
Acute PDE4 Inhibition Induces a Transient Increase in Blood Glucose in Mice
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
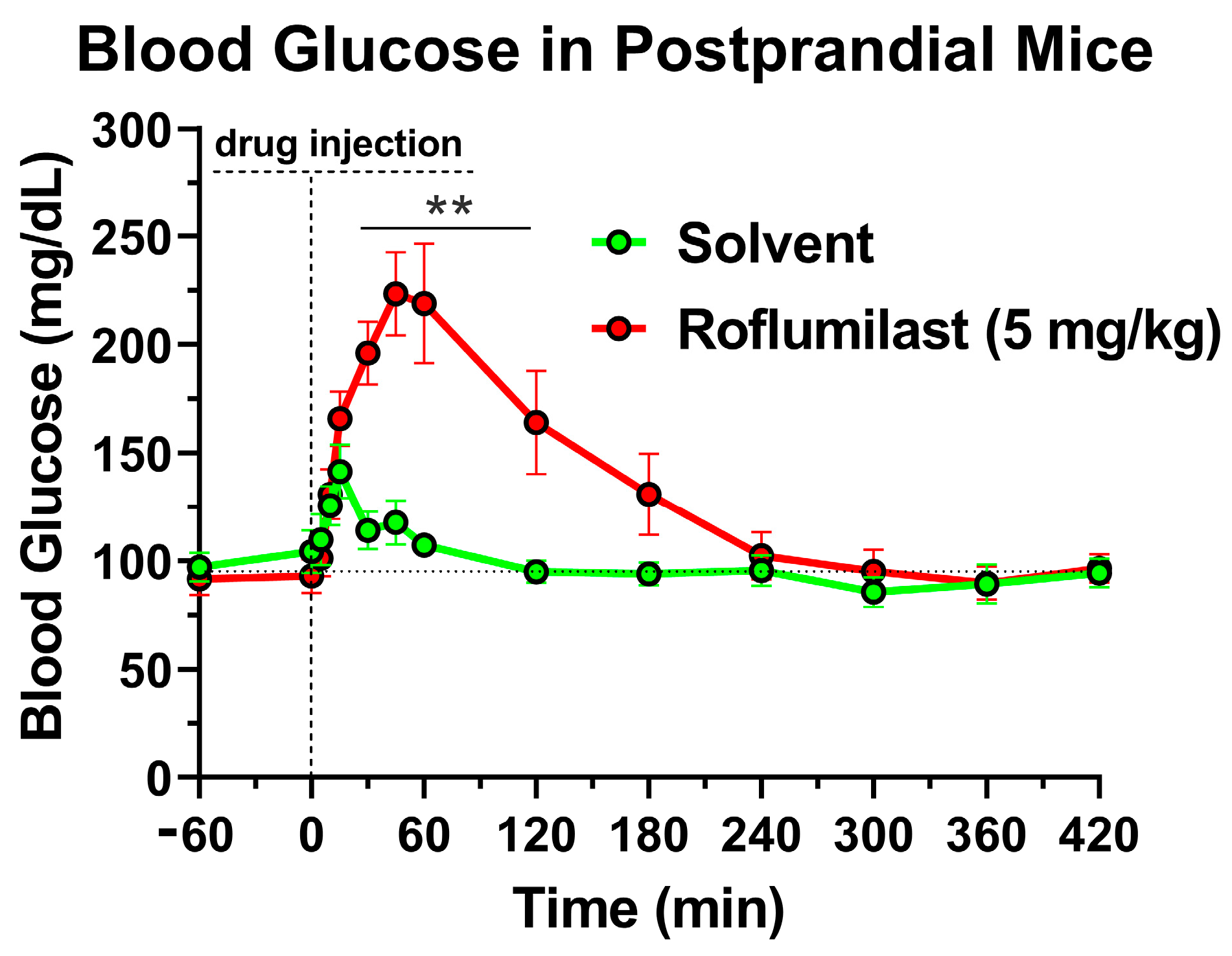
2.1. Treatment with the PDE4 Inhibitor Roflumilast Produces a Transient Increase in Blood Glucose Levels in Mice
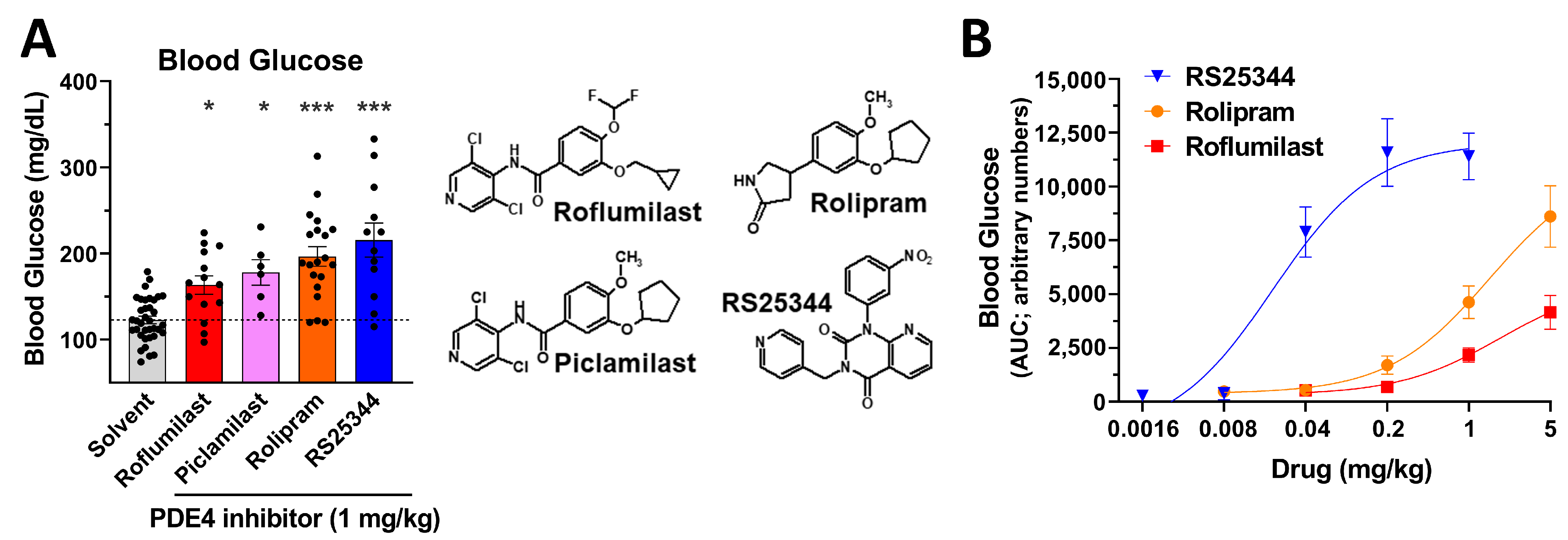
2.2. An Acute Increase in Blood Glucose Levels Is a Class Effect of PAN-PDE4 Inhibitors
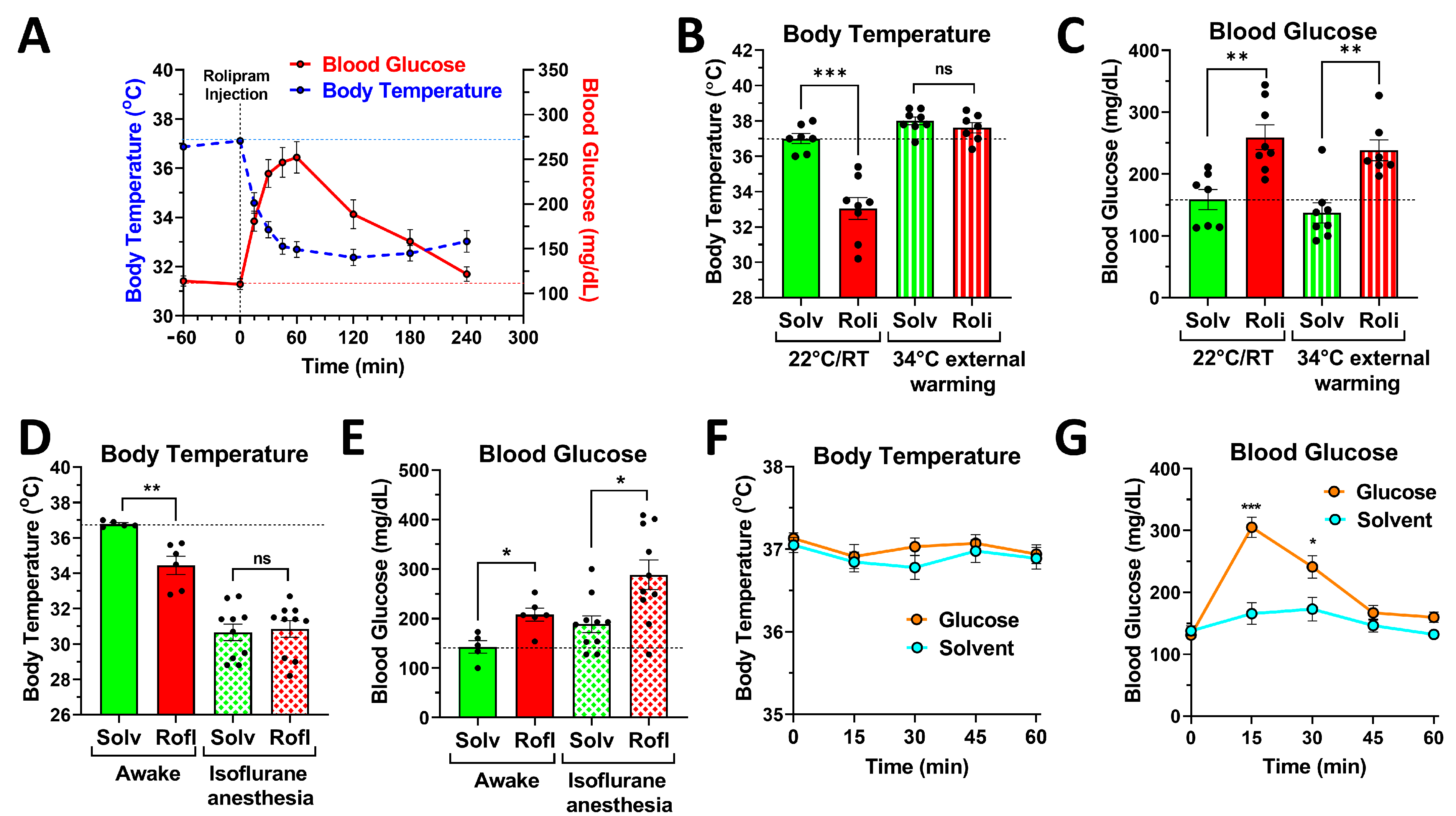
2.3. PDE4 Controls Blood Glucose Levels, Independent of Its Effects on Body Temperature
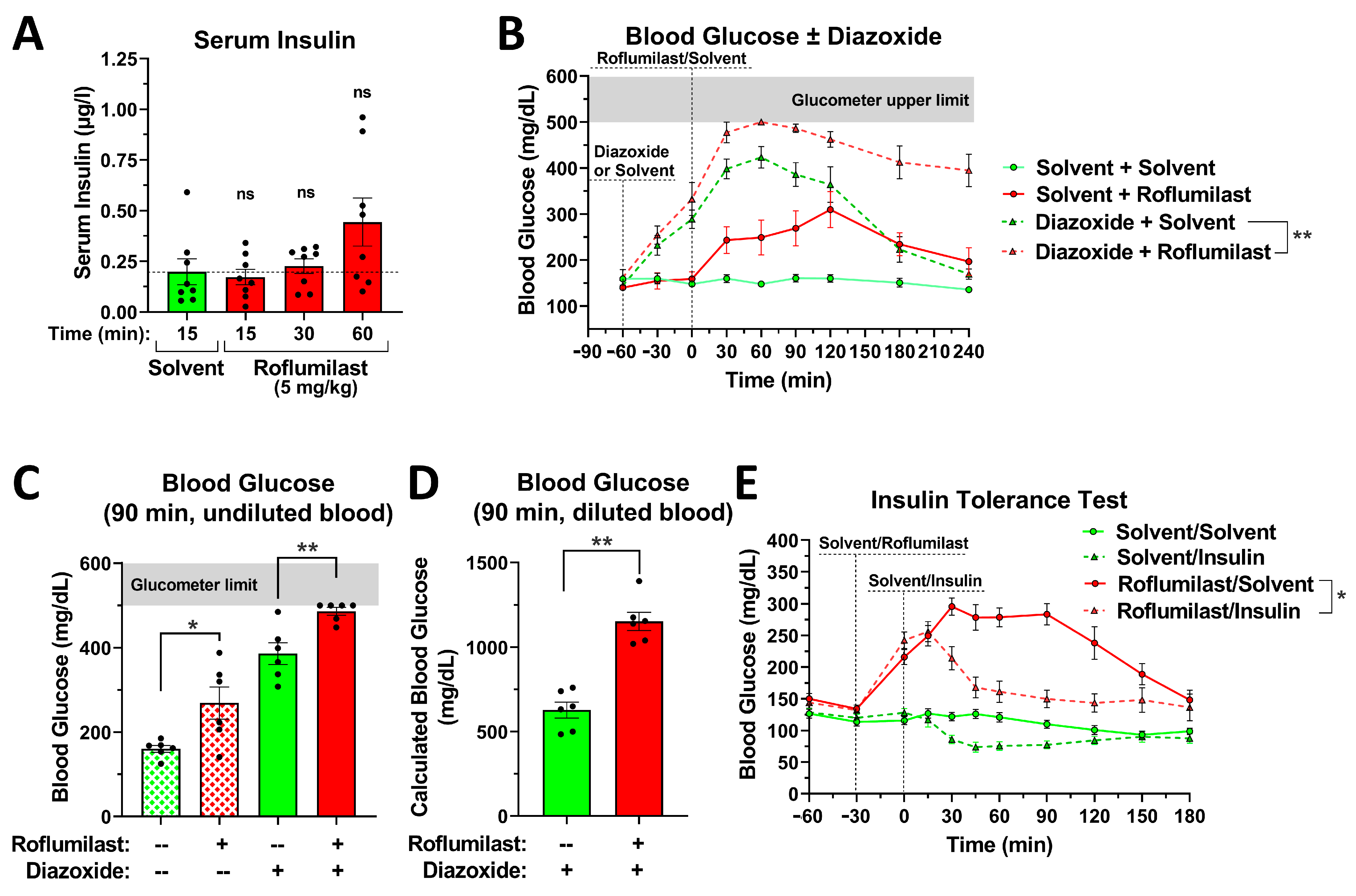
2.4. The Effect of PDE4 Inhibition on Blood Glucose Levels Is Independent of Changes in Insulin Secretion or Insulin Sensitivity
2.5. Blockade of α2- or β-Adrenoceptors Alleviates the Acute Glycemic Effects of PAN-PDE4 Inhibition
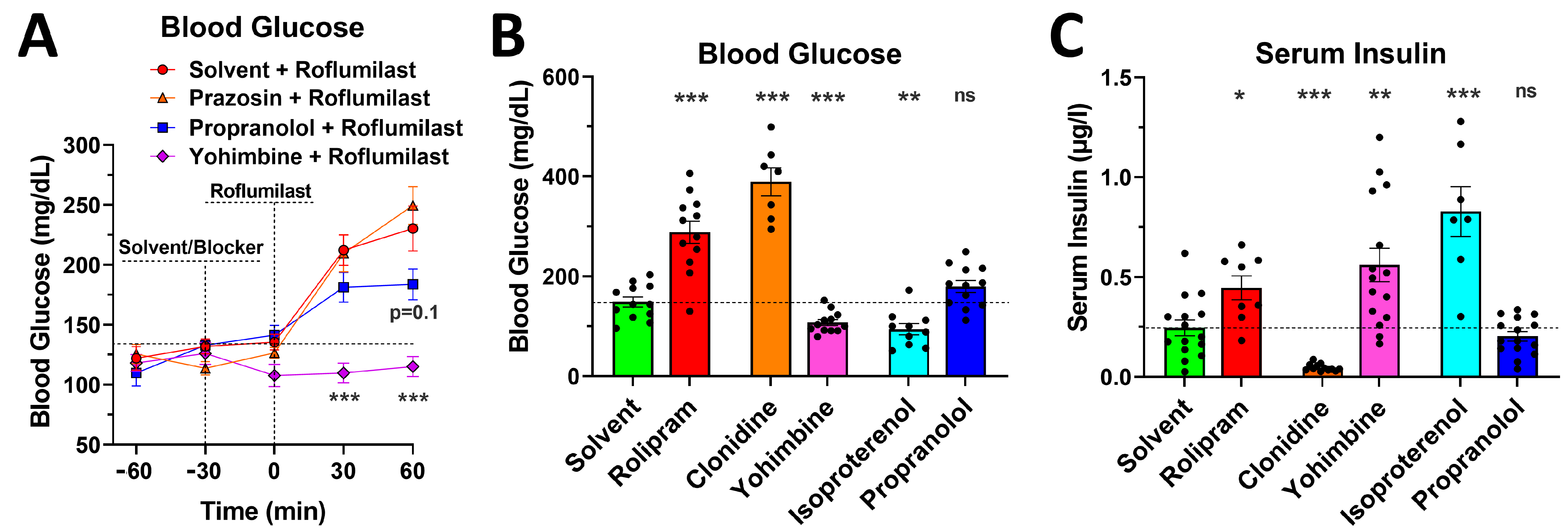
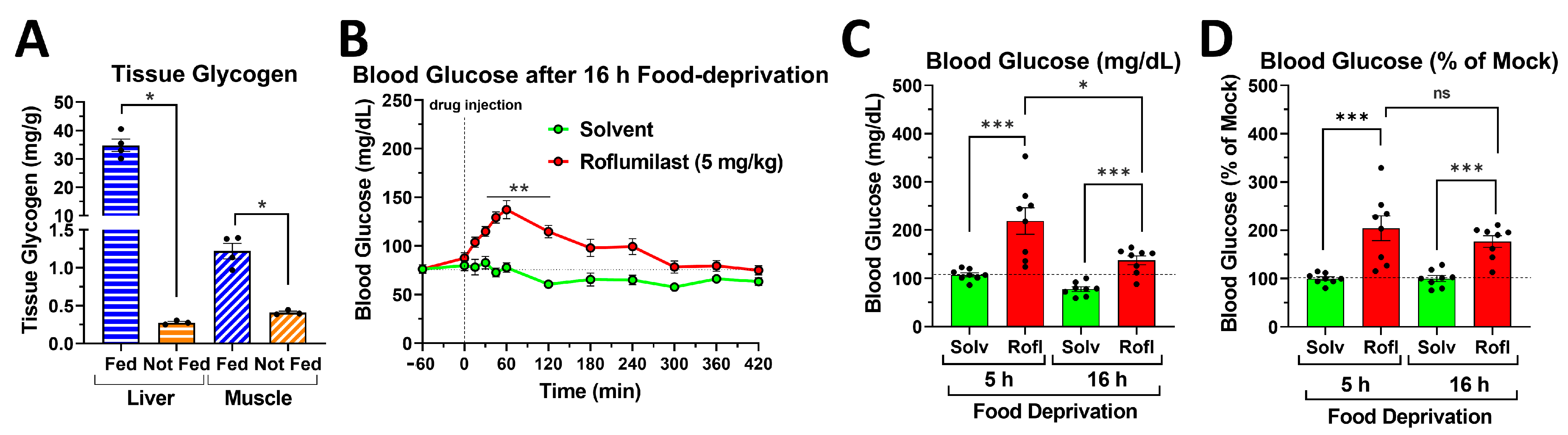
2.6. PDE4 Inhibition Induces Glycemic Effects in Mice Deprived of Food for 5 h or for 16 h Overnight
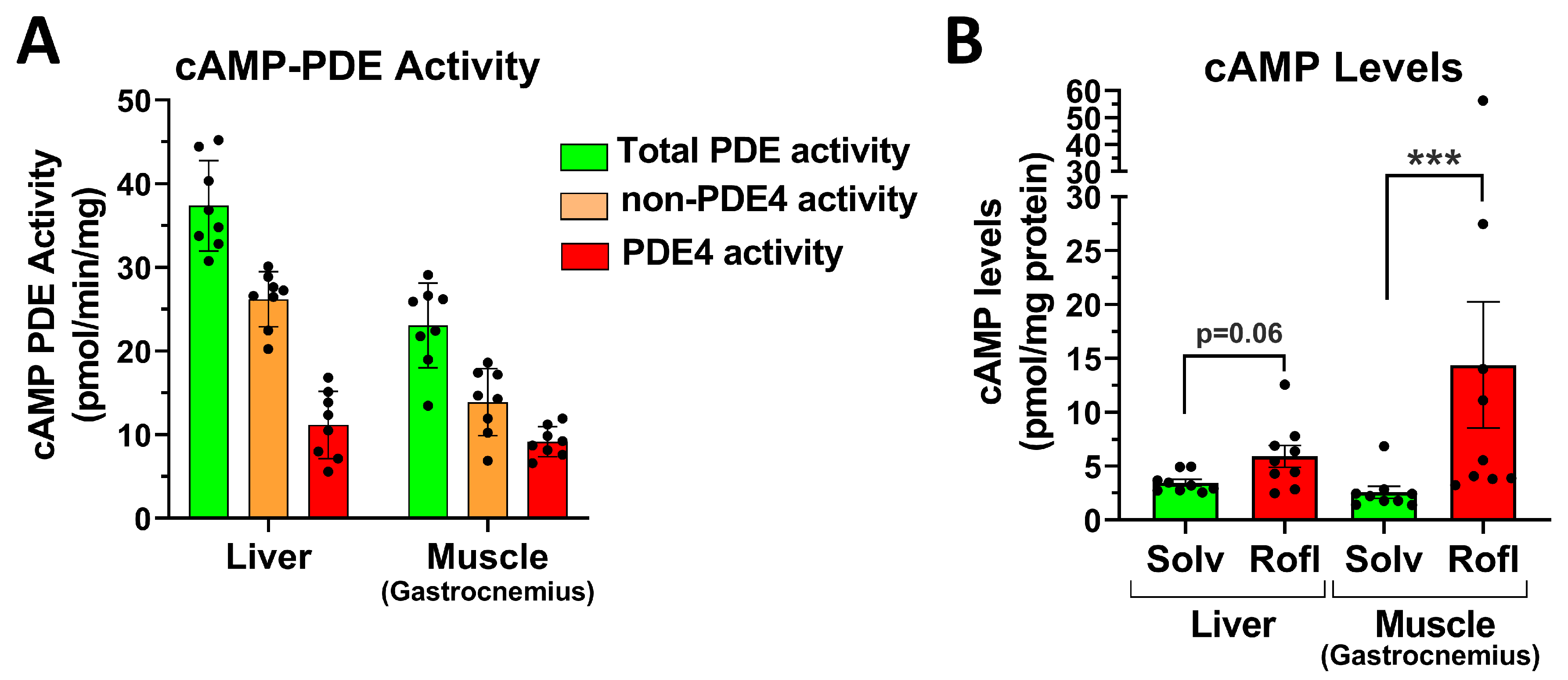
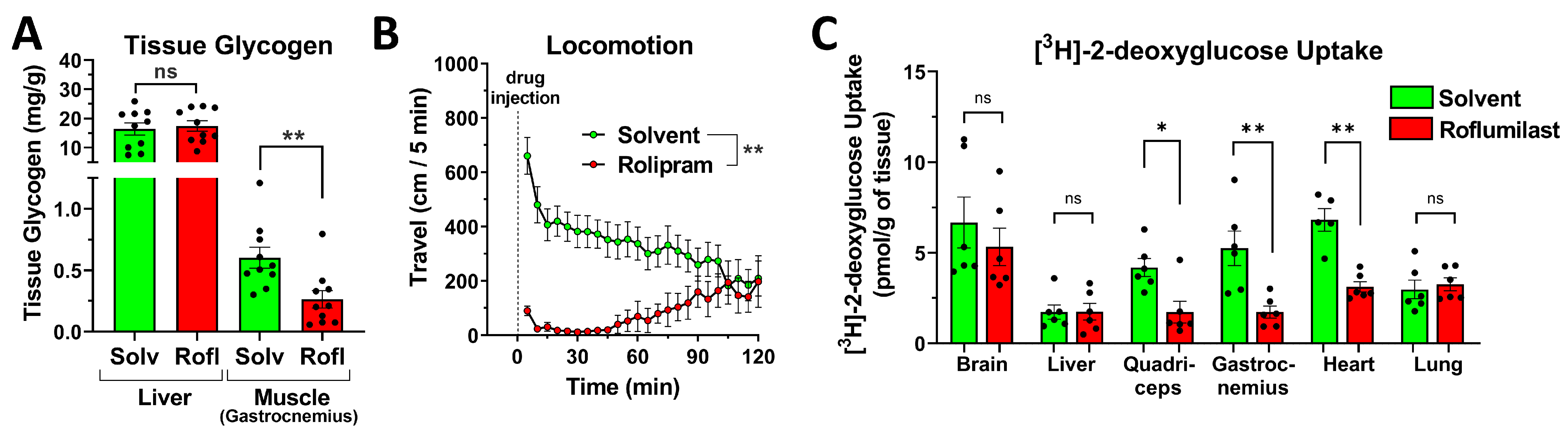
2.7. PDE4 Inhibition Promotes Skeletal Muscle Glycogenolysis in the Face of Reduced Glucose Uptake
3. Discussion
3.1. An Acute, Transient Increase in Blood Glucose Levels Is a Class Effect of PAN-PDE4 Inhibitors in Mice
3.2. PDE4 Inhibition Does Not Reduce Insulin Secretion and/or Sensitivity
3.3. PDE4 Inhibition and Glucose Production and Utilization
3.4. Clinical Relevance of the Acute Glycemic Effects of PDE4 Inhibitors
4. Materials and Methods
4.1. Drugs
4.2. Animals
4.3. Measurement of Blood Glucose Levels and Insulin Tolerance Tests (ITT)
4.4. Measurement of Core Body Temperature, Isoflurane Anesthesia, and the External Warming of Cages
4.5. ELISA and cAMP-Enzyme Immunoassay (EIA)
4.6. Measurement of Tissue Glycogen Content
4.7. Measurement of [3H]-2-Deoxy-Glucose Uptake
4.8. Measurement of cAMP-PDE Activity
4.9. Measurement of Locomotion Using SmartCageTM Technology
