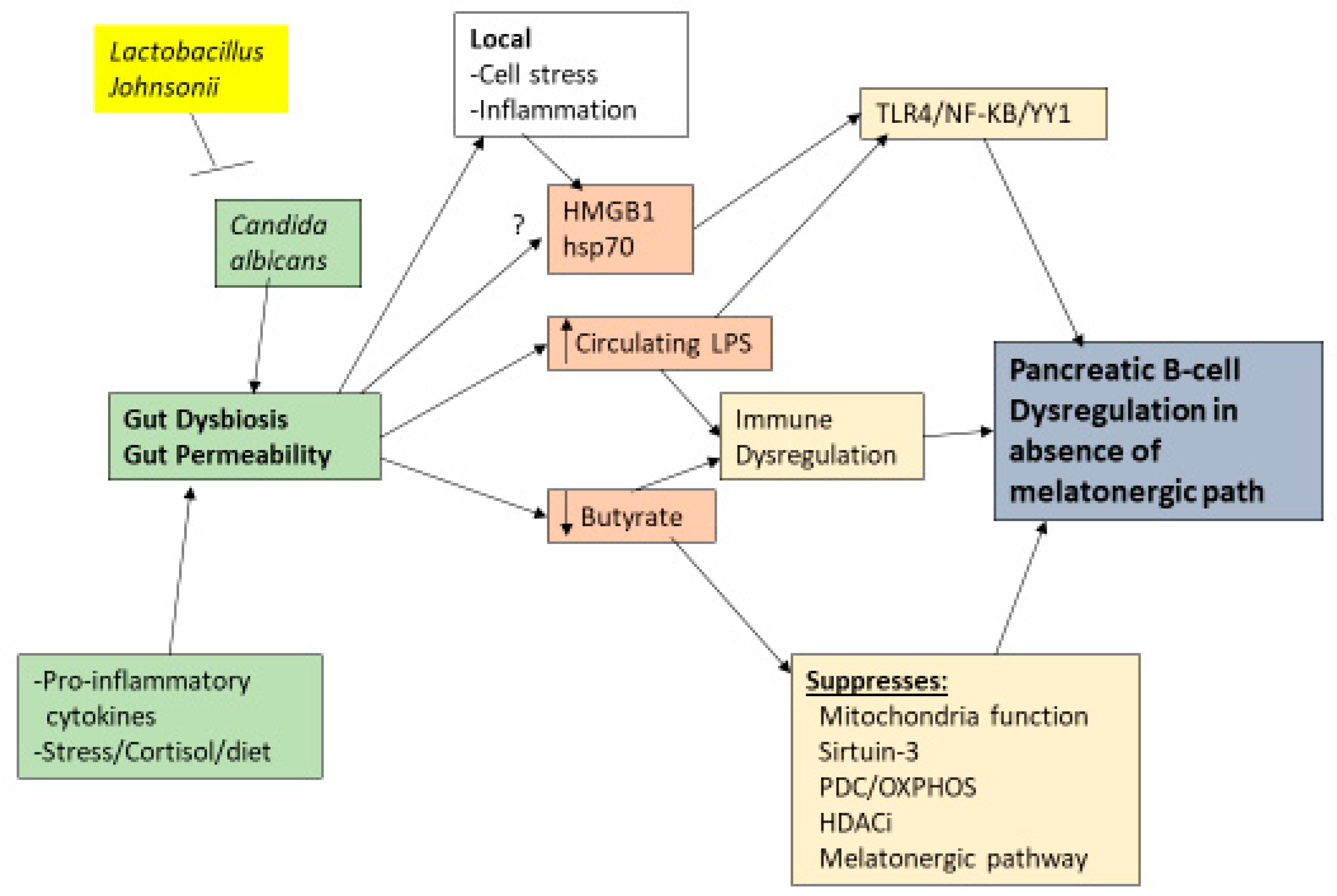
3. Wider T1DM Pathophysiology
4. Pancreatic β-Cell Mitochondria and Metabolism
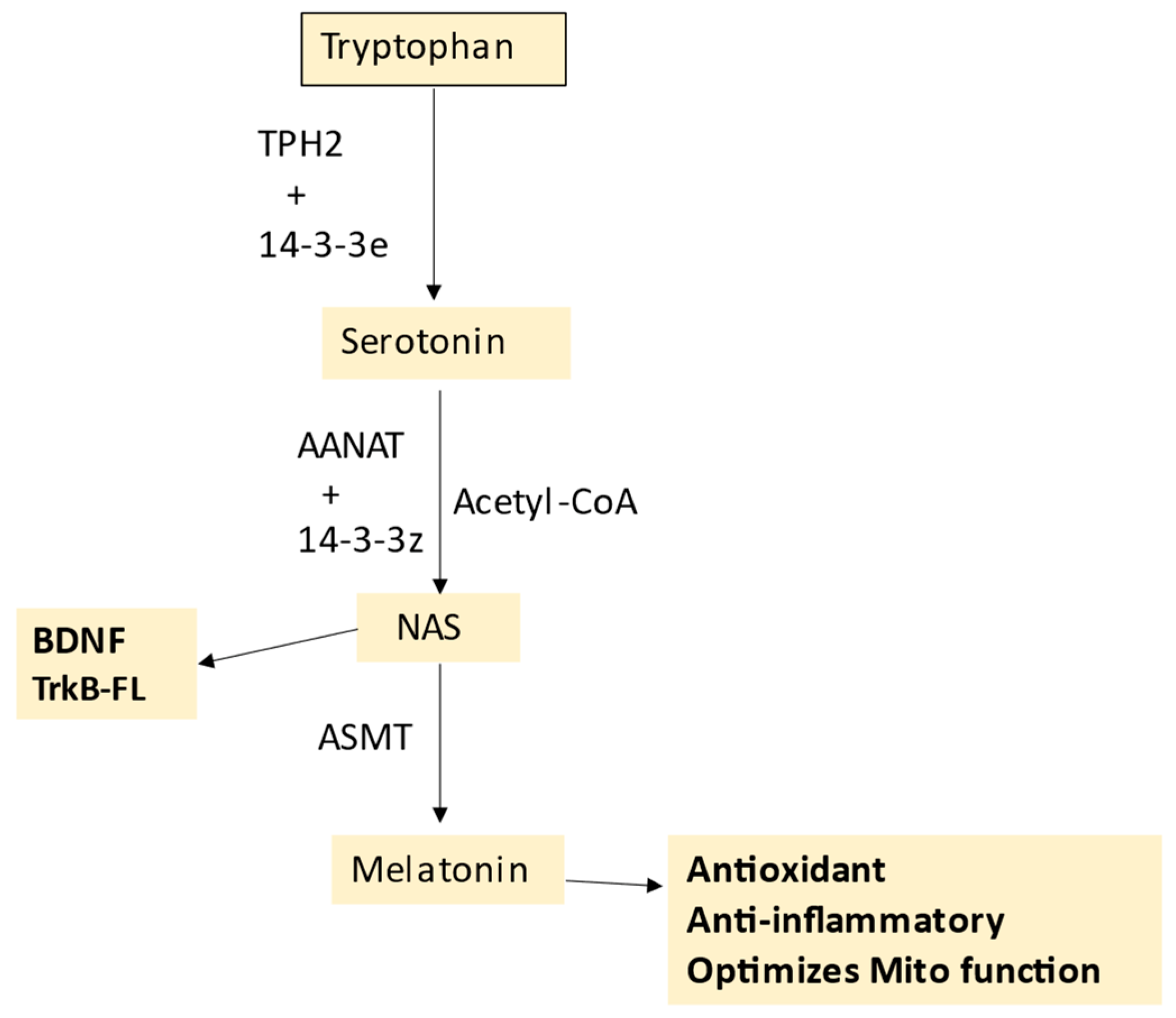
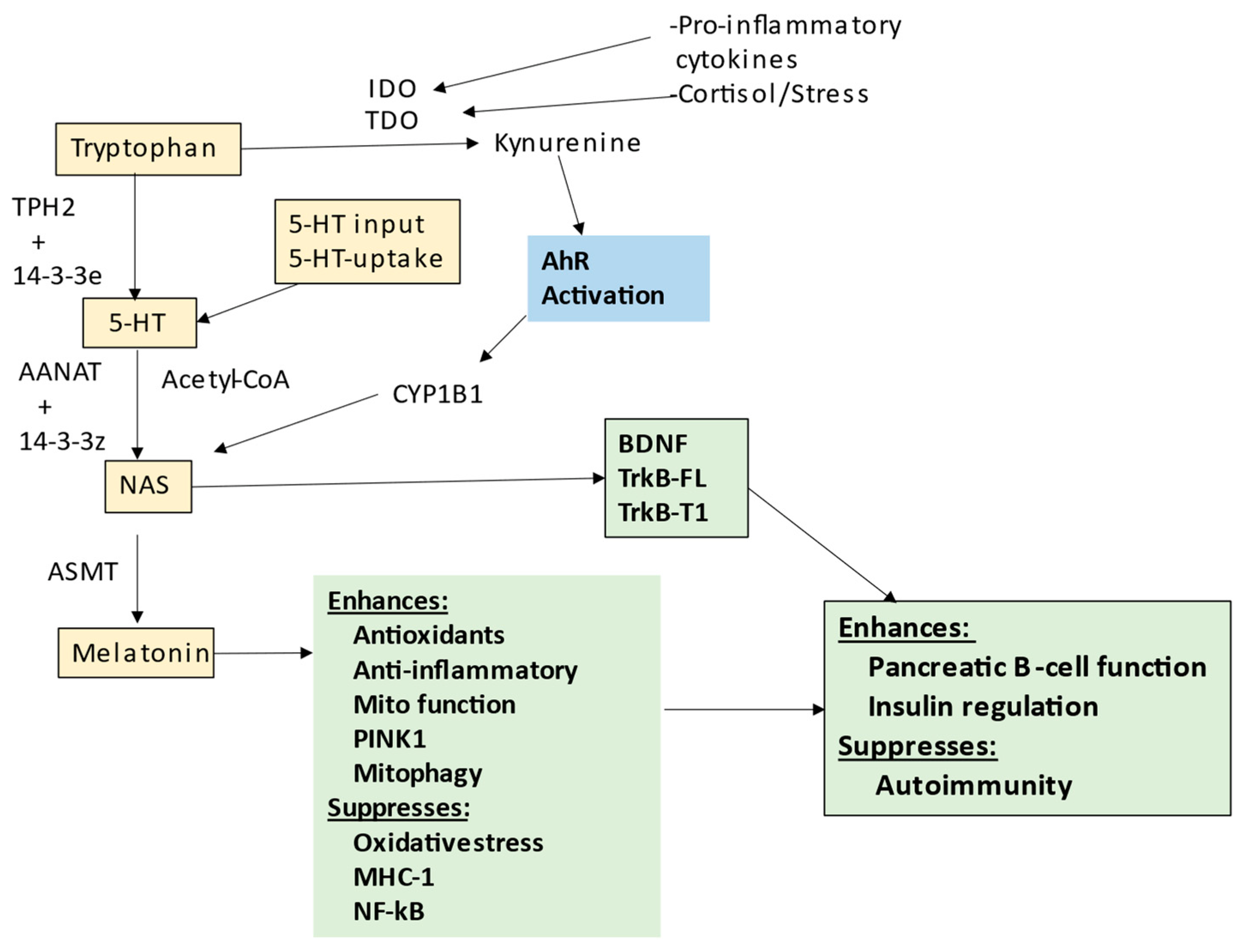
5. Melatonergic Pathway and T1DM
Melatonergic Pathway and Wider T1DM Pathophysiology
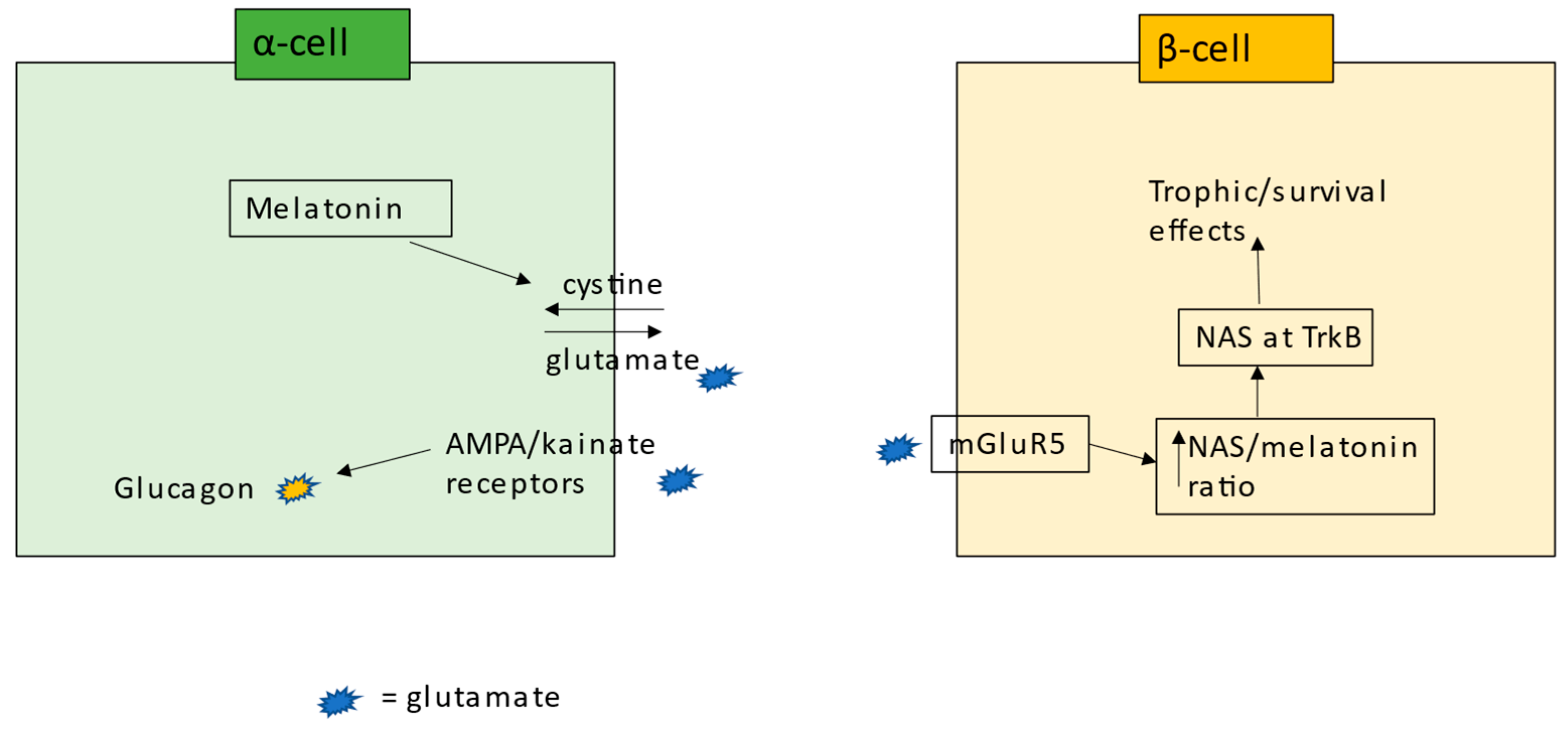
6. Pancreatic Cells and Interactions
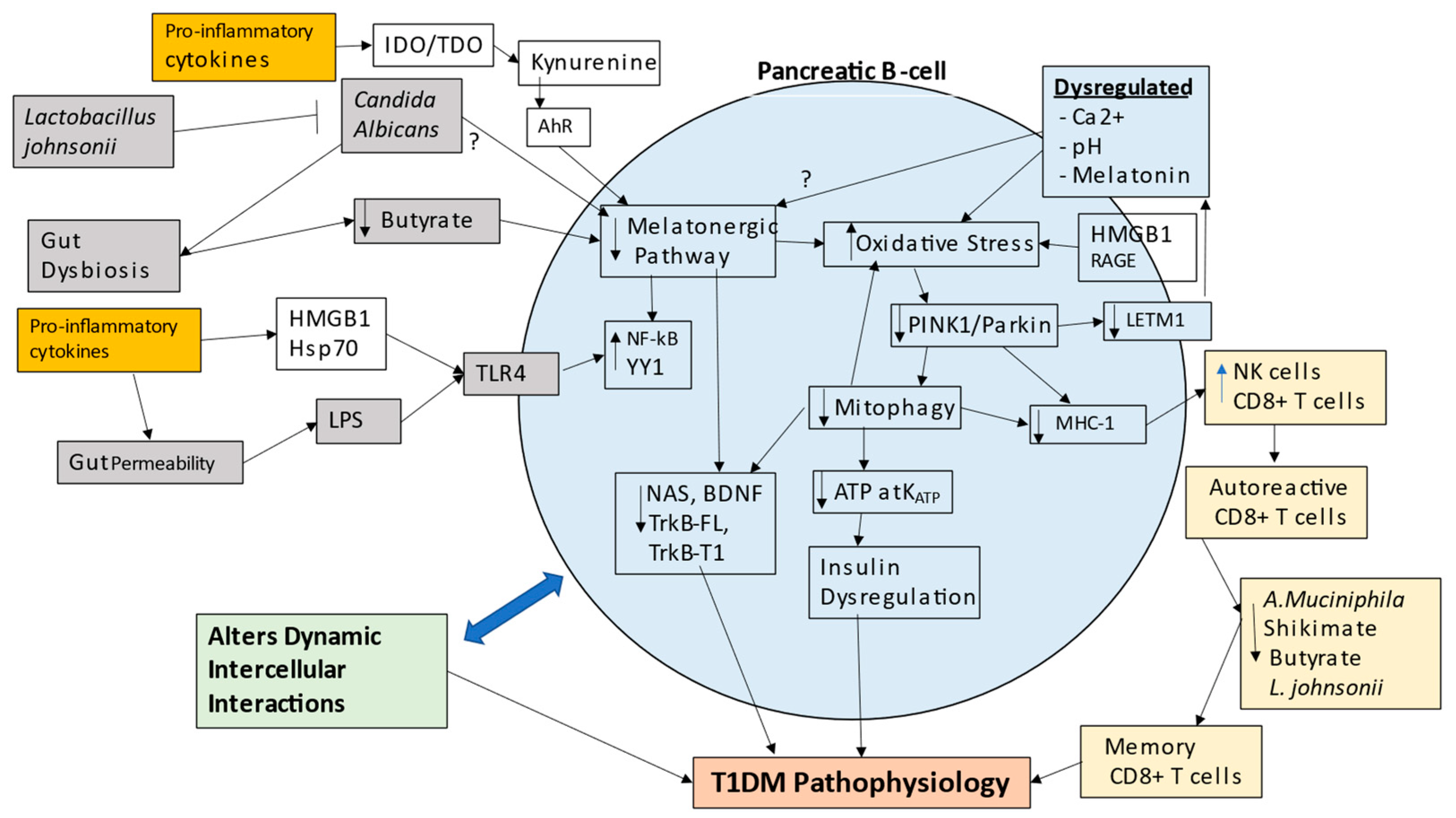
7. Integrating T1DM Pathogenesis and Pathophysiology
8. Future Research
- Is the melatonergic pathway evident in pancreatic β-cells? Is the pancreatic β-cell melatonergic pathway suppressed in T1DM?
- Is the suppression of the mitochondrial melatonergic pathway relevant in diverse tissues and organs associated with T1DM comorbidities, either directly suppressed in these organs/tissues or via alterations in patterned immune responses?
- How does the suppression of the mitochondrial melatonergic pathway link to, or drive, immune-mediated processes underpinning classical concepts of autoimmunity? Is a suppressed mitochondrial melatonergic pathway relevant in other immune-mediated, autoimmune disorders, redefining these conditions as a subtype of mitochondrial disorders?
- Are there parallels to the tumor microenvironment, where other cells (as with cancer cells) act to suppress the mitochondrial melatonergic pathway in pancreatic β-cells, thereby allowing local intercellular processes to drive ‘autoimmunity’ via miRNA-induced MHC-1?
- What is the relative importance of the TrkB-FL vs. TrkB-T1 in pancreatic β-cells? Does NAS, vs. BDNF, have any differential effects at TrkB-FL vs. TrkB-T1 in pancreatic β-cells? Is the TrkB-FL/TrkB-T1 ratio determined by mitochondrial ROS driving miRNA patterning and/or in association with the NAS/melatonin ratio?
- Skin symptoms are evident in T1DM, with streptozotocin altering skin function, including decreasing fibroblast growth factors (FGFs), especially during wound healing [160]. As melatonin can increase FGFs [161], are the effects of streptozotocin in the skin driven by streptozotocin (and T1DM?) suppression of the tryptophan-melatonin pathway in skin cells?
