
Interfering with the Ubiquitin-Mediated Regulation of Akt as a Strategy for Cancer Treatment


{kind=link}
{kind=link}
{kind=link}
Abstract
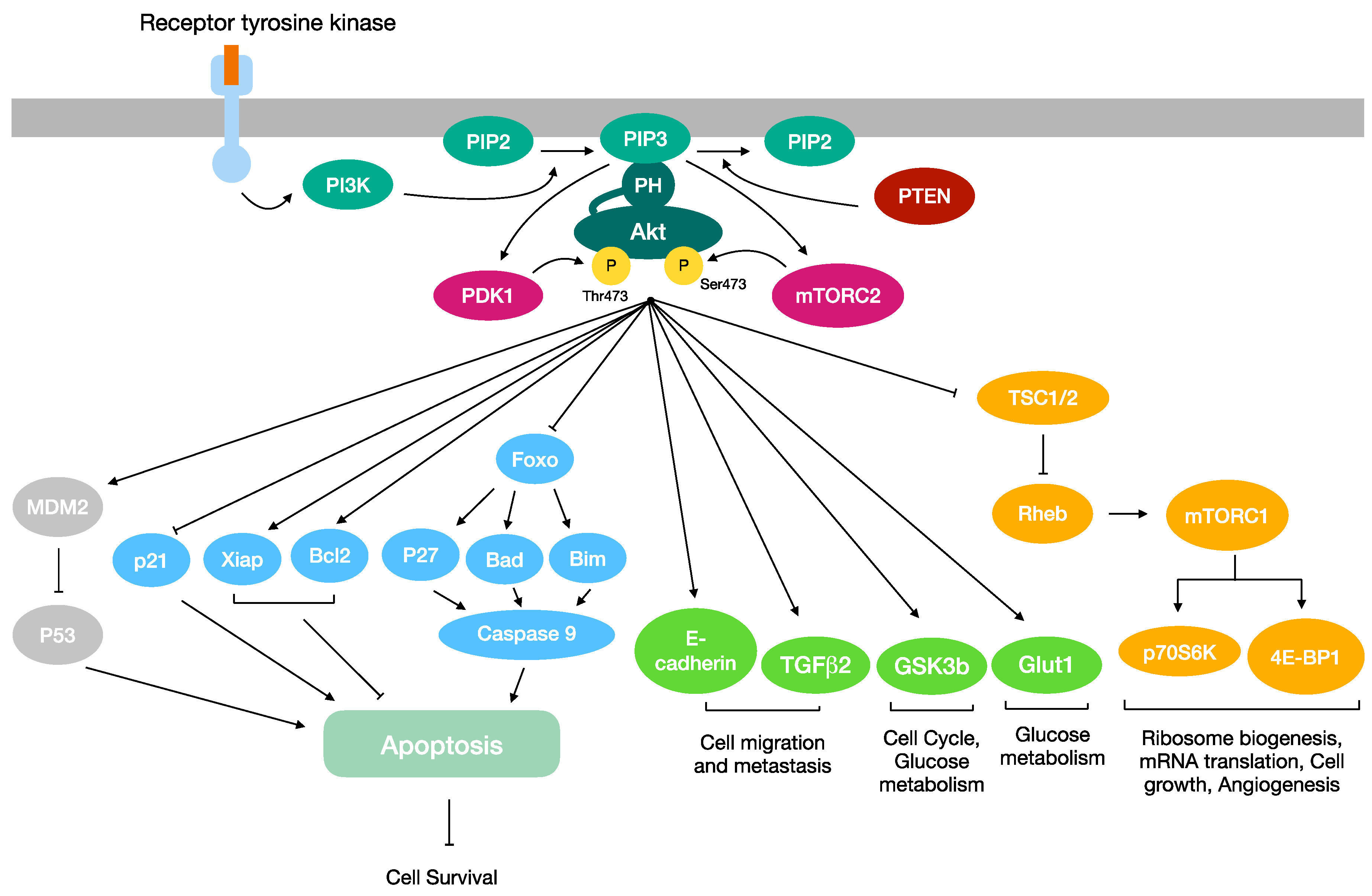
1. The Akt Signaling Pathway in Cancer
1.1. Akt: An Overview
1.2. Akt in Proliferation and Cell Survival
1.3. Akt in Cell Migration and Metastasis
1.4. Akt in Cancer Metabolism Regulation
2. Two Distinct Ubiquitination Processes Regulate the Activation and Inactivation of Akt Kinase
2.1. K63-Linked Polyubiquitination of Akt
2.2. K48-Linked Polyubiquitination of Akt
2.3. The Role of the Deubiquitination of Akt
3. Akt as a Target for Cancer Therapy
3.1. Targeting Akt Kinase
3.2. Targeting the Ubiquitin Pathway
3.3. Proteolysis-Targeting Chimeras
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shi, Y.; Liu, X.; Han, E.K.; Guan, R.; Shoemaker, A.R.; Oleksijew, A.; Woods, K.W.; Fisher, J.P.; Klinghofer, V.; Lasko, L.; et al. Optimal classes of chemotherapeutic agents sensitized by specific small-molecule inhibitors of Akt in vitro and in vivo. Neoplasia 2005, 7, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Pandolfi, P.P. The PTEN–PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. Akt/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Al-Bazz, Y.O.; Underwood, J.C.; Brown, B.L.; Dobson, P.R. Prognostic significance of Akt, phospho-Akt and BAD expression in primary breast cancer. Eur. J. Cancer 2009, 45, 694–704. [Google Scholar] [CrossRef]
- Xue, G.; Hemmings, B.A. PKB/Akt-dependent regulation of cell motility. J. Natl. Cancer Inst. 2013, 105, 393–404. [Google Scholar] [CrossRef]
- Staal, S.P. Molecular cloning of the Akt oncogene and its human homologues Akt1 and Akt2: Amplification of Akt1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef]
- Altomare, D.A.; Tanno, S.; De Rienzo, A.; Klein-Szanto, A.J.; Tanno, S.; Skele, K.L.; Hoffman, J.P.; Testa, J.R. Frequent activation of Akt2 kinase in human pancreatic carcinomas. J. Cell Biochem. 2002, 87, 470–476. [Google Scholar] [CrossRef]
- Dobashi, Y.; Kimura, M.; Matsubara, H.; Endo, S.; Inazawa, J.; Ooi, A. Molecular alterations in Akt and its protein activation in human lung carcinomas. Hum. Pathol. 2012, 43, 2229–2240. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, B.A.; Huang, L.; Wood, M.; Cheng, J.Q.; Testa, J.R. Amplification and overexpression of the Akt2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol. Carcinog. 1998, 21, 81–86. [Google Scholar] [CrossRef]
- Bellacosa, A.; De Feo, D.; Godwin, A.K.; Bell, D.W.; Cheng, J.Q.; Altomare, D.A.; Wan, M.; Dubeau, L.; Scambia, G.; Masciullo, V.; et al. Molecular alterations of the Akt2 oncogene in ovarian and breast carcinomas. Int. J. Cancer 1995, 64, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M.; et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. USA. 2015, 112, 3421–3426. [Google Scholar] [CrossRef]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004, 64, 7002–7010. [Google Scholar] [CrossRef]
- Bellacosa, A.; Testa, J.R.; Moore, R.; Larue, L. A portrait of Akt kinases: Human cancer and animal models depict a family with strong individualities. Cancer Biol. Ther. 2004, 3, 268–275. [Google Scholar] [CrossRef]