4.10. Data and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rogne, M.; Taskén, K. Compartmentalization of cAMP signaling in adipogenesis, lipogenesis, and lipolysis. Horm. Metab. Res. 2014, 46, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, L. Targeting cAMP/PKA pathway for glycemic control and type 2 diabetes therapy. J. Mol. Endocrinol. 2016, 57, R93–R108. [Google Scholar] [CrossRef]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. Control of glycaemia. Baillières Clin. Endocrinol. Metab. 1993, 7, 551–596. [Google Scholar] [CrossRef] [PubMed]
- Hers, H. Mechanisms of blood glucose homeostasis. J. Inherit. Metab. Dis. 1990, 13, 395–410. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Maratou, E.; Kountouri, A.; Board, M.; Lambadiari, V. Regulation of Postabsorptive and Postprandial Glucose Metabolism by Insulin-Dependent and Insulin-Independent Mechanisms: An Integrative Approach. Nutrients 2021, 13, 159. [Google Scholar] [CrossRef]
- Furman, B.; Ong, W.K.; Pyne, N.J. Cyclic AMP signaling in pancreatic islets. Adv. Exp. Med. Biol. 2010, 654, 281–304. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug. Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Richter, W.; Jin, S.L.; Conti, M. Splice variants of the cyclic nucleotide phosphodiesterase PDE4D are differentially expressed and regulated in rat tissue. Biochem. J. 2005, 388, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Richter, W.; Mehats, C.; Livera, G.; Park, J.Y.; Jin, C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J. Biol. Chem. 2003, 278, 5493–5496. [Google Scholar] [CrossRef]
- Paes, D.; Schepers, M.; Rombaut, B.; van den Hove, D.; Vanmierlo, T.; Prickaerts, J. The Molecular Biology of Phosphodiesterase 4 Enzymes as Pharmacological Targets: An Interplay of Isoforms, Conformational States, and Inhibitors. Pharmacol. Rev. 2021, 73, 1016–1049. [Google Scholar] [CrossRef]
- Peng, T.; Qi, B.; He, J.; Ke, H.; Shi, J. Advances in the Development of Phosphodiesterase-4 Inhibitors. J. Med. Chem. 2020, 63, 10594–10617. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zuo, J.; Tang, W. Phosphodiesterase-4 Inhibitors for the Treatment of Inflammatory Diseases. Front Pharm. 2018, 9, 1048. [Google Scholar] [CrossRef]
- Richter, W.; Menniti, F.S.; Zhang, H.T.; Conti, M. PDE4 as a target for cognition enhancement. Expert Opin. Ther. Targets 2013, 17, 1011–1027. [Google Scholar] [CrossRef]