- As human amylin increases TLR-4 in rodents [162], does this indicate an amylin-driven increase in the TLR-4/NF-κB-YY1 induction of inflammatory changes in pancreatic β-cells, with the effects of amylin dependent upon the capacity of NF-κB and YY1 to upregulate the melatonergic pathway?
- Given that amylin is normally secreted with insulin, would YY1, like NF-κB [134], upregulate amylin in pancreatic β-cells?
- Does gut microbiome-derived butyrate, including as regulated by Lactobacillus johnsonii and its suppression of Candida albicans, have a role in the pathoetiology and pathophysiology of T1DM? Do the effects of butyrate in pancreatic β-cells require a functional tryptophan-melatonin pathway?
- Does TPH1 require stabilization by a specific 14-3-3 isoform, as with TPH2 by 14-3-3eta, to convert tryptophan to serotonin in pancreatic β-cells?
- Prenatal serotonin effects in pancreatic β-cells have been proposed to upregulate adult pancreatic β-cell numbers [163], although this is not supported by data using TPH1 knockout [164]. Is there a relevant role for the tryptophan–melatonin pathway in pancreatic β-cells during development? Would the survival and proliferative/functional importance be lost when no neighboring cells express the melatonergic pathway, indicating the importance of the tryptophan–melatonin pathway to challenging, if not competitive, intercellular interactions?
- In other cell types, exogenous melatonin (local, autocrine, paracrine, circadian) can be taken up into mitochondria via the organic anion transporter (OAT)3 and the peptide transporters (PEPT)1/2 [165]. OAT is expressed in pancreatic β-cells, and so whether OAT3 and/or PEPT1/2 are present in the mitochondrial membrane in pancreatic β-cells will be important to determine.