- Cheng, J.Q.; Lindsley, C.W.; Cheng, G.Z.; Yang, H.; Nicosia, S.V. The Akt/PKB pathway: Molecular target for cancer drug discovery. Oncogene 2005, 24, 7482–7492. [Google Scholar] [CrossRef]
- Altomare, D.A.; Testa, J.R. Perturbations of the Akt signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef]
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of Akt kinases in cancer: Implications for therapeutic targeting. Adv. Cancer Res. 2005, 94, 29–86. [Google Scholar]
- Sobočan, M.; Bračič, S.; Knez, J.; Takač, I.; Haybaeck, J. The communication between the PI3K/Akt/mTOR pathway and Y-box binding protein-1 in gynecological cancer. Cancers 2020, 12, 205. [Google Scholar] [CrossRef]
- Ma, X.L.; Shen, M.N.; Hu, B.; Wang, B.L.; Yang, W.J.; Lv, L.H.; Wang, H.; Zhou, Y.; Jin, A.-L.; Sun, Y.-F.; et al. CD73 promotes hepatocellular carcinoma progression and metastasis via activating PI3K/Akt signaling by inducing Rap1-mediated membrane localization of P110β and predicts poor prognosis. J. Hematol. Oncol. 2019, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Gallyas, F., Jr.; Sumegi, B.; Szabo, C. Role of Akt activation in PARP inhibitor resistance in cancer. Cancers 2020, 12, 532. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adhes. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-G.; Ai, Y.-W.; Yu, L.-L.; Zhou, X.-D.; Liu, J.; Li, J.-H.; Xu, X.-M.; Liu, S.; Chen, J.; Liu, F.; et al. Phosphoinositide 3-kinase/Akt pathway plays an important role in chemoresistance of gastric cancer cells against etoposide and doxorubicin induced cell death. Int. J. Cancer 2008, 122, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.L.; Dai, N.; Yu, H.G.; Sun, L.M.; Si, J.M. Akt associates with nuclear factor κB and plays an important role in chemoresistance of gastric cancer cells. Oncol. Rep. 2010, 24, 113–119. [Google Scholar] [CrossRef]
- Page, C.; Lin, H.J.; Jin, Y.; Castle, V.P.; Nunez, G.; Huang, M.; Lin, J. Overexpression of Akt/Akt can modulate chemotherapy-induced apoptosis. Anticancer Res. 2000, 20, 407–416. [Google Scholar]
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; De Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/Akt/mTOR Pathway Alterations. Cancer Cell. 2017, 12, 820–832. [Google Scholar] [CrossRef]
- Alwhaibi, A.; Kolhe, R.; Gao, F.; Cobran, E.K.; Somanath, P.R. Genome atlas analysis based profiling of Akt pathway genes in the early and advanced human prostate cancer. Oncoscience 2019, 6, 317–336. [Google Scholar] [CrossRef]
- Xu, J.; Wang, X.; Ke, Q.; Liao, K.; Wan, Y.; Zhang, K.; Zhang, G.; Wang, X. Combined bioinformatics technology to explore pivot genes and related clinical prognosis in the development of gastric cancer. Sci. Rep. 2021, 11, 15412. [Google Scholar] [CrossRef]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef]