- Jin, S.L.; Ding, S.L.; Lin, S.C. Phosphodiesterase 4 and its inhibitors in inflammatory diseases. Chang. Gung. Med. J. 2012, 35, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D.; Baillie, G.S.; Maurice, D.H. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: A molecular toolbox for generating compartmentalized cAMP signaling. Circ. Res. 2007, 100, 950–966. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D.; Schafer, P.; Zhang, K.Y. Keynote review: Phosphodiesterase-4 as a therapeutic target. Drug Discov. Today 2005, 10, 1503–1519. [Google Scholar] [CrossRef]
- Zhang, K.Y.; Ibrahim, P.N.; Gillette, S.; Bollag, G. Phosphodiesterase-4 as a potential drug target. Expert Opin. Targets 2005, 9, 1283–1305. [Google Scholar] [CrossRef]
- Wu, C.; Rajagopalan, S. Phosphodiesterase-4 inhibition as a therapeutic strategy for metabolic disorders. Obes. Rev. 2016, 17, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Muo, I.M.; MacDonald, S.D.; Madan, R.; Park, S.J.; Gharib, A.M.; Martinez, P.E.; Walter, M.F.; Yang, S.B.; Rodante, J.A.; Courville, A.B.; et al. Early effects of roflumilast on insulin sensitivity in adults with prediabetes and overweight/obesity involve age-associated fat mass loss-results of an exploratory study. Diabetes Metab. Syndr. Obes. 2019, 12, 743–759. [Google Scholar] [CrossRef]
- Wouters, E.F.; Bredenbröker, D.; Teichmann, P.; Brose, M.; Rabe, K.F.; Fabbri, L.M.; Göke, B. Effect of the phosphodiesterase 4 inhibitor roflumilast on glucose metabolism in patients with treatment-naive, newly diagnosed type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, E1720–E1725. [Google Scholar] [CrossRef]
- Ferguson, L.D.; Cathcart, S.; Rimmer, D.; Semple, G.; Brooksbank, K.; Paterson, C.; Brown, R.; Harvie, J.; Gao, X.; Radjenovic, A.; et al. Effect of the phosphodiesterase 4 inhibitor apremilast on cardiometabolic outcomes in psoriatic disease—Results of the Immune Metabolic Associations in Psoriatic Arthritis study. Rheumatology 2021, 61, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Mazzilli, S.; Lanna, C.; Chiaramonte, C.; Cesaroni, G.M.; Zangrilli, A.; Palumbo, V.; Cosio, T.; Dattola, A.; Gaziano, R.; Galluzzo, M.; et al. Real life experience