- T1DM is associated with an increased risk of amyotrophic lateral sclerosis (ALS) in people aged < 50 years of age [166], with streptozotocin-induced T1DM also leading to neuromuscular junction retraction and muscle atrophy [167]. There also seems genome-wide genetic overlaps between T1DM and ALS [168], whilst glyphosate-based herbicides, a proposed risk factor for ALS [7], induces a T2DM phenotype when combined with a high fructose diet [169]. Chronic glyphosate causes severe degeneration in pancreatic acinar cells and islets of Langerhans [170]. This could suggest epigenetic and genetic overlaps of ALS and T1DM. Is this mediated via gut, immune, and/or mitochondrial melatonergic-related factors? Are glyphosate-based herbicides an environmental risk factor for T1DM? Glyphosate-based herbicides can inhibit the shikimate pathway, which is a relevant provider of tryptophan to the body, with the inhibition of the shikimate pathway increasing gut permeability and gut dysbiosis, including decreased butyrate producing gut bacteria [7]. Do enteroviruses and/or bacteriophages in T1DM suppress the shikimate pathway and Akkermansia muciniphila, in association with decreased L. johnsonii and butyrate, to contribute not only to alterations in the mitochondrial melatonergic pathway in pancreatic β-cells, but also to the ‘bystander activation’ of memory CD8+ T cells in Peyer’s patches? Do such alterations in the gut enhance the cytotoxicity of autoreactive CD8+ T cells and prevent their elimination/deselection in the thymus?
- How relevant is the modulation of PINK1 by melatonin to the interactions of LETM1, PINK1, and Parkin in the mitochondrial membrane? Does the 14-3-3-like domain of LETM1 bind 14-3-3 and/or AANAT to regulate the mitochondrial melatonergic pathway? Would this more directly link autophagy with mitochondrial melatonergic pathway regulation?
- By limiting the oxidative stress-induced DNA damage, and therefore the induction of PARP1, does melatonin increase the availability of NAD+ for sirtuin induction, thereby increasing PDC and mitochondrial OXPHOS [171]?
9. Treatment Implications
- Preclinical data show that the administration of soluble RAGE dramatically suppresses T1DM via Treg upregulation, and associated inhibition of conventional T cell division [27]. Such data would implicate a significant clinical impact of RAGE ligands that can be suppressed by soluble RAGE. As melatonin inhibits RAGE ligands and RAGE activation in diabetes models [28], this could implicate the utilization of melatonin in the suppression of RAGE-driven T1DM pathophysiology.
- Would Lactobacillus johnsonii prove useful in suppressing progressive pancreatic β-cell loss at initial T1DM diagnosis? Is the efficacy of Lactobacillus johnsonii only evident when Candida albicans is present in the gut?
- An array of different pharmaceuticals and nutriceuticals afford protection in pancreatic β-cells via the suppression of the NLRP3 inflammasome [176,177,178]. Although not investigated in pancreatic β-cells, melatonin suppresses the NLRP3 inflammasome in different cell types across the body [179], indicating another aspect to melatonin’s potential utility in T1DM treatment, as well as from locally produced pancreatic melatonin.
- Stem cell development now allows for the priming of stem cells to have the contents of their exosome/vesicles shaped to provide targeted treatments (such as miRNAs and 14-3-3 proteins) to particular cells. This requires the identification of relevant targets. Would targeting the melatonergic pathway in pancreatic β-cells optimize mitochondrial function, whilst decreasing oxidative stress-induced MHC-1 and there preventing NK cell and CD8+ T cell induced apoptosis?
- A peptide-based therapy, JC-1 ScFv, has recently been found to bind specifically to the Candida albicans cell wall, where it inhibits the growth and viability of Candida albicans, both in vitro and in vivo [180]. As well as the utilization of Lactobacillus johnsonii for the management of Candida albicans in T1DM, such peptide-based therapies may provide another treatment option.
- A plethora of preclinical studies indicate the utility of melatonin in attenuating many of the consequences of T1DM, including the following: cardiovascular disorders [181], pancreatic β-cell regeneration [109], renal impairment [182], bone loss [183], cognitive deficits [110], erectile dysfunction [184], and body temperature circadian rhythm [185]. This is supported by clinical data showing lower melatonin levels in T1DM children [186]. The capacity of melatonin to decrease gut dysbiosis and gut permeability, as well optimize mitochondrial function, will be relevant to the management of many aspects of T1DM pathophysiology.
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AANAT | aralkylamine N-acetyltransferase |
| AhR | aryl hydrocarbon receptor |
| ASMT | acetylserotonin methytransferase |
| BACE1 | beta-site amyloid precursor protein cleaving enzyme 1 |
| BDNF | rain-derived neurotrophic factor |
| Bmal1 | brain and muscle ARNT-Like 1 |
| CYP | cytochrome P450 |
| GBH | glyphosate-based herbicides |
| HDAC | histone deacetylase |
| HMGB | high-mobility group box |
| hsp | heat shock protein |
| IDO | indoleamine 2,3-dioxygenase |
| LETM1 | leucine zipper-EF hand-containing transmembrane protein 1 |
| LPS | lipopolysaccharide |
| mGluR5 | metabotropic glutamate receptor |
| MHC-1 | major histocompatibility complex, class 1 |
| NAS | N-acetylserotonin |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | natural killer cell |
| OXPHOS | oxidative phosphorylation |
| PDC | pyruvate dehydrogenase complex |
| PINK1 | PTEN-induced kinase 1 |
| RAGE | receptor for advanced glycation end products |
| ROS | reactive oxygen species |
| SNP | single nucleotide polymorphisms |
| TCA | tricarboxylic acid |
| TDO | tryptophan 2,3-dioxygenase |
| TLR | Toll-like receptor |
| TPH2 | tryptophan hydroxylase 2 |
| Treg | regulatory T cell |
| TrkB | tyrosine receptor kinase B |
| VDAC1 | voltage-dependent anion channel |
References
- Ferrannini, E.; Mari, A.; Monaco, G.S.F.; Skyler, J.S.; Evans-Molina, C. The effect of age on longitudinal measures of beta cell function and insulin sensitivity during the progression of early stage type 1 diabetes. Diabetologia. 2023, 66, 508–519. [Google Scholar] [CrossRef]
- Zhang, Y.R.; Yang, L.; Wang, H.F.; Wu, B.S.; Huang, S.Y.; Cheng, W.; Feng, J.F.; Yu, J.T. Immune-mediated diseases are associated with a higher incidence of dementia: A prospective cohort study of 375,894 individuals. Alzheimers Res Ther. 2022, 14, 130. [Google Scholar] [CrossRef] [PubMed]
- Smolina, K.; Wotton, C.J.; Goldacre, M.J. Risk of dementia in patients hospitalised with type 1 and type 2 diabetes in England, 1998-2011: A retrospective national record linkage cohort study. Diabetologia 2015, 58, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Longinetti, E.; Larsson, H.; Andersson, J.; Pawitan, Y.; Piehl, F.; Fang, F. Associations between autoimmune diseases and amyotrophic lateral sclerosis: A register-based study. Amyotroph Lateral Scler Front. Degener. 2021, 22, 211–219. [Google Scholar] [CrossRef]
- Smidtslund, P.; Jansson Sigfrids, F.; Ylinen, A.; Elonen, N.; Harjutsalo, V.; Groop, P.H.; Thorn, L.M.; FinnDiane Study Group. Prognosis After First-Ever Myocardial Infarction in Type 1 Diabetes Is Strongly Affected by Chronic Kidney Disease. Diabetes Care 2023, 46, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Sacchetta, L.; Chiriacò, M.; Nesti, L.; Leonetti, S.; Forotti, G.; Natali, A.; Solini, A.; Tricò, D. Synergistic effect of chronic kidney disease, neuropathy, and retinopathy on all-cause mortality in type 1 and type 2 diabetes: A 21-year longitudinal study. Cardiovasc Diabetol. 2022, 21, 233. [Google Scholar] [CrossRef]
- Anderson, G. Amyotrophic Lateral Sclerosis Pathoetiology and Pathophysiology: Roles of Astrocytes, Gut microbiome and muscle interactions via the Mitochondrial melatonergic pathway, with disruption by Glyphosate-based herbicides. Int. J. Mol. Sci. 2023, 24, 587. [Google Scholar] [CrossRef]
- Qian, B.; Zhang, K.; Li, Y.; Sun, K. Update on gut microbiota in cardiovascular diseases. Front. Cell. Infect. Microbiol. 2022, 12, 1059349. [Google Scholar] [CrossRef]
- Ismail, H.M.; Evans-Molina, C. Does the Gut Microbiome Play a Role in Obesity in Type 1 Diabetes? Unanswered Questions and Review of the Literature. Front. Cell. Infect. Microbiol. 2022, 12, 892291. [Google Scholar] [CrossRef]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat Commun. 2019, 10, 2474. [Google Scholar] [CrossRef]
- Makam, A.A.; Biswas, A.; Kothegala, L.; Gandasi, N.R. Setting the Stage for Insulin Granule Dysfunction during Type-1-Diabetes: Is ER Stress the Culprit? Biomedicines 2022, 10, 2695. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.J.; Scrocchi, L.A.; Warnock, G.L.; Ao, Z.; Younker, M.A.; Rosenberg, L.; Lipsett, M.; Verchere, C.B.; Fraser, P.E. Amyloid inhibitors enhance survival of cultured human islets. Biochim. Biophys. Acta 2009, 1790, 566–574. [Google Scholar] [CrossRef]
- Guillemain, G.; Lacapere, J.J.; Khemtemourian, L. Targeting hIAPP fibrillation: A new paradigm to prevent β-cell death? Biochim. Biophys. Acta Biomembr. 2022, 1864, 184002. [Google Scholar] [CrossRef]
- Khan, I.A.; Moretto, M. Nfkbid-mediated humoral immunity during secondary toxoplasmosis. Trends Parasitol. 2022, 38, 272–273. [Google Scholar] [CrossRef]