- Datta, K.; Franke, T.F.; Chan, T.O.; Makris, A.; Yang, S.I.; Kaplan, D.R.; Morrison, D.K.; Golemis, E.A.; Tsichlis, P.N. AH/PH domain-mediated interaction between Akt molecules and its potential role in Akt regulation. Mol. Cell Biol. 1995, 15, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Du-Cuny, L.; Chen, L.; Meuillet, E.J.; Mash, E.A.; Powis, G.; Zhang, S. Recent development of anticancer therapeutics targeting Akt. Recent Pat. Anticancer Drug Discov. 2011, 6, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Franke, T.F.; Gonzalez-Portal, M.E.; Datta, K.; Taguchi, T.; Gardner, J.; Cheng, J.Q.; Testa, J.R.; Tsichlis, P.N. Structure, expression and chromosomal mapping of c-Akt: Relationship to v-Akt and its implications. Oncogene 1993, 8, 745–754. [Google Scholar]
- Chen, W.S.; Xu, P.Z.; Gottlob, K.; Chen, M.L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K.; et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001, 15, 2203–2208. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, R.S.; Orena, S.J.; Rafidi, K.; Torchia, A.J.; Stock, J.L.; Hildebrandt, A.L.; Coskran, T.; Black, S.C.; Brees, D.J.; Wicks, J.R.; et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKBβ. J. Clin. Investig. 2003, 112, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, O.; Yang, Z.Z.; Brodbeck, D.; Dummler, B.A.; Hemmings-Mieszczak, M.; Watanabe, T.; Michaelis, T.; Frahm, J.; Hemmings, B.A. Essential role of protein kinase Bγ (PKBγ/Akt3) in postnatal brain development but not in glucose homeostasis. Development 2005, 132, 2943–2954. [Google Scholar] [CrossRef]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef]
- Stambolic, V.; Woodgett, J.R. Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol. 2006, 16, 461–466. [Google Scholar] [CrossRef]
- Chu, N.; Salguero, A.L.; Liu, A.Z.; Chen, Z.; Dempsey, D.R.; Ficarro, S.B.; Alexander, W.M.; Marto, J.A.; Li, Y.; Amzel, L.M.; et al. Akt kinase activation mechanisms revealed using protein semisynthesis. Cell 2018, 174, 897–907. [Google Scholar] [CrossRef]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef]
- Guo, J.; Dai, X.; Laurent, B.; Zheng, N.; Gan, W.; Zhang, J.; Guo, A.; Yuan, M.; Liu, P.; Asara, J.M.; et al. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat. Cell Biol. 2019, 21, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-H.; Jo, U.; Kohrman, A.; Rezaeian, A.H.; Chou, P.-C.; Logothetis, C.; Lin, H.-K. Posttranslational regulation of Akt in human cancer. Cell Biosci. 2014, 4, 59. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Kozlowski, M.T.; Weng, Q.P.; Morrice, N.; Avruch, J. Avruch 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 1998, 8, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. Signaling—2000 and beyond. Cell 2000, 100, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.; Brandhuber, B.J. Crystal structure of human Akt1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- James, S.R.; Downes, C.P.; Gigg, R.; Grove, S.J.; Holmes, A.B.; Alessi, D.R. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem. J. 1996, 315, 709–713. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Singh, S.S.; Yap, W.N.; Arfuso, F.; Kar, S.; Wang, C.; Cai, W.; Dharmarajan, A.M.; Sethi, G.; Kumar, A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015, 21, 12261–12273. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef]