of apremilast in psoriasis and arthritis psoriatic patients: Preliminary results on metabolic biomarkers. J. Derm. 2020, 47, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Jensterle, M.; Salamun, V.; Kocjan, T.; Vrtacnik Bokal, E.; Janez, A. Short term monotherapy with GLP-1 receptor agonist liraglutide or PDE 4 inhibitor roflumilast is superior to metformin in weight loss in obese PCOS women: A pilot randomized study. J. Ovarian Res. 2015, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Mollmann, J.; Kahles, F.; Lebherz, C.; Kappel, B.; Baeck, C.; Tacke, F.; Werner, C.; Federici, M.; Marx, N.; Lehrke, M. The PDE4 inhibitor roflumilast reduces weight gain by increasing energy expenditure and leads to improved glucose metabolism. Diabetes Obes. Metab. 2017, 19, 496–508. [Google Scholar] [CrossRef]
- Vollert, S.; Kaessner, N.; Heuser, A.; Hanauer, G.; Dieckmann, A.; Knaack, D.; Kley, H.P.; Beume, R.; Weiss-Haljiti, C. The glucose-lowering effects of the PDE4 inhibitors roflumilast and roflumilast-N-oxide in db/db mice. Diabetologia 2012, 55, 2779–2788. [Google Scholar] [CrossRef]
- Aragon, I.V.; Boyd, A.; Abou Saleh, L.; Rich, J.; McDonough, W.; Koloteva, A.; Richter, W. Inhibition of cAMP-phosphodiesterase 4 (PDE4) potentiates the anesthetic effects of Isoflurane in mice. Biochem. Pharmacol. 2021, 186, 114477. [Google Scholar] [CrossRef]
- Boyd, A.; Aragon, I.V.; Abou Saleh, L.; Southers, D.; Richter, W. The cAMP-phosphodiesterase 4 (PDE4) controls β-adrenoceptor- and CFTR-dependent saliva secretion in mice. Biochem. J. 2021, 478, 1891–1906. [Google Scholar] [CrossRef]
- McDonough, W.; Aragon, I.V.; Rich, J.; Murphy, J.M.; Abou Saleh, L.; Boyd, A.; Koloteva, A.; Richter, W. PAN-selective inhibition of cAMP-phosphodiesterase 4 (PDE4) induces gastroparesis in mice. FASEB J. 2020, 34, 12533–12548. [Google Scholar] [CrossRef] [PubMed]
- McDonough, W.; Rich, J.; Aragon, I.V.; Abou Saleh, L.; Boyd, A.; Richter, A.; Koloteva, A.; Richter, W. Inhibition of type 4 cAMP-phosphodiesterases (PDE4s) in mice induces hypothermia via effects on behavioral and central autonomous thermoregulation. Biochem. Pharmacol. 2020, 180, 114158. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.; Aragon, I.V.; Rich, J.; McDonough, W.; Oditt, M.; Irelan, D.; Fiedler, E.; Abou Saleh, L.; Richter, W. Assessment of PDE4 Inhibitor-Induced Hypothermia as a Correlate of Nausea in Mice. Biology 2021, 10, 1355. [Google Scholar] [CrossRef] [PubMed]