- Dwyer, J.R.; Racine, J.J.; Chapman, H.D.; Quinlan, A.; Presa, M.; Stafford, G.A.; Schmitz, I.; Serreze, D.V. Nfkbid Overexpression in Nonobese Diabetic Mice Elicits Complete Type 1 Diabetes Resistance in Part Associated with Enhanced Thymic Deletion of Pathogenic CD8 T Cells and Increased Numbers and Activity of Regulatory T Cells. J. Immunol. 2022, 209, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Presa, M.; Racine, J.J.; Dwyer, J.R.; Lamont, D.J.; Ratiu, J.J.; Sarsani, V.K.; Chen, Y.G.; Geurts, A.; Schmitz, I.; Stearns, T.; et al. Hypermorphic Nfkbid Allele Contributes to Impaired Thymic Deletion of Autoreactive Diabetogenic CD8+ T Cells in NOD Mice. J. Immunol. 2018, 201, 1907–1917. [Google Scholar] [CrossRef]
- Kieleväinen, V.; Turtinen, M.; Luopajärvi, K.; Härkönen, T.; Ilonen, J.; Knip, M.; Finnish Pediatric Diabetes Register. Increased HLA class II risk is associated with a more aggressive presentation of clinical type 1 diabetes. Acta Paediatr. 2022. [CrossRef] [PubMed]
- Haghnazari, L.; Sabzi, R. Relationship between TP53 and interleukin-6 gene variants and the risk of types 1 and 2 diabetes mellitus development in the Kermanshah province. J. Med. Life 2021, 14, 37–44. [Google Scholar] [CrossRef]
- Guo, D.; Li, M.; Zhang, Y.; Yang, P.; Eckenrode, S.; Hopkins, D.; Zheng, W.; Purohit, S.; Podolsky, R.H.; Muir, A.; et al. A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat. Genet. 2004, 36, 837–841. [Google Scholar] [CrossRef]
- Gui, Y.; Lei, X.; Huang, S. Collective effects of common single nucleotide polymorphisms and genetic risk prediction in type 1 diabetes. Clin. Genet. 2018, 93, 1069–1074. [Google Scholar] [CrossRef]
- Gootjes, C.; Zwaginga, J.J.; Roep, B.O.; Nikolic, T. Functional Impact of Risk Gene Variants on the Autoimmune Responses in Type 1 Diabetes. Front. Immunol. 2022, 13, 886736. [Google Scholar] [CrossRef]
- Pearson, G.; Chai, B.; Vozheiko, T.; Liu, X.; Kandarpa, M.; Piper, R.C.; Soleimanpour, S.A. Clec16a, Nrdp1, and USP8 Form a Ubiquitin-Dependent Tripartite Complex That Regulates β-Cell Mitophagy. Diabetes 2018, 67, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and functional specialisations of the pancreatic beta cell: Gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 2020, 63, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Whiddett, R.O.; Buckle, I.; Chen, C.; Forbes, J.M.; Fotheringham, A.K. Advanced Glycation End Products and Inflammation in Type 1 Diabetes Development. Cells 2022, 11, 3503. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, Y.; Huang, Y.; Deng, H. Pathophysiology of RAGE in inflammatory diseases. Front. Immunol. 2022, 13, 931473. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Derk, J.; Schmidt, A.M. The receptor for advanced glycation endproducts is a mediator of toxicity by IAPP and other proteotoxic aggregates: Establishing and exploiting common ground for novel amyloidosis therapies. Protein Sci. 2018, 27, 1166–1180. [Google Scholar] [CrossRef]
- Leung, S.S.; Borg, D.J.; McCarthy, D.A.; Boursalian, T.E.; Cracraft, J.; Zhuang, A.; Fotheringham, A.K.; Flemming, N.; Watkins, T.; Miles, J.J.; et al. Soluble RAGE Prevents Type 1 Diabetes Expanding Functional Regulatory T Cells. Diabetes 2022, 71, 1994–2008. [Google Scholar] [CrossRef]
- Ergenc, M.; Ozacmak, H.S.; Turan, I.; Ozacmak, V.H. Melatonin reverses depressive and anxiety like-behaviours induced by diabetes: Involvement of oxidative stress, age, rage and S100B levels in the hippocampus and prefrontal cortex of rats. Arch. Physiol. Biochem. 2022, 128, 402–410. [Google Scholar] [CrossRef]
- Yue, T.; Sun, F.; Yang, C.; Wang, F.; Luo, J.; Yang, P.; Xiong, F.; Zhang, S.; Yu, Q.; Wang, C.Y. The AHR Signaling Attenuates Autoimmune Responses During the Development of Type 1 Diabetes. Front. Immunol. 2020, 11, 1510. [Google Scholar] [CrossRef]
- Xu, Y.N.; Wang, Z.; Zhang, S.K.; Xu, J.R.; Pan, Z.X.; Wei, X.; Wen, H.H.; Luo, Y.S.; Guo, M.J.; Zhu, Q. Low-grade elevation of palmitate and lipopolysaccharide synergistically induced β-cell damage via inhibition of neutral ceramidase. Mol. Cell. Endocrinol. 2022, 539, 111473. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.M.; Burke, S.J.; Batdorf, H.M.; Burk, D.H.; Ghosh, S.; Dupuy, S.D.; Karlstad, M.D.; Collier, J.J. ICAM-1 Abundance Is Increased in Pancreatic Islets of Hyperglycemic Female NOD Mice and Is Rapidly Upregulated by NF-κB in Pancreatic β-Cells. J. Immunol. 2022, ji2200065. [Google Scholar] [CrossRef]
- Liu, D.; Yang, K.Y.; Chan, V.W.; Ye, W.; Chong, C.C.N.; Wang, C.C.; Wang, H.; Zhou, B.; Cheng, K.K.Y.; Lui, K.O. YY1 Regulates Glucose Homeostasis Through Controlling Insulin Transcription in Pancreatic Beta Cells. Diabetes 2022, 71, db210695. [Google Scholar] [CrossRef]
- Jung, E.M.; Joo, S.S.; Yoo, Y.M. Nanomolar melatonin influences insulin synthesis and secretion in rat insulinoma INS-1E cells. J. Physiol. Pharmacol. 2020, 71, 705–716. [Google Scholar] [CrossRef]
- Lee, J.; Liu, R.; de Jesus, D.; Kim, B.S.; Ma, K.; Moulik, M.; Yechoor, V. Circadian control of β-cell function and stress responses. Diabetes Obes. Metab. 2015, 17 (Suppl. 1), 123–133. [Google Scholar] [CrossRef]
- Mousa, W.K.; Chehadeh, F.; Husband, S. Microbial dysbiosis in the gut drives systemic autoimmune diseases. Front. Immunol. 2022, 13, 906258. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Depression Pathophysiology: Astrocyte Mitochondrial Melatonergic Pathway as Crucial Hub. Int. J. Mol. Sci. 2023, 24, 350. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Interactions of Tryptophan and Its Catabolites With Melatonin and the Alpha 7 Nicotinic Receptor in Central Nervous System and Psychiatric Disorders: Role of the Aryl Hydrocarbon Receptor and Direct Mitochondria Regulation. Int. J. Tryptophan. Res. 2017, 10, 1178646917691738. [Google Scholar] [CrossRef]
- Anderson, G. Tumour Microenvironment: Roles of the Aryl Hydrocarbon Receptor, O-GlcNAcylation, Acetyl-CoA and Melatonergic Pathway in Regulating Dynamic Metabolic Interactions across Cell Types-Tumour Microenvironment and Metabolism. Int. J. Mol. Sci. 2020, 22, 141. [Google Scholar] [CrossRef]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 19376–19387. [Google Scholar] [CrossRef]
- Kim, Y.H.; Shim, Y.J.; Shin, Y.J.; Sul, D.; Lee, E.; Min, B.H. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces calcium influx through T-type calcium channel and enhances lysosomal exocytosis and insulin secretion in INS-1 cells. Int. J. Toxicol. 2009, 28, 151–161. [Google Scholar] [CrossRef]
- Mukhuty, A.; Fouzder, C.; Kundu, R. Blocking TLR4-NF-κB pathway protects mouse islets from the combinatorial impact of high fat and fetuin-A mediated dysfunction and restores ability for insulin secretion. Mol. Cell. Endocrinol. 2021, 532, 111314. [Google Scholar] [CrossRef]
- Cheng, X.; Huang, Y.; Yang, P.; Bu, L. miR-383 ameliorates high glucose-induced β-cells apoptosis and hyperglycemia in high-fat induced diabetic mice. Life Sci. 2020, 263, 118571. [Google Scholar] [CrossRef]
- Yan, S.; Jiang, Z.; Cheng, L.; Lin, Y.; Fan, B.; Luo, L.; Yan, Y.; Yang, L.; Shen, X. TLR4 knockout can improve dysfunction of β-cell by rebalancing proteomics disorders in pancreas of obese rats. Endocrine 2020, 67, 67–79. [Google Scholar] [CrossRef]
- Taskin, E.; Guven, C.; Kaya, S.T.; Sahin, L.; Kocahan, S.; Degirmencioglu, A.Z.; Gur, F.M.; Sevgiler, Y. The role of toll-like receptors in the protective effect of melatonin against doxorubicin-induced pancreatic beta cell toxicity. Life Sci. 2019, 233, 116704. [Google Scholar] [CrossRef]
- Zhou, L.; Song, X.D.; Xu, H.; Liang, G.Q.; Wang, F.; Zhang, L.R.; Huang, F.; Cai, J.; Jiang, G.R. Exogenous 3-Deoxyglucosone-Induced Carbonyl and Oxidative Stress Causes β-Cells Dysfunction by Impairing Gut Permeability in Rats. Biochemistry 2018, 83, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Ghezzal, S.; Postal, B.G.; Quevrain, E.; Brot, L.; Seksik, P.; Leturque, A.; Thenet, S.; Carrière, V. Palmitic acid damages gut epithelium integrity and initiates inflammatory cytokine production. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2020, 1865, 158530. [Google Scholar] [CrossRef]
- Khan, H.N.; Perlee, D.; Schoenmaker, L.; van der Meer, A.J.; Franitza, M.; Toliat, M.R.; Nürnberg, P.; Zwinderman, A.H.; van der Poll, T.; Scicluna, B.P. Leukocyte transcriptional signatures dependent on LPS dosage in human endotoxemia. J. Leukoc. Biol. 2019, 106, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Sang, T.; Chen, C.; Peng, H.; Lin, X.; Zhao, Q.; Chen, S.; Eling, T.; Wang, X. NAG-1/GDF15 protects against streptozotocin-induced type 1 diabetes by inhibiting apoptosis, preserving beta-cell function, and suppressing inflammation in pancreatic islets. Mol. Cell. Endocrinol. 2022, 549, 111643. [Google Scholar] [CrossRef] [PubMed]
- Darville, M.I.; Eizirik, D.L. Cytokine induction of Fas gene expression in insulin-producing cells requires the transcription factors NF-kappaB and C/EBP. Diabetes 2001, 50, 1741–1748. [Google Scholar] [CrossRef]
- Bernard, M.; Voisin, P. Photoreceptor-specific expression, light-dependent localization, and transcriptional targets of the zinc-finger protein Yin Yang 1 in the chicken retina. J. Neurochem. 2008, 105, 595–604. [Google Scholar] [CrossRef]
- Markus, R.P.; Fernandes, P.A.; Kinker, G.S.; da Silveira Cruz-Machado, S.; Marçola, M. Immune-pineal axis—Acute inflammatory responses coordinate melatonin synthesis by pinealocytes and phagocytes. Br. J. Pharmacol. 2018, 175, 3239–3250. [Google Scholar] [CrossRef]