- Gkountakos, A.; Sartori, G.; Falcone, I.; Piro, G.; Ciuffreda, L.; Carbone, C.; Tortora, G.; Scarpa, A.; Bria, E.; Milella, M.; et al. PTEN in lung cancer: Dealing with the problem, building on new knowledge and turning the game around. Cancers 2019, 11, 1141. [Google Scholar] [CrossRef] [PubMed]
- Luongo, F.; Colonna, F.; Calapà, F.; Vitale, S.; Fiori, M.E.; De Maria, R. PTEN tumor-suppressor: The dam of stemness in cancer. Cancers 2019, 11, 1076. [Google Scholar] [CrossRef]
- Yang, W.L.; Wang, J.; Chan, C.H.; Lee, S.W.; Campos, A.D.; Lamothe, B.; Hur, L.; Grabiner, B.C.; Lin, X.; Darnay, B.G.; et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 2009, 325, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-L.; Wu, C.-Y.; Wu, J.; Lin, H.-K. Regulation of Akt signalling activation by ubiquitination. Cell Cycle 2010, 9, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Li, C.F.; Yang, W.L.; Gao, Y.; Lee, S.W.; Feng, Z.; Huang, H.-Y.; Tsai, K.K.; Flores, L.G.; Shao, Y.; et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012, 149, 1098–1111. [Google Scholar] [CrossRef]
- Li, W.; Peng, C.; Lee, M.H.; Lim, D.; Zhu, F.; Fu, Y.; Yang, G.; Sheng, Y.; Xiao, L.; Dong, X.; et al. TRAF4 is a critical molecule for Akt activation in lung cancer. Cancer Res. 2013, 73, 6938–6950. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. Akt/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 25, 11598–11603. [Google Scholar] [CrossRef]
- Zhou, B.P.; Liao, Y.; Xia, W.; Zou, Y.; Spohn, B.; Hung, M.C. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001, 3, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Heron-Milhavet, L.; Franckhauser, C.; Rana, V.; Berthenet, C.; Fisher, D.; Hemmings, B.A.; Fernandez, A.; Lamb, N.J.C. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol. Cell Biol. 2006, 26, 8267–8280. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.L.; Marcotte, R.; Hennessy, B.T.; Woodgett, J.R.; Mills, G.B.; Muller, W.J. Akt1 and Akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res. 2009, 69, 5057–5064. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann-Zeh, A.; Rodriguez-Viciana, P.; Ulrich, E.; Gilbert, C.; Coffer, P.; Downward, J.; Evan, G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 1997, 385, 544. [Google Scholar] [CrossRef]
- Deng, L.; Chen, L.; Zhao, L.; Xu, Y.; Peng, X.; Wang, X.; Ding, L.; Jin, J.; Teng, H.; Wang, Y.; et al. Ubiquitination of Rheb governs growth factor-induced mTORC1 activation. Cell Res. 2019, 29, 136–150. [Google Scholar] [CrossRef]
- Gentilella, A.; Kozma, S.C.; Thomas, G. A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2015, 1849, 812–820. [Google Scholar] [CrossRef]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/Akt pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Wee, K.B.; Aguda, B.D. Akt versus p53 in a network of oncogenes and tumour suppressor genes regulating cell survival and death. Biophys. J. 2006, 91, 857–865. [Google Scholar] [CrossRef]