- Lenhardt, R. Body temperature regulation and anesthesia. Handb. Clin. Neurol. 2018, 157, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.E.; Egan, J.M. Pharmacological Agents That Directly Modulate Insulin Secretion. Pharmacol. Rev. 2003, 55, 105–131. [Google Scholar] [CrossRef] [PubMed]
- Fagerholm, V.; Haaparanta, M.; Scheinin, M. α2-Adrenoceptor Regulation of Blood Glucose Homeostasis. Basic Clin. Pharmacol. Toxicol. 2011, 108, 365–370. [Google Scholar] [CrossRef]
- Ito, K.; Dezaki, K.; Yoshida, M.; Yamada, H.; Miura, R.; Rita, R.S.; Ookawara, S.; Tabei, K.; Kawakami, M.; Hara, K.; et al. Endogenous α2A-Adrenoceptor–Operated Sympathoadrenergic Tones Attenuate Insulin Secretion via cAMP/TRPM2 Signaling. Diabetes 2017, 66, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.Z.; Wu, J.M.; Hu, G.M.; Gu, H.J.; Feng, Y.N.; Wang, S.X.; Cong, W.W.; Li, M.Z.; Xu, W.L.; Song, Y.; et al. α 1-AR overactivation induces cardiac inflammation through NLRP3 inflammasome activation. Acta Pharmacol. Sin. 2020, 41, 311–318. [Google Scholar] [CrossRef]
- Hamamdzic, D.; Duzic, E.; Sherlock, J.D.; Lanier, S.M. Regulation of alpha 2-adrenergic receptor expression and signaling in pancreatic beta-cells. Am. J. Physiol. 1995, 269, E162–E171. [Google Scholar] [CrossRef]
- Bruss, M.D.; Richter, W.; Horner, K.; Jin, S.L.; Conti, M. Critical role of PDE4D in beta2-adrenoceptor-dependent cAMP signaling in mouse embryonic fibroblasts. J Biol. Chem. 2008, 283, 22430–22442. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Abi-Gerges, A.; Nikolaev, V.O.; Richter, W.; Lechêne, P.; Mazet, J.L.; Conti, M.; Fischmeister, R.; Vandecasteele, G. Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: Role of phosphodiesterases. Circ. Res. 2008, 102, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Rochais, F.; Abi-Gerges, A.; Horner, K.; Lefebvre, F.; Cooper, D.M.; Conti, M.; Fischmeister, R.; Vandecasteele, G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res 2006, 98, 1081–1088. [Google Scholar] [CrossRef]