- Lee, Y.H.; Jung, H.S.; Kwon, M.J.; Jang, J.E.; Kim, T.N.; Lee, S.H.; Kim, M.K.; Park, J.H. Melatonin protects INS-1 pancreatic β-cells from apoptosis and senescence induced by glucotoxicity and glucolipotoxicity. Islets 2020, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Pan, W.; Xu, X.; Tian, X.; Zhang, M.; Hu, Q. NF-κB inhibits the occurrence of type 1 diabetes through microRNA-150-dependent PUMA degradation. Life Sci. 2020, 255, 117724. [Google Scholar] [CrossRef]
- Muxel, S.M.; Pires-Lapa, M.A.; Monteiro, A.W.; Cecon, E.; Tamura, E.K.; Floeter-Winter, L.M.; Markus, R.P. NF-κB drives the synthesis of melatonin in RAW 264.7 macrophages by inducing the transcription of the arylalkylamine-N-acetyltransferase (AA-NAT) gene. PLoS ONE 2012, 7, e52010. [Google Scholar] [CrossRef]
- do Carmo Buonfiglio, D.; Peliciari-Garcia, R.A.; do Amaral, F.G.; Peres, R.; Nogueira, T.C.; Afeche, S.C.; Cipolla-Neto, J. Early-stage retinal melatonin synthesis impairment in streptozotocin-induced diabetic wistar rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7416–7422. [Google Scholar] [CrossRef] [PubMed]
- Uhlemeyer, C.; Müller, N.; Rieck, M.; Kuboth, J.; Schlegel, C.; Grieß, K.; Dorweiler, T.F.; Heiduschka, S.; Eckel, J.; Roden, M.; et al. Selective ablation of P53 in pancreatic beta cells fails to ameliorate glucose metabolism in genetic, dietary and pharmacological models of diabetes mellitus. Mol. Metab. 2022, 67, 101650. [Google Scholar] [CrossRef]
- Tao, S.; Yang, Y.; Fan, Y.; Chu, K.; Sun, J.; Wu, Q.; Wang, A.; Wan, J.; Tian, H. Melatonin protects against nonylphenol caused pancreatic β-cells damage through MDM2-P53-P21 axis. Toxicol. Res. 2022, 11, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, D.O.; Evsyukova, I.I.; Mazzoccoli, G.; Anderson, G.; Polyakova, V.O.; Kvetnoy, I.M.; Carbone, A.; Nasyrov, R.A. The Role of Prenatal Melatonin in the Regulation of Childhood Obesity. Biology 2020, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Rutters, F.; Nefs, G. Sleep and Circadian Rhythm Disturbances in Diabetes: A Narrative Review. Diabetes Metab. Syndr. Obes. 2022, 15, 3627–3637. [Google Scholar] [CrossRef]
- Hjortkjær, H.Ø.; Persson, F.; Theilade, S.; Winther, S.A.; Tofte, N.; Ahluwalia, T.S.; Rossing, P. Non-dipping and higher nocturnal blood pressure are associated with risk of mortality and development of kidney disease in type 1 diabetes. J. Diabetes Complicat. 2022, 36, 108270. [Google Scholar] [CrossRef]
- Chiriacò, M.; Sacchetta, L.; Forotti, G.; Leonetti, S.; Nesti, L.; Taddei, S.; Natali, A.; Solini, A.; Tricò, D. Prognostic value of 24-hour ambulatory blood pressure patterns in diabetes: A 21-year longitudinal study. Diabetes Obes. Metab. 2022, 24, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Kulecki, M.; Naskret, D.; Kaminski, M.; Kasprzak, D.; Lachowski, P.; Klause, D.; Kozlowska, M.; Flotynska, J.; Uruska, A.; Zozulinska-Ziolkiewicz, D. Association between central non-dipping pattern and platelet morphology in adults with type 1 diabetes without cardiovascular disease: A cross-sectional study. Sci. Rep. 2021, 11, 15416. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lee, Y.J.; Lee, Y.A.; Shin, C.H. Effects of circadian blood pressure patterns on development of microvascular complications in pediatric patients with type 1 diabetes mellitus. Ann. Pediatr. Endocrinol. Metab. 2022, 27, 44–51. [Google Scholar] [CrossRef]
- Beam, C.A.; Beli, E.; Wasserfall, C.H.; Woerner, S.E.; Legge, M.T.; Evans-Molina, C.; McGrail, K.M.; Silk, R.; Grant, M.B.; Atkinson, M.A.; et al. Peripheral immune circadian variation, synchronisation and possible dysrhythmia in established type 1 diabetes. Diabetologia 2021, 64, 1822–1833. [Google Scholar] [CrossRef]
- Pallayova, M.; Breznoscakova, D. The altered circadian pattern of basal insulin Requirements—An early marker of autoimmune polyendocrine syndromes in type 1 diabetes mellitus. Endocr. Regul. 2020, 54, 126–132. [Google Scholar] [CrossRef]
- Anderson, G.; Seo, M.; Berk, M.; Carvalho, A.F.; Maes, M. Gut Permeability and Microbiota in Parkinson’s Disease: Role of Depression, Tryptophan Catabolites, Oxidative and Nitrosative Stress and Melatonergic Pathways. Curr. Pharm. Des. 2016, 22, 6142–6151. [Google Scholar] [CrossRef]
- Bankole, T.; Winn, H.; Li, Y. Dietary Impacts on Gestational Diabetes: Connection between Gut Microbiome and Epigenetic Mechanisms. Nutrients 2022, 14, 5269. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Chi, J.; Tan, X.; Hu, J.; Ma, X.; Sun, X.; Che, K.; Lv, W.; Wang, Y. hUCMSCs carrying exenatide prevent T1DM by improving intestinal microflora composition and islet tissue damage repair. Mol. Med. 2022, 28, 155. [Google Scholar] [CrossRef]
- Teixeira, L.D.; Harrison, N.A.; da Silva, D.R.; Mathews, C.E.; Gonzalez, C.F.; Lorca, G.L. Nanovesicles From Lactobacillus johnsonii N6.2 Reduce Apoptosis in Human Beta Cells by Promoting AHR Translocation and IL10 Secretion. Front. Immunol. 2022, 13, 899413. [Google Scholar] [CrossRef]
- Teixeira, L.D.; Kling, D.N.; Lorca, G.L.; Gonzalez, C.F. Lactobacillus johnsonii N6.2 diminishes caspase-1 maturation in the gastrointestinal system of diabetes prone rats. Benef. Microbes 2018, 9, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Kingma, S.D.; Li, N.; Sun, F.; Valladares, R.B.; Neu, J.; Lorca, G.L. Lactobacillus johnsonii N6.2 stimulates the innate immune response through Toll-like receptor 9 in Caco-2 cells and increases intestinal crypt Paneth cell number in biobreeding diabetes-prone rats. J. Nutr. 2011, 141, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.; Benitez, P.; Ardissone, A.; Wilson, T.D.; Collins, E.L.; Lorca, G.; Li, N.; Sankar, D.; Wasserfall, C.; Neu, J.; et al. Inhibition of type 1 diabetes correlated to a Lactobacillus johnsonii N6.2-mediated Th17 bias. J. Immunol. 2011, 186, 3538–3546. [Google Scholar] [CrossRef] [PubMed]
- Marcial, G.E.; Ford, A.L.; Haller, M.J.; Gezan, S.A.; Harrison, N.A.; Cai, D.; Meyer, J.L.; Perry, D.J.; Atkinson, M.A.; Wasserfall, C.H.; et al. Lactobacillus johnsonii N6.2 Modulates the Host Immune Responses: A Double-Blind, Randomized Trial in Healthy Adults. Front. Immunol. 2017, 8, 655. [Google Scholar] [CrossRef]
- He, T.; Zhu, Y.H.; Yu, J.; Xia, B.; Liu, X.; Yang, G.Y.; Su, J.H.; Guo, L.; Wang, M.L.; Wang, J.F. Lactobacillus johnsonii L531 reduces pathogen load and helps maintain short-chain fatty acid levels in the intestines of pigs challenged with Salmonella enterica Infantis. Vet. Microbiol. 2019, 230, 187–194. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Zhang, L.; Zhou, F.; Zhu, K.; Zhu, Q.; Liu, Q.; Liu, Y.; Jiang, L.; Ning, G.; et al. Protein acetylation derepresses Serotonin Synthesis to potentiate Pancreatic Beta-Cell Function through HDAC1-PKA-Tph1 signaling. Theranostics 2020, 10, 7351–7368. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.J.; Engstler, A.J.; Sellmann, C.; Ziegenhardt, D.; Landmann, M.; Kanuri, G.; Lounis, H.; Schröder, M.; Vetter, W.; Bergheim, I. Sodium butyrate protects mice from the development of the early signs of non-alcoholic fatty liver disease: Role of melatonin and lipid peroxidation. Br. J. Nutr. 2016, 116, 1682–1693. [Google Scholar] [CrossRef]
- Gavin, P.G.; Kim, K.W.; Craig, M.E.; Hill, M.M.; Hamilton-Williams, E.E. Multi-omic interactions in the gut of children at the onset of islet autoimmunity. Microbiome 2022, 10, 230. [Google Scholar] [CrossRef]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Härkönen, T.; Hämäläinen, A.M.; et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef]
- Shilo, S.; Godneva, A.; Rachmiel, M.; Korem, T.; Bussi, Y.; Kolobkov, D.; Karady, T.; Bar, N.; Wolf, B.C.; Glantz-Gashai, Y.; et al. The Gut Microbiome of Adults With Type 1 Diabetes and Its Association With the Host Glycemic Control. Diabetes Care 2022, 45, 555–563. [Google Scholar] [CrossRef] [PubMed]
- van Heck, J.I.P.; Gacesa, R.; Stienstra, R.; Fu, J.; Zhernakova, A.; Harmsen, H.J.M.; Weersma, R.K.; Joosten, L.A.B.; Tack, C.J. The Gut Microbiome Composition Is Altered in Long-standing Type 1 Diabetes and Associates With Glycemic Control and Disease-Related Complications. Diabetes Care 2022, 45, 2084–2094. [Google Scholar] [CrossRef]
- Zheng, P.; Li, Z.; Zhou, Z. Gut microbiome in type 1 diabetes: A comprehensive review. Diabetes Metab. Res. Rev. 2018, 34, e3043. [Google Scholar] [CrossRef]
- Tetz, G.; Brown, S.M.; Hao, Y.; Tetz, V. Type 1 Diabetes: An Association Between Autoimmunity, the Dynamics of Gut Amyloid-producing E. coli and Their Phages. Sci. Rep. 2019, 9, 9685. [Google Scholar] [CrossRef]
- Park, A.; Zhao, G. Mining the Virome for Insights into Type 1 Diabetes. DNA Cell Biol. 2018, 37, 422–425. [Google Scholar] [CrossRef]
- Alnek, K.; Talja, I.; Laht, B.; Metsküla, K.; Mandel, M.; Reppo, I.; Lubi, M.; Uibo, R. IgA-Type Enterovirus Antibodies Are Increased among Adults and Children with Recently Diagnosed Type 1 Diabetes. BioMed Res. Int. 2022, 2022, 7603062. [Google Scholar] [CrossRef]
- Gürsoy, S.; Koçkar, T.; Atik, S.U.; Önal, Z.; Önal, H.; Adal, E. Autoimmunity and intestinal colonization by Candida albicans in patients with type 1 diabetes at the time of the diagnosis. Korean J. Pediatr. 2018, 61, 217–220. [Google Scholar] [CrossRef]
- Vazquez-Munoz, R.; Thompson, A.; Russell, J.T.; Sobue, T.; Zhou, Y.; Dongari-Bagtzoglou, A. Insights From the Lactobacillus johnsonii Genome Suggest the Production of Metabolites With Antibiofilm Activity Against the Pathobiont Candida albicans. Front. Microbiol. 2022, 13, 853762. [Google Scholar] [CrossRef]