- Singh, S.; Ramamoorthy, M.; Vaughan, C.; Yeudall, W.A.; Deb, S. Human oncoprotein MDM2 activates the Akt signalling pathway through an interaction with the repressor element-1 silencing transcription factor conferring a survival advantage to cancer cells. Cell Death Differ. 2013, 20, 558. [Google Scholar] [CrossRef]
- Georgescu, M.M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef]
- Zhou, B.P.; Hung, M.C. Novel targets of Akt, p21(Cipl/WAF1), and MDM2. Semin. Oncol. 2002, 29, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, X.; Wang, H.; Liu, S.; Hu, N.; Li, X. Akt Regulated Phosphorylation of GSK-3β/Cyclin D1, p21 and p27 Contributes to Cell Proliferation Through Cell Cycle Progression From G1 to S/G2M Phase in Low-Dose Arsenite Exposed HaCat Cells. Front Pharmacol. 2019, 11, 1176. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.C.; Yuan, T.; Rui, B.Y.; Zhu, Z.Z.; Guo, S.C.; Zhang, C.Q. Exosomes derived from human platelet-rich plasma prevent apoptosis induced by glucocorticoid-associated endoplasmic reticulum stress in rat osteonecrosis of the femoral head via the Akt/Bad/Bcl-2 signal pathway. Theranostics 2017, 15, 733–750. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Tran, H.; Brunet, A.; Griffith, E.C.; Greenberg, M.E. The many forks in FOXO’s road. Sci. STKE 2003, 2003, RE5. [Google Scholar] [CrossRef]
- Herold, M.J.; Rohrbeck, L.; Lang, M.J.; Grumont, R.; Gerondakis, S.; Tai, L.; Bouillet, P.; Kaufmann, T.; Strasser, A. Foxo-mediated Bim transcription is dispensable for the apoptosis of hematopoietic cells that is mediated by this BH3-only protein. EMBO Rep. 2013, 14, 992–998. [Google Scholar] [CrossRef]
- Dan, H.C.; Sun, M.; Kaneko, S.; Feldman, R.I.; Nicosia, S.V.; Wang, H.-G.; Tsang, B.K.; Cheng, J.Q. Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP). J. Biol. Chem. 2004, 279, 5405–5412. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Sangawa, A.; Shintani, M.; Yamao, N.; Kamoshida, S. Phosphorylation status of Akt and caspase-9 in gastric and colorectal carcinomas. Int. J. Clin. Exp. Pathol. 2014, 7, 3312–3317. [Google Scholar]
- Chin, Y.R.; Toker, A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell. Signal. 2009, 21, 470–476. [Google Scholar] [CrossRef]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling in control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Jiang, B.H.; Liu, L.Z. Akt signaling in regulating angiogenesis. Curr. Cancer Drug Targets 2008, 8, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Miyakoda, G.; Hirose, Y.; Mori, T. Activation of endothelial nitric oxide synthase by cilostazol via a cAMP/protein kinase A- and phosphatidylinositol 3-kinase/Akt-dependent mechanism. Atherosclerosis 2006, 189, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Restuccia, D.F.; Lan, Q.; Hynx, D.; Dirnhofer, S.; Hess, D.; Ruegg, C.; Hemmings, B.A. Akt/ PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Discov. 2012, 2, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor cell invadopodia: Invasive protrusions that orchestrate metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef]