- Baillie, G.S.; Sood, A.; McPhee, I.; Gall, I.; Perry, S.J.; Lefkowitz, R.J.; Houslay, M.D. beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc. Natl. Acad. Sci. USA 2003, 100, 940–945. [Google Scholar] [CrossRef]
- Pratt, E.P.S.; Harvey, K.E.; Salyer, A.E.; Hockerman, G.H. Regulation of cAMP accumulation and activity by distinct phosphodiesterase subtypes in INS-1 cells and human pancreatic β-cells. PLoS ONE 2019, 14, e0215188. [Google Scholar] [CrossRef]
- Furman, B.; Pyne, N. Modulation of cyclic nucleotides and cyclic nucleotide phosphodiesterases in pancreatic islet beta-cells and intestinal L-cells as targets for treating diabetes mellitus. Curr. Opin. Investig. Drugs 2006, 7, 898–905. [Google Scholar]
- Pyne, N.J.; Furman, B.L. Cyclic nucleotide phosphodiesterases in pancreatic islets. Diabetologia 2003, 46, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Mutel, E.; Gautier-Stein, A.; Abdul-Wahed, A.; Amigó-Correig, M.; Zitoun, C.; Stefanutti, A.; Houberdon, I.; Tourette, J.A.; Mithieux, G.; Rajas, F. Control of blood glucose in the absence of hepatic glucose production during prolonged fasting in mice: Induction of renal and intestinal gluconeogenesis by glucagon. Diabetes 2011, 60, 3121–3131. [Google Scholar] [CrossRef]
- Miller, R.A.; Birnbaum, M.J. Glucagon: Acute actions on hepatic metabolism. Diabetologia 2016, 59, 1376–1381. [Google Scholar] [CrossRef]
- Imahashi, K.; Yoshioka, J.; Yamakita, T.; Yamano, S.; Kusuoka, H.; Nishimura, T. Type IV Phosphodiesterase Inhibitor Suppresses Insulin-Dependent Myocardial Glucose Uptake. Clin. Exp. Pharmacol. Physiol. 2001, 28, 290–291. [Google Scholar] [CrossRef]
- Ishikawa, M.; Hosoi, R.; Kobayashi, K.; Nishimura, T.; Inoue, O. Rolipram depresses [3H]2-deoxyglucose uptake in mouse brain and heart in vivo. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Ngala, R.A.; O’Dowd, J.F.; Stocker, C.J.; Cawthorne, M.A.; Arch, J.R.S. β2-adrenoceptor agonists can both stimulate and inhibit glucose uptake in mouse soleus muscle through ligand-directed signalling. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2013, 386, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, J.; Evans, B.A.; Bengtsson, T.; Summers, R.J. Multiple signalling pathways involved inβ2-adrenoceptor-mediated glucose uptake in rat skeletal muscle cells. Br. J. Pharmacol. 2006, 147, 446–454. [Google Scholar] [CrossRef]