- Janssen, A.W.M.; Stienstra, R.; Jaeger, M.; van Gool, A.J.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P.; Tack, C.J. Understanding the increased risk of infections in diabetes: Innate and adaptive immune responses in type 1 diabetes. Metabolism 2021, 121, 154795. [Google Scholar] [CrossRef]
- Charlet, R.; Le Danvic, C.; Sendid, B.; Nagnan-Le Meillour, P.; Jawhara, S. Oleic Acid and Palmitic Acid from Bacteroides thetaiotaomicron and Lactobacillus johnsonii Exhibit Anti-Inflammatory and Antifungal Properties. Microorganisms 2022, 10, 1803. [Google Scholar] [CrossRef]
- Pullen, T.J.; da Silva Xavier, G.; Kelsey, G.; Rutter, G.A. miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol. Cell Biol. 2011, 31, 3182–3194. [Google Scholar] [CrossRef]
- Patterson, J.N.; Cousteils, K.; Lou, J.W.; Manning Fox, J.E.; MacDonald, P.E.; Joseph, J.W. Mitochondrial metabolism of pyruvate is essential for regulating glucose-stimulated insulin secretion. J. Biol. Chem. 2014, 289, 13335–13346. [Google Scholar] [CrossRef]
- Sharoyko, V.V.; Abels, M.; Sun, J.; Nicholas, L.M.; Mollet, I.G.; Stamenkovic, J.A.; Göhring, I.; Malmgren, S.; Storm, P.; Fadista, J.; et al. Loss of TFB1M results in mitochondrial dysfunction that leads to impaired insulin secretion and diabetes. Hum. Mol. Genet. 2014, 23, 5733–5749. [Google Scholar] [CrossRef]
- Verma, G.; Bowen, A.; Gheibi, S.; Hamilton, A.; Muthukumar, S.; Cataldo, L.R.; Asplund, O.; Esguerra, J.; Karagiannopoulos, A.; Lyons, C.; et al. Ribosomal biogenesis regulator DIMT1 controls β-cell protein synthesis, mitochondrial function, and insulin secretion. J. Biol. Chem. 2022, 298, 101692. [Google Scholar] [CrossRef]
- Jesinkey, S.R.; Madiraju, A.K.; Alves, T.C.; Yarborough, O.H.; Cardone, R.L.; Zhao, X.; Parsaei, Y.; Nasiri, A.R.; Butrico, G.; Liu, X.; et al. Mitochondrial GTP Links Nutrient Sensing to β Cell Health, Mitochondrial Morphology, and Insulin Secretion Independent of OxPhos. Cell Rep. 2019, 28, 759–772-e10. [Google Scholar] [CrossRef] [PubMed]
- Foster, H.R.; Ho, T.; Potapenko, E.; Sdao, S.M.; Huang, S.M.; Lewandowski, S.L.; VanDeusen, H.R.; Davidson, S.M.; Cardone, R.L.; Prentki, M.; et al. β-cell deletion of the PKm1 and PKm2 isoforms of pyruvate kinase in mice reveals their essential role as nutrient sensors for the KATP channel. eLife 2022, 11, e79422. [Google Scholar] [CrossRef]
- Medini, H.; Cohen, T.; Mishmar, D. Mitochondrial gene expression in single cells shape pancreatic beta cells’ sub-populations and explain variation in insulin pathway. Sci. Rep. 2021, 11, 466. [Google Scholar] [CrossRef]
- Ferraz, R.S.; Santos, L.C.B.; da-Silva-Cruz, R.L.; Braga-da-Silva, C.H.; Magalhães, L.; Ribeiro-Dos-Santos, A.; Vidal, A.; Vinasco-Sandoval, T.; Reis-das-Mercês, L.; Sena-Dos-Santos, C.; et al. Global miRNA expression reveals novel nuclear and mitochondrial interactions in Type 1 diabetes mellitus. Front. Endocrinol. 2022, 13, 1033809. [Google Scholar] [CrossRef]
- Anderson, G. Why Do Anti-Amyloid Beta Antibodies not work? Time to reconceptualize Dementia Pathophysiology by incorporating astrocyte melatonergic pathway desynchronization from amyloid-beta production. Braz. J. Psychiatry. 2022. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Liao, Q.; Wong, Y.K.; Chen, X.; Yang, C.; Xu, C.; Sun, J.; Wang, J. The role of melatonin in the treatment of type 2 diabetes mellitus and Alzheimer’s disease. Int. J. Biol. Sci. 2022, 18, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Rodriguez, M.; Reiter, R.J. Multiple Sclerosis: Melatonin, Orexin, and Ceramide Interact with Platelet Activation Coagulation Factors and Gut-Microbiome-Derived Butyrate in the Circadian Dysregulation of Mitochondria in Glia and Immune Cells. Int. J. Mol. Sci. 2019, 20, 5500. [Google Scholar] [CrossRef] [PubMed]
- Tobore, T.O. Towards a comprehensive etiopathogenetic and pathophysiological theory of multiple sclerosis. Int. J. Neurosci. 2020, 130, 279–300. [Google Scholar] [CrossRef]
- Anderson, G.; Reiter, R.J. Glioblastoma: Role of Mitochondria N-acetylserotonin/Melatonin Ratio in Mediating Effects of miR-451 and Aryl Hydrocarbon Receptor and in Coordinating Wider Biochemical Changes. Int. J. Tryptophan. Res. 2019, 12, 1178646919855942. [Google Scholar] [CrossRef]
- Franco, D.G.; Moretti, I.F.; Marie, S.K.N. Mitochondria Transcription Factor A: A Putative Target for the Effect of Melatonin on U87MG Malignant Glioma Cell Line. Molecules 2018, 23, 1129. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Sharma, R.; Rodriguez, C.; Martin, V.; Rosales-Corral, S.; Zuccari, D.A.P.C.; Chuffa, L.G.A. Part-time cancers and role of melatonin in determining their metabolic phenotype. Life Sci. 2021, 278, 119597. [Google Scholar] [CrossRef]
- Anderson, G. Breast cancer: Occluded role of mitochondria N-acetylserotonin/melatonin ratio in co-ordinating pathophysiology. Biochem. Pharmacol. 2019, 168, 259–268. [Google Scholar] [CrossRef]
- Anderson, G. Linking the biological underpinnings of depression: Role of mitochondria interactions with melatonin, inflammation, sirtuins, tryptophan catabolites, DNA repair and oxidative and nitrosative stress, with consequences for classification and cognition. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 255–266. [Google Scholar] [CrossRef]
- Uguz, A.C.; Demirci, K.; Espino, J. The Importance of Melatonin and Mitochondria Interaction in Mood Disorders and Schizophrenia: A Current Assessment. Curr. Med. Chem. 2016, 23, 2146–2158. [Google Scholar] [CrossRef]
- Maestro, I.; de la Ballina, L.R.; Porras, G.; Corrochano, S.; De Lago, E.; Simonsen, A.; Boya, P.; Martinez, A. Discovery of Mitophagy Inhibitors with Therapeutic Potential in Different Familial Amyotrophic Lateral Sclerosis Mutations. Int. J. Mol. Sci. 2022, 23, 12676. [Google Scholar] [CrossRef]
- Zhang, Y.; Cook, A.; Kim, J.; Baranov, S.V.; Jiang, J.; Smith, K.; Cormier, K.; Bennett, E.; Browser, R.P.; Day, A.L.; et al. Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits MT1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 55, 26–35. [Google Scholar] [CrossRef]
- Patel, R.; Parmar, N.; Rathwa, N.; Palit, S.P.; Li, Y.; Garcia-Ocaña, A.; Begum, R. A novel therapeutic combination of sitagliptin and melatonin regenerates pancreatic β-cells in mouse and human islets. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119263. [Google Scholar] [CrossRef]
- Albazal, A.; Delshad, A.A.; Roghani, M. Melatonin reverses cognitive deficits in streptozotocin-induced type 1 diabetes in the rat through attenuation of oxidative stress and inflammation. J. Chem. Neuroanat. 2021, 112, 101902. [Google Scholar] [CrossRef]
- Amaral, F.G.; Turati, A.O.; Barone, M.; Scialfa, J.H.; do Carmo Buonfiglio, D.; Peres, R.; Peliciari-Garcia, R.A.; Afeche, S.C.; Lima, L.; Scavone, C.; et al. Melatonin synthesis impairment as a new deleterious outcome of diabetes-derived hyperglycemia. J. Pineal Res. 2014, 57, 67–79. [Google Scholar] [CrossRef]
- Luchetti, F.; Carloni, S.; Nasoni, M.G.; Reiter, R.J.; Balduini, W. Melatonin, tunneling nanotubes, mesenchymal cells, and tissue regeneration. Neural Regen. Res. 2023, 18, 760–762. [Google Scholar] [CrossRef]
- Cucielo, M.S.; Cesário, R.C.; Silveira, H.S.; Gaiotte, L.B.; Dos Santos, S.A.A.; de Campos Zuccari, D.A.P.; Seiva, F.R.F.; Reiter, R.J.; de Almeida Chuffa, L.G. Melatonin Reverses the Warburg-Type Metabolism and Reduces Mitochondrial Membrane Potential of Ovarian Cancer Cells Independent of MT1 Receptor Activation. Molecules 2022, 27, 4350. [Google Scholar] [CrossRef]
- Williams, B.B.; Van Benschoten, A.H.; Cimermancic, P.; Donia, M.S.; Zimmermann, M.; Taketani, M.; Ishihara, A.; Kashyap, P.C.; Fraser, J.S.; Fischbach, M.A. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 2014, 16, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zheng, M.; Liu, J.; Fan, W.; He, H.; Huang, F. Melatonin promoted osteogenesis of human periodontal ligament cells by regulating mitochondrial functions through the translocase of the outer mitochondrial membrane 20. J. Periodontal. Res. 2022, 58, 53–69. [Google Scholar] [CrossRef]
- Gonzalez, A.; Estaras, M.; Martinez-Morcillo, S.; Martinez, R.; García, A.; Estévez, M.; Santofimia-Castaño, P.; Tapia, J.A.; Moreno, N.; Pérez-López, M.; et al. Melatonin modulates red-ox state and decreases viability of rat pancreatic stellate cells. Sci. Rep. 2020, 10, 6352. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Pradoldej, S.; Tosini, G.; Chang, Q.; Iuvone, P.M.; Ye, K. N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. USA 2010, 107, 3876–3881. [Google Scholar] [CrossRef]
- Fulgenzi, G.; Hong, Z.; Tomassoni-Ardori, F.; Barella, L.F.; Becker, J.; Barrick, C.; Swing, D.; Yanpallewar, S.; Croix, B.S.; Wess, J.; et al. Novel metabolic role for BDNF in pancreatic β-cell insulin secretion. Nat. Commun. 2020, 11, 1950. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, Z.; Wang, Y.; Yao, W.; Liao, S.; Yu, B.; Zhang, J.; Zhang, Y.; Zheng, B.; Ren, B.; et al. Sodium Butyrate Ameliorates Streptozotocin-Induced Type 1 Diabetes in Mice by Inhibiting the HMGB1 Expression. Front. Endocrinol. 2018, 9, 630. [Google Scholar] [CrossRef]