- Fukumoto, M.; Ijuin, T.; Takenawa, T. PI(3,4)P2 plays critical roles in the regulation of focal adhesion dynamics of MDA-MB-231 breast cancer cells. Cancer Sci. 2017, 108, 941–951. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Yoshida, S.; Muroi, E.; Yoshida, N.; Kawamura, M.; Kouchi, Z.; Nakamura, Y.; Sakai, R.; Fukami, K. Phosphoinositide 3-kinase signaling pathway mediated by p110α regulates invadopodia formation. J. Cell Biol. 2011, 193, 1275–1288. [Google Scholar] [CrossRef]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-Akt network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Papadopoli, D.; Pollak, M.; Topisirovic, I. The role of GSK3 in metabolic pathway perturbations in cancer. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2021, 1868, 119059. [Google Scholar] [CrossRef] [PubMed]
- Taha, C.; Liu, Z.; Jin, J.; Al-Hasani, H.; Sonenberg, N.; Klip, A. Opposite Translational Control of glut1 and glut4 glucose transporter mRNAs in response to insulin role of mammalian target of rapamycin, protein kinase b, and phosphatidylinositol 3-kinase in glut1 mRNA TRANSLATION. J. Biol. Chem. 1999, 274, 33085–33091. [Google Scholar] [CrossRef] [PubMed]
- Picton, C.; Woodgett, J.; Hemmings, B.; Cohen, P. Multisite phosphorylation of glycogen synthase from rabbit skeletal muscle. Phosphorylation of site 5 by glycogen synthase kinase-5 (casein kinase-II) is a prerequisite for phosphorylation of sites 3 by glycogen synthase kinase-3. FEBS Lett. 1982, 150, 191–196. [Google Scholar] [CrossRef]
- Hunter, R.W.; Treebak, J.T.; Wojtaszewski, J.F.; Sakamoto, K. Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 2011, 60, 766–774. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P.; Biondi, R.M. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 2001, 7, 1321–1327. [Google Scholar] [CrossRef]
- Hochstrasser, M. Evolution and function of ubiquitin-like protein-conjugation systems. Nat. Cell Biol. 2000, 2, E153–E157. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- Merbl, Y.; Refour, P.; Patel, H.; Springer, M.; Kirschner, M.W. Profiling of ubiquitin-like modifications reveals features of mitotic control. Cell 2013, 152, 1160. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Z.; Ou, J.; Xia, X.; Zhi, F.; Cui, J. The F-box protein FBXL18 promotes glioma progression by promoting K63-linked ubiquitination of Akt. FEBS Lett. 2017, 591, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Long, J.; Gao, Y.; Zhang, W.; Han, F.; Xu, C.; Sun, L.; Yang, S.C.; Lan, J.; Hou, Z.; et al. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat. Cell Biol. 2019, 21, 214–225. [Google Scholar] [CrossRef]
- Li, H.; Lan, J.; Wang, G.; Guo, K.; Han, C.; Li, X.; Hu, J.; Cao, Z.; Luo, X. KDM4B facilitates colorectal cancer growth and glucose metabolism by stimulating TRAF6-mediated AKT activation. J. Exp. Clin. Cancer Res. 2020, 39, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Balzerano, A.; Pisani, A.; Paccosi, E.; Filippi, S.; Sugoni, C.; Balajee, A.S.; Proietti-De-Santis, L. The novel role of E3-ubiquitin ligase CSA in Akt activation highlights the implication of CSA-Akt axis in premature aging and cancer. Proc. Natl. Acad. Sci. USA 2023. submitted for publication. [Google Scholar]