- Potter, D.E.; Ellis, S. Isoproterenol- and epinephrine-induced changes in blood glucose and tissue glycogen levels in normal and diabetic rats: The influence of alteration in endogenous insulin levels and state of nourishment. J. Pharm. Exp. 1975, 193, 576–584. [Google Scholar]
- Piper, R.C.; James, D.E.; Slot, J.W.; Puri, C.; Lawrence, J.C. GLUT4 phosphorylation and inhibition of glucose transport by dibutyryl cAMP. J. Biol. Chem. 1993, 268, 16557–16563. [Google Scholar] [CrossRef]
- Lawrence, J.C.; Piper, R.C.; Robinson, L.J.; James, D.E. GLUT4 facilitates insulin stimulation and cAMP-mediated inhibition of glucose transport. Proc. Natl. Acad. Sci. USA 1992, 89, 3493–3497. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Bilan, P.J.; Hayashi, M.; Da, Y.; Yao, Z. Insulin sensitivity and inhibition by forskolin, dipyridamole and pentobarbital of glucose transport in three L6 muscle cell lines. Sci. China Ser. C Life Sci. 2007, 50, 739–747. [Google Scholar] [CrossRef]
- Tan, V.P.; Miyamoto, S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy 2015, 11, 963–964. [Google Scholar] [CrossRef]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef]
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef]
- Ritov, V.B.; Kelley, D.E. Hexokinase Isozyme Distribution in Human Skeletal Muscle. Diabetes 2001, 50, 1253–1262. [Google Scholar] [CrossRef]
- Brás, N.F.; Fernandes, P.A.; Ramos, M.J.; Schwartz, S.D. Mechanistic Insights on Human Phosphoglucomutase Revealed by Transition Path Sampling and Molecular Dynamics Calculations. Chem.–A Eur. J. 2017, 24, 1978–1987. [Google Scholar] [CrossRef]
- Fueger, P.T. GLUCOSE PHOSPHORYLATION AS A BARRIER TO MUSCLE GLUCOSE UPTAKE. Clin. Exp. Pharmacol. Physiol. 2005, 32, 314–318. [Google Scholar] [CrossRef]
- Jeukendrup, A.E. Regulation of Fat Metabolism in Skeletal Muscle. Ann. N. Y. Acad. Sci. 2006, 967, 217–235. [Google Scholar] [CrossRef] [PubMed]