- Chung, H.; Hong, S.J.; Choi, S.W.; Park, C.G. The effect of preexisting HMGB1 within fetal bovine serum on murine pancreatic beta cell biology. Islets 2020, 12, 1–8. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis via Suboptimal Mitochondrial Function: Assessment, Treatment and Classification Implications. Curr. Top. Med. Chem. 2020, 20, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Kalwat, M.A.; Huang, Z.; Binns, D.D.; McGlynn, K.; Cobb, M.H. α2-Adrenergic Disruption of β Cell BDNF-TrkB Receptor Tyrosine Kinase Signaling. Front. Cell Dev. Biol. 2020, 8, 576396. [Google Scholar] [CrossRef]
- Yoo, D.Y.; Nam, S.M.; Kim, W.; Lee, C.H.; Won, M.H.; Hwang, I.K.; Yoon, Y.S. N-acetylserotonin increases cell proliferation and differentiating neuroblasts with tertiary dendrites through upregulation of brain-derived neurotrophic factor in the mouse dentate gyrus. J. Vet. Med. Sci. 2011, 73, 1411–1416. [Google Scholar] [CrossRef]
- Anquetil, F.; Mondanelli, G.; Gonzalez, N.; Rodriguez Calvo, T.; Zapardiel Gonzalo, J.; Krogvold, L.; Dahl-Jørgensen, K.; Van den Eynde, B.; Orabona, C.; Grohmann, U.; et al. Loss of IDO1 Expression From Human Pancreatic β-Cells Precedes Their Destruction During the Development of Type 1 Diabetes. Diabetes 2018, 67, 1858–1866. [Google Scholar] [CrossRef]
- Marcheva, B.; Weidemann, B.J.; Taguchi, A.; Perelis, M.; Ramsey, K.M.; Newman, M.V.; Kobayashi, Y.; Omura, C.; Manning Fox, J.E.; Lin, H.; et al. P2Y1 purinergic receptor identified as a diabetes target in a small-molecule screen to reverse circadian β-cell failure. eLife 2022, 11, e75132. [Google Scholar] [CrossRef]
- Brice, N.L.; Varadi, A.; Ashcroft, S.J.; Molnar, E. Metabotropic glutamate and GABA(B) receptors contribute to the modulation of glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia 2002, 45, 242–252. [Google Scholar] [CrossRef]
- Panzer, J.K.; Tamayo, A.; Caicedo, A. Restoring glutamate receptor signaling in pancreatic alpha cells rescues glucagon responses in type 1 diabetes. Cell Rep. 2022, 41, 111792. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Bae, J.Y.; Moon, J.; Koh, G. System χc-overexpression prevents 2-deoxy-d-ribose-induced β-cell damage. Free Radic. Biol. Med. 2020, 153, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Resch, J.; Rush, T.; Lobner, D. Functional upregulation of system xc- by fibroblast growth factor-2. Neuropharmacology 2012, 62, 901–906. [Google Scholar] [CrossRef]
- Regeenes, R.; Silva, P.N.; Chang, H.H.; Arany, E.J.; Shukalyuk, A.I.; Audet, J.; Kilkenny, D.M.; Rocheleau, J.V. Fibroblast growth factor receptor 5 (FGFR5) is a co-receptor for FGFR1 that is up-regulated in beta-cells by cytokine-induced inflammation. J. Biol. Chem. 2018, 293, 17218–17228. [Google Scholar] [CrossRef]
- Mias, C.; Trouche, E.; Seguelas, M.H.; Calcagno, F.; Dignat-George, F.; Sabatier, F.; Piercecchi-Marti, M.D.; Daniel, L.; Bianchi, P.; Calise, D.; et al. Ex vivo pretreatment with melatonin improves survival, proangiogenic/mitogenic activity, and efficiency of mesenchymal stem cells injected into ischemic kidney. Stem. Cells 2008, 26, 1749–1757. [Google Scholar] [CrossRef]
- Gardner, G.; Fraker, C.A. Natural Killer Cells as Key Mediators in Type I Diabetes Immunopathology. Front. Immunol. 2021, 12, 722979. [Google Scholar] [CrossRef]
- Wang, G.; Zhu, X.; Song, X.; Zhang, Q.; Qian, Z. Melatonin Inhibits hIAPP Oligomerization by Preventing β-Sheet and Hydrogen Bond Formation of the Amyloidogenic Region Revealed by Replica-Exchange Molecular Dynamics Simulation. Int. J. Mol. Sci. 2022, 23, 10264. [Google Scholar] [CrossRef]
- Cai, K.; Qi, D.; Hou, X.; Wang, O.; Chen, J.; Deng, B.; Qian, L.; Liu, X.; Le, Y. MCP-1 upregulates amylin expression in murine pancreatic β cells through ERK/JNK-AP1 and NF-κB related signaling pathways independent of CCR2. PLoS ONE 2011, 6, e19559. [Google Scholar] [CrossRef]
- Li, W.; Liu, H.; Fu, L.; Li, D.; Zhao, Y. Identification of Yin Yang 1-interacting partners at -1026C/A in the human iNOS promoter. Arch Biochem. Biophys. 2010, 498, 119–126. [Google Scholar] [CrossRef]
- Hoffmeister, A.; Tuennemann, J.; Sommerer, I.; Mössner, J.; Rittger, A.; Schleinitz, D.; Kratzsch, J.; Rosendahl, J.; Klöting, N.; Stahl, T.; et al. Genetic and biochemical evidence for a functional role of BACE1 in the regulation of insulin mRNA expression. Obes. (Silver Spring) 2013, 21, E626-33. [Google Scholar] [CrossRef]
- Esterházy, D.; Stützer, I.; Wang, H.; Rechsteiner, M.P.; Beauchamp, J.; Döbeli, H.; Hilpert, H.; Matile, H.; Prummer, M.; Schmidt, A.; et al. Bace2 is a β cell-enriched protease that regulates pancreatic β cell function and mass. Cell Metab. 2011, 14, 365–377. [Google Scholar] [CrossRef]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.-O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef]
- Anderson, G. Tumour microenvironment and metabolism: Role of the mitochondrial melatonergic pathway in determining Intercellular Interactions in a new dynamic homeostasis. Int. J. Mol. Sci. 2023, 24, 311. [Google Scholar] [CrossRef]
- Maussion, G.; Yang, J.; Yerko, V.; Barker, P.; Mechawar, N.; Ernst, C.; Turecki, G. Regulation of a truncated form of tropomyosin-related kinase B (TrkB) by Hsa-miR-185* in frontal cortex of suicide completers. PLoS ONE 2012, 7, e39301. [Google Scholar] [CrossRef]
- Shamblott, M.J.; O’Driscoll, M.L.; Gomez, D.L.; McGuire, D.L. Neurogenin 3 is regulated by neurotrophic tyrosine kinase receptor type 2 (TRKB) signaling in the adult human exocrine pancreas. Cell. Commun. Signal. 2016, 14, 23. [Google Scholar] [CrossRef]
- Kim, Y.K.; Walters, J.A.; Moss, N.D.; Wells, K.L.; Sheridan, R.; Miranda, J.G.; Benninger, R.K.P.; Pyle, L.L.; O’Brien, R.M.; Sussel, L.; et al. Zinc transporter 8 haploinsufficiency protects against beta cell dysfunction in type 1 diabetes by increasing mitochondrial respiration. Mol. Metab. 2022, 66, 101632. [Google Scholar] [CrossRef]
- Wang, B.Y.; Ye, Y.Y.; Qian, C.; Zhang, H.B.; Mao, H.X.; Yao, L.P.; Sun, X.; Lu, G.H.; Zhang, S.Z. Stress increases MHC-I expression in dopaminergic neurons and induces autoimmune activation in Parkinson’s disease. Neural Regen. Res. 2021, 16, 2521–2527. [Google Scholar] [CrossRef] [PubMed]
- Salemi, M.; Lanza, G.; Mogavero, M.P.; Cosentino, F.I.I.; Borgione, E.; Iorio, R.; Ventola, G.M.; Marchese, G.; Salluzzo, M.G.; Ravo, M.; et al. A Transcriptome Analysis of mRNAs and Long Non-Coding RNAs in Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 1535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hou, L.; Liu, Z.; Huang, S.; Meng, Z.; Liang, L. A mitophagic response to iron overload-induced oxidative damage associated with the PINK1/Parkin pathway in pancreatic beta cells. J. Trace Elem. Med. Biol. 2020, 60, 126493. [Google Scholar] [CrossRef]
- Huang, E.; Qu, D.; Huang, T.; Rizzi, N.; Boonying, W.; Krolak, D.; Ciana, P.; Woulfe, J.; Klein, C.; Slack, R.S.; et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat. Commun. 2017, 8, 1399. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Dadar, M.; Anderson, G.; Chirumbolo, S.; Maes, M. Preventive treatments to slow substantia nigra damage and Parkinson’s disease progression: A critical perspective review. Pharmacol. Res. 2020, 161, 105065. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, J.; Sun, C.; Li, G.; Li, S.; Zhang, L.; Di, C.; Gan, L.; Wang, Y.; Zhou, R.; et al. Ameliorating mitochondrial dysfunction restores carbon ion-induced cognitive deficits via co-activation of NRF2 and PINK1 signaling pathway. Redox Biol. 2018, 17, 143–157. [Google Scholar] [CrossRef]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef]
- Mesnage, R.; Antoniou, M.N. Computational modelling provides insight into the effects of glyphosate on the shikimate pathway in the human gut microbiome. Curr. Res. Toxicol. 2020, 1, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Fréville, M.; Estienne, A.; Ramé, C.; Lefort, G.; Chahnamian, M.; Staub, C.; Venturi, E.; Lemarchand, J.; Maximin, E.; Hondelatte, A.; et al. Chronic dietary exposure to a glyphosate-based herbicide results in total or partial reversibility of plasma oxidative stress, cecal microbiota abundance and short-chain fatty acid composition in broiler hens. Front. Physiol. 2022, 13, 974688. [Google Scholar] [CrossRef] [PubMed]
- Mesnage, R.; Teixeira, M.; Mandrioli, D.; Falcioni, L.; Ducarmon, Q.R.; Zwittink, R.D.; Mazzacuva, F.; Caldwell, A.; Halket, J.; Amiel, C.; et al. Use of Shotgun Metagenomics and Metabolomics to Evaluate the Impact of Glyphosate or Roundup MON 52276 on the Gut Microbiota and Serum Metabolome of Sprague-Dawley Rats. Environ. Health Perspect. 2021, 129, 17005. [Google Scholar] [CrossRef]
- Hansen, C.H.; Krych, L.; Nielsen, D.S.; Vogensen, F.K.; Hansen, L.H.; Sørensen, S.J.; Buschard, K.; Hansen, A.K. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia 2012, 55, 2285–2294. [Google Scholar] [CrossRef] [PubMed]
- Hänninen, A.; Toivonen, R.; Pöysti, S.; Belzer, C.; Plovier, H.; Ouwerkerk, J.P.; Emani, R.; Cani, P.D.; De Vos, W.M. Akkermansia muciniphila induces gut microbiota remodelling and controls islet autoimmunity in NOD mice. Gut 2018, 67, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Fassatoui, M.; Lopez-Siles, M.; Díaz-Rizzolo, D.A.; Jmel, H.; Naouali, C.; Abdessalem, G.; Chikhaoui, A.; Nadal, B.; Jamoussi, H.; Abid, A.; et al. Gut microbiota imbalances in Tunisian participants with type 1 and type 2 diabetes mellitus. BioSci. Rep. 2019, 39, BSR20182348. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Yen, H.R.; Lu, W.L.; Ho, H.H.; Lin, W.Y.; Kuo, Y.W.; Huang, Y.Y.; Tsai, S.Y.; Lin, H.C. Adjuvant Probiotics of Lactobacillus salivarius subsp. salicinius AP-32, L. johnsonii MH-68, and Bifidobacterium animalis subsp. lactis CP-9 Attenuate Glycemic Levels and Inflammatory Cytokines in Patients With Type 1 Diabetes Mellitus. Front. Endocrinol. 2022, 13, 754401. [Google Scholar] [CrossRef]