- Niu, T.; Wu, Z.; Xiao, W. Uev1A promotes breast cancer cell migration by up-regulating CT45A expression via the AKT pathway. BMC Cancer 2021, 21, 1012. [Google Scholar] [CrossRef]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.D.; Lum, M.A.; Xu, C.; Black, J.D.; Wang, X. Ubiquitin-dependent regulation of phospho-AKT dynamics by the ubiquitin E3 ligase, NEDD4-1, in the insulin-like growth factor-1 response. J. Biol. Chem. 2013, 288, 1674–1684. [Google Scholar] [CrossRef]
- Xiang, T.; Ohashi, A.; Huang, Y.; Pandita, T.K.; Ludwig, T.; Powell, S.N.; Yang, Q. Negative Regulation of AKT Activation by BRCA1. Cancer Res. 2008, 68, 10040–10044. [Google Scholar] [CrossRef]
- Bae, S.; Kim, S.Y.; Jung, J.H.; Yoon, Y.; Cha, H.J.; Lee, H.; Kim, K.; Kim, J.; An, I.S.; Kim, J.; et al. Akt is negatively regulated by the MULAN E3 ligase. Cell Res. 2012, 22, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.M.; Kim, J.Y.; Jeong, J.B.; Seong, K.M.; Nam, S.Y.; Yang, K.H.; Kim, C.S.; Kim, H.S.; Jeong, M.; An, S.; et al. Ret finger protein 2 enhances ionizing radiation-induced apoptosis via degradation of AKT and MDM2. Eur. J. Cell Biol. 2011, 90, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Su, C.H.; Wang, C.Y.; Lan, K.H.; Li, C.P.; Chao, Y.; Lin, H.C.; Lee, S.D.; Lee, W.P. Akt phosphorylation at Thr308 and Ser473 is required for CHIP-mediated ubiquitination of the kinase. Cell. Signal. 2011, 23, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Suizu, F.; Hiramuki, Y.; Okumura, F.; Matsuda, M.; Okumura, A.J.; Hirata, N.; Narita, M.; Kohno, T.; Yokota, J.; Bohgaki, M.; et al. The E3 Ligase TTC3 Facilitates Ubiquitination and Degradation of Phosphorylated Akt. Dev. Cell 2009, 17, 800–810. [Google Scholar] [CrossRef]
- Wakatsuki, S.; Saitoh, F.; Araki, T. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat. Cell Biol. 2011, 13, 1415–1423. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Y.; Gao, C.; Sun, Y.; Zhou, K.; Wang, P.; Cheng, J.; Guo, W.; Ya, C.; Fan, J.; et al. USP1 Maintains the Survival of Liver Circulating Tumor Cells by Deubiquitinating and Stabilizing TBLR1. Front. Oncol. 2020, 25, 554809. [Google Scholar] [CrossRef]
- Goldbraikh, D.; Neufeld, D.; Eid-Mutlak, Y.; Lasry, I.; Gilda, J.E.; Parnis, A.; Cohen, S. USP1 deubiquitinates Akt to inhibit PI3K-Akt-FoxO signaling in muscle during prolonged starvation. EMBO Rep. 2020, 21, e48791. [Google Scholar] [CrossRef]
- Lim, J.H.; Jono, H.; Komatsu, K.; Woo, C.H.; Lee, J.; Miyata, M.; Matsuno, T.; Xu, X.; Huang, Y.; Zhang, W.; et al. CYLD negatively regulates transforming growth factor-β-signalling via deubiquitinating Akt. Nat. Commun. 2012, 3, 771. [Google Scholar] [CrossRef]
- Yin, F.; He, H.; Zhang, B.; Zheng, J.; Wang, M.; Zhang, M.; Cui, H. Effect of Deubiquitinase Ovarian Tumor Domain-Containing Protein 5 (OTUD5) on Radiosensitivity of Cervical Cancer by Regulating the Ubiquitination of Akt and its Mechanism. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 3469–3475. [Google Scholar] [CrossRef]