- Reitman, M.L. Of mice and men—Environmental temperature, body temperature, and treatment of obesity. FEBS Lett. 2018, 592, 2098–2107. [Google Scholar] [CrossRef] [PubMed]
- Demetrius, L. Of mice and men. EMBO Rep. 2005, 6, S39–S44. [Google Scholar] [CrossRef] [PubMed]
- Jensterle, M.; Kocjan, T.; Janez, A. Phosphodiesterase 4 Inhibition as a Potential New Therapeutic Target in Obese Women With Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2014, 99, E1476–E1481. [Google Scholar] [CrossRef]
- Morley, L.A.; Gomez, T.H.; Goldman, J.L.; Flores, R.; Robinson, M.A. Accuracy of 5 Point-of-Care Glucometers in C57BL/6J Mice. JAALAS 2018, 57, 44–50. [Google Scholar]
- Togashi, Y.; Shirakawa, J.; Okuyama, T.; Yamazaki, S.; Kyohara, M.; Miyazawa, A.; Suzuki, T.; Hamada, M.; Terauchi, Y. Evaluation of the appropriateness of using glucometers for measuring the blood glucose levels in mice. Sci. Rep. 2016, 6, 25465. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irelan, D.; Boyd, A.; Fiedler, E.; Lochmaier, P.; McDonough, W.; Aragon, I.V.; Rachek, L.; Abou Saleh, L.; Richter, W. Acute PDE4 Inhibition Induces a Transient Increase in Blood Glucose in Mice. Int. J. Mol. Sci. 2023, 24, 3260. https://doi.org/10.3390/ijms24043260
Irelan D, Boyd A, Fiedler E, Lochmaier P, McDonough W, Aragon IV, Rachek L, Abou Saleh L, Richter W. Acute PDE4 Inhibition Induces a Transient Increase in Blood Glucose in Mice. International Journal of Molecular Sciences. 2023; 24(4):3260. https://doi.org/10.3390/ijms24043260
Chicago/Turabian StyleIrelan, Daniel, Abigail Boyd, Edward Fiedler, Peter Lochmaier, Will McDonough, Ileana V. Aragon, Lyudmila Rachek, Lina Abou Saleh, and Wito Richter. 2023. "Acute PDE4 Inhibition Induces a Transient Increase in Blood Glucose in Mice" International Journal of Molecular Sciences 24, no. 4: 3260. https://doi.org/10.3390/ijms24043260
APA StyleIrelan, D., Boyd, A., Fiedler, E., Lochmaier, P., McDonough, W., Aragon, I. V., Rachek, L., Abou Saleh, L., & Richter, W. (2023). Acute PDE4 Inhibition Induces a Transient Increase in Blood Glucose in Mice. International Journal of Molecular Sciences, 24(4), 3260. https://doi.org/10.3390/ijms24043260