- Karcher, N.; Nigro, E.; Punčochář, M.; Blanco-Míguez, A.; Ciciani, M.; Manghi, P.; Zolfo, M.; Cumbo, F.; Manara, S.; Golzato, D.; et al. Genomic diversity and ecology of human-associated Akkermansia species in the gut microbiome revealed by extensive metagenomic assembly. Genome Biol. 2021, 22, 209. [Google Scholar] [CrossRef]
- Dean, J.W.; Helm, E.Y.; Fu, Z.; Xiong, L.; Sun, N.; Oliff, K.N.; Muehlbauer, M.; Avram, D.; Zhou, L. The aryl hydrocarbon receptor cell intrinsically promotes resident memory CD8+ T cell differentiation and function. Cell Rep. 2023, 42, 111963. [Google Scholar] [CrossRef]
- Okada, M.; Zhang, V.; Loaiza Naranjo, J.D.; Tillett, B.J.; Wong, F.S.; Steptoe, R.J.; Bergot, A.S.; Hamilton-Williams, E.E. Islet-specific CD8+ T cells gain effector function in the gut lymphoid tissues via bystander activation not molecular mimicry. Immunol. Cell Biol. 2023, 101, 36–48. [Google Scholar] [CrossRef]
- Wang, N.Q.; Jia, W.H.; Yin, L.; Li, N.; Liang, M.D.; Shang, J.M.; Hou, B.Y.; Zhang, L.; Qiang, G.F.; Du, G.H.; et al. Sex difference on fibroblast growth factors (FGFs) expression in skin and wound of streptozotocin(STZ)-induced type 1 diabetic mice. Mol. Biol. Rep. 2022, 1–11. [Google Scholar] [CrossRef]
- Uslu, S.; Oktem, G.; Uysal, A.; Soner, B.C.; Arbak, S.; Ince, U. Stem cell and extracellular matrix-related molecules increase following melatonin treatment in the skin of postmenopausal rats. Cell Biol. Int. 2014, 38, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.X.; Qiao, Y.C.; Li, W.; Zou, X.; Chen, Y.L.; Shen, J.; Liao, Q.Y.; Zhang, Q.J.; He, L.; Zhao, H.L. Human amylin induces CD4+Foxp3+ regulatory T cells in the protection from autoimmune diabetes. Immunol. Res. 2018, 66, 179–186. [Google Scholar] [CrossRef]
- Moon, J.H.; Kim, Y.G.; Kim, K.; Osonoi, S.; Wang, S.; Saunders, D.C.; Wang, J.; Yang, K.; Kim, H.; Lee, J.; et al. Serotonin Regulates Adult β-Cell Mass by Stimulating Perinatal β-Cell Proliferation. Diabetes 2020, 69, 205–214. [Google Scholar] [CrossRef]
- Goyvaerts, L.; Schraenen, A.; Lemaire, K.; Veld, P.I.; Smolders, I.; Maroteaux, L.; Schuit, F. Normal Pregnancy-Induced Islet Beta Cell Proliferation in Mouse Models That Are Deficient in Serotonin-Signaling. Int. J. Mol. Sci. 2022, 23, 15816. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Wang, C.; Yu, Z.; Peng, Y.; Wang, S.; Feng, S.; Zhang, S.; Tian, X.; Sun, C.; Liu, K.; et al. Human transporters, PEPT1/2, facilitate melatonin transportation into mitochondria of cancer cells: An implication of the therapeutic potential. J. Pineal Res. 2017, 62, e12390. [Google Scholar] [CrossRef]
- Mariosa, D.; Kamel, F.; Bellocco, R.; Ye, W.; Fang, F. Association between diabetes and amyotrophic lateral sclerosis in Sweden. Eur. J. Neurol. 2015, 22, 1436–1442. [Google Scholar] [CrossRef]
- Estrada-Bonilla, Y.C.; Castro, P.A.T.S.; Luna, G.L.F.; Souza, A.B.A.; Santos, G.S.; Salvini, T.F.; Leal, A.M.O.; Russo, T.L. Reaching task performance is associated to neuromuscular junction adaptations in rats with induced diabetes mellitus. Braz. J. Med. Biol. Res. 2020, 53, e8763. [Google Scholar] [CrossRef]
- Li, C.Y.; Yang, T.M.; Ou, R.W.; Wei, Q.Q.; Shang, H.F. Genome-wide genetic links between amyotrophic lateral sclerosis and autoimmune diseases. BMC Med. 2021, 19, 27. [Google Scholar] [CrossRef]
- Kale, O.E.; Vongdip, M.; Ogundare, T.F.; Osilesi, O. The use of combined high-fructose diet and glyphosate to model rats type 2 diabetes symptomatology. Toxicol. Mech. Methods 2021, 31, 126–137. [Google Scholar] [CrossRef]
- Tizhe, E.; Ibrahim, N.; Fatihu, M.; Ambali, S.; Igbokwe, I.; Tizhe, U. Pancreatic function and histoarchitecture in Wistar rats following chronic exposure to Bushfire®: The mitigating role of zinc. J. Int. Med. Res. 2018, 46, 3296–3305. [Google Scholar] [CrossRef]
- Czarny, P.; Kwiatkowski, D.; Toma, M.; Gałecki, P.; Orzechowska, A.; Bobińska, K.; Bielecka-Kowalska, A.; Szemraj, J.; Berk, M.; Anderson, G.; et al. Single-Nucleotide Polymorphisms of Genes Involved in Repair of Oxidative DNA Damage and the Risk of Recurrent Depressive Disorder. Med. Sci. Monit. 2016, 22, 4455–4474. [Google Scholar] [CrossRef]
- Leitner, M.; Fragner, L.; Danner, S.; Holeschofsky, N.; Leitner, K.; Tischler, S.; Doerfler, H.; Bachmann, G.; Sun, X.; Jaeger, W.; et al. Combined Metabolomic Analysis of Plasma and Urine Reveals AHBA, Tryptophan and Serotonin Metabolism as Potential Risk Factors in Gestational Diabetes Mellitus (GDM). Front Mol. Biosci. 2017, 4, 84. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Feng, Z.; Cheng, H.W. Perspective: Gestational Tryptophan Fluctuation Altering Neuroembryogenesis and Psychosocial Development. Cells 2022, 11, 1270. [Google Scholar] [CrossRef]
- Young, L.M.; Cao, P.; Raleigh, D.P.; Ashcroft, A.E.; Radford, S.E. Ion mobility spectrometry-mass spectrometry defines the oligomeric intermediates in amylin amyloid formation and the mode of action of inhibitors. J. Am. Chem. Soc. 2014, 136, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Press, M.; Jung, T.; König, J.; Grune, T.; Höhn, A. Protein aggregates and proteostasis in aging: Amylin and β-cell function. Mech. Ageing Dev. 2019, 177, 46–54. [Google Scholar] [CrossRef]
- Lei, D.; Sun, Y.; Liu, J.; Chi, J. Bergenin inhibits palmitic acid-induced pancreatic β-cell inflammatory death via regulating NLRP3 inflammasome activation. Ann. Transl. Med. 2022, 10, 1058. [Google Scholar] [CrossRef]
- Yuan, J.; Li, S.; Peng, H.; Ma, Y.; Li, L.; Fu, L.; Liu, J.; Jiang, H. Artesunate protects pancreatic β-cells from streptozotocin-induced diabetes via inhibition of the NLRP3/caspase-1/GSDMD pathway. Gen. Comp. Endocrinol. 2022, 326, 114068. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, Z.; Wang, J.; Zhang, X.; Yu, X.; Li, Y. Empagliflozin protects diabetic pancreatic tissue from damage by inhibiting the activation of the NLRP3/caspase-1/GSDMD pathway in pancreatic β cells: In Vitro and In Vivo studies. Bioengineered 2021, 12, 9356–9366. [Google Scholar] [CrossRef]
- Saha, M.; Manna, K.; Das Saha, K. Melatonin Suppresses NLRP3 Inflammasome Activation via TLR4/NF-κB and P2X7R Signaling in High-Fat Diet-Induced Murine NASH Model. J Inflamm. Res. 2022, 15, 3235–3258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, S.; Huang, Y.; Shi, H.; Chen, X.; Wang, Y.; Li, Y.; Cao, D.; Wang, L. A scFv phage targeting the C. albicans cell wall screened from a bacteriophage-based library of induced immune protection in mice. Infect. Genet. Evol. 2022, 102, 105303. [Google Scholar] [CrossRef]
- Rahbarghazi, R.; Farhoudi, M.; Rahmani-Youshanlouei, H.; Hassanpour, M.; Rahbarghazi, A.; Rezaie, J.; Ahmadi, M. Putative effect of melatonin on cardiomyocyte senescence in mice with type 1 diabetes mellitus. J. Diabetes Metab. Disord. 2022, 21, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Hajam, Y.A.; Rai, S.; Pandi-Perumal, S.R.; Brown, G.M.; Reiter, R.J.; Cardinali, D.P. Coadministration of Melatonin and Insulin Improves Diabetes-Induced Impairment of Rat Kidney Function. Neuroendocrinology 2022, 112, 807–822. [Google Scholar] [CrossRef]
- Gong, Z.; Da, W.; Tian, Y.; Zhao, R.; Qiu, S.; Wu, Q.; Wen, K.; Shen, L.; Zhou, R.; Tao, L.; et al. Exogenous melatonin prevents type 1 diabetes mellitus-induced bone loss, probably by inhibiting senescence. Osteoporos. Int. 2022, 33, 453–466. [Google Scholar] [CrossRef]
- Sahan, A.; Akbal, C.; Tavukcu, H.H.; Cevik, O.; Cetinel, S.; Sekerci, C.A.; Sener, T.E.; Sener, G.; Tanidir, Y. Melatonin prevents deterioration of erectile function in streptozotocin-induced diabetic rats via sirtuin-1 expression. Andrologia 2020, 52, e13639. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Lobo, A.M.; Buonfiglio, D.C.; Cipolla-Neto, J. Streptozotocin-induced diabetes disrupts the body temperature daily rhythm in rats. Diabetol. Metab. Syndr. 2015, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Kor, Y.; Geyikli, I.; Keskin, M.; Akan, M. Preliminary study: Evaluation of melatonin secretion in children and adolescents with type 1 diabetes mellitus. Indian J. Endocrinol. Metab. 2014, 18, 565–568. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, G. Type I Diabetes Pathoetiology and Pathophysiology: Roles of the Gut Microbiome, Pancreatic Cellular Interactions, and the ‘Bystander’ Activation of Memory CD8+ T Cells. Int. J. Mol. Sci. 2023, 24, 3300. https://doi.org/10.3390/ijms24043300
Anderson G. Type I Diabetes Pathoetiology and Pathophysiology: Roles of the Gut Microbiome, Pancreatic Cellular Interactions, and the ‘Bystander’ Activation of Memory CD8+ T Cells. International Journal of Molecular Sciences. 2023; 24(4):3300. https://doi.org/10.3390/ijms24043300
Chicago/Turabian StyleAnderson, George. 2023. "Type I Diabetes Pathoetiology and Pathophysiology: Roles of the Gut Microbiome, Pancreatic Cellular Interactions, and the ‘Bystander’ Activation of Memory CD8+ T Cells" International Journal of Molecular Sciences 24, no. 4: 3300. https://doi.org/10.3390/ijms24043300
APA StyleAnderson, G. (2023). Type I Diabetes Pathoetiology and Pathophysiology: Roles of the Gut Microbiome, Pancreatic Cellular Interactions, and the ‘Bystander’ Activation of Memory CD8+ T Cells. International Journal of Molecular Sciences, 24(4), 3300. https://doi.org/10.3390/ijms24043300