- Yang, W.L.; Jin, G.; Li, C.F.; Jeong, Y.S.; Moten, A.; Xu, D.; Feng, Z.; Chen, W.; Cai, Z.; Darnay, B.; et al. Cycles of ubiquitination and deubiquitination critically regulate growth factor-mediated activation of Akt signaling. Sci. Signal. 2013, 6, ra3. [Google Scholar] [CrossRef]
- Qiu, C.; Liu, K.; Zhang, S.; Gao, S.; Chen, W.; Li, D.; Huang, Y. Bisdemethoxycurcumin Inhibits Hepatocellular Carcinoma Proliferation Through Akt Inactivation via CYLD-Mediated Deubiquitination. Drug Des. Dev. Ther. 2020, 14, 993–1001. [Google Scholar] [CrossRef]
- Garcia-Echeverria, C.; Sellers, W.R. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene 2008, 27, 5511–5526. [Google Scholar] [CrossRef]
- Hua, H.; Zhang, H.; Chen, J.; Wang, J.; Liu, J.; Jiang, Y. Targeting Akt in cancer for precision therapy. J. Hematol. Oncol. 2021, 14, 128. [Google Scholar] [CrossRef]
- Tsai, P.J.; Lai, Y.H.; Manne, R.K.; Tsai, Y.S.; Sarbassov, D.; Lin, H.K. Akt: A key transducer in cancer. J. Biomed. Sci. 2022, 29, 76. [Google Scholar] [CrossRef] [PubMed]
- Kondapaka, S.B.; Singh, S.S.; Dasmahapatra, G.P.; Sausville, E.A.; Roy, K.K. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol. Cancer Ther. 2003, 2, 1093–1103. [Google Scholar]
- Song, Z.; Tu, X.; Zhou, Q.; Huang, J.; Chen, Y.; Liu, J.; Lee, S.; Kim, W.; Nowsheen, S.; Luo, K.; et al. A novel UCHL3 inhibitor, perifosine, enhances PARP inhibitor cytotoxicity through inhibition of homologous recombination-mediated DNA double strand break repair. Cell Death Dis. 2019, 10, 398. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Jabeen, I. Pharmacoinformatic Approaches to Design Novel Inhibitors of Protein Kinase B Pathways in Cancer. Curr. Cancer Drug Targets 2018, 18, 830–846. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Lo, P.K.; Li, Y.; Loison, F.; Green, S.; Wang, J.; Silberstein, L.E.; Ye, K.; Chen, H.; Luo, H.R. Deactivation of Akt by a small molecule inhibitor targeting pleckstrin homology domain and facilitating Akt ubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 6486–6491. [Google Scholar] [CrossRef]
- Li, J.; Liu, N.; Tang, L.; Yan, B.; Chen, X.; Zhang, J.; Peng, C. The relationship between TRAF6 and tumors. Cancer Cell Int. 2020, 20, 429. [Google Scholar] [CrossRef]
- Hou, J.; Wang, P.; Lin, L.; Liu, X.; Ma, F.; An, H.; Wang, Z.; Cao, X. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 2009, 183, 2150–2158. [Google Scholar] [CrossRef]
- Hurst, D.R.; Edmonds, M.D.; Scott, G.K.; Benz, C.C.; Vaidya, K.S.; Welch, D.R. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer Res. 2009, 69, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; Wells, R.A.; et al. Identification of miR-145 and miR-146a as mediators of the 5q-syndrome phenotype. Nat. Med. 2010, 16, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, J.; Li, H.; Sun, C.; Yu, L.; Li, Y.; Shi, C.; Zhou, X.; Bian, X.; Ping, Y.; et al. miR-146b-5p functions as a tumor suppressor by targeting TRAF6 and predicts the prognosis of human gliomas. Oncotarget 2015, 6, 29129–29142. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Zhang, W.; Xu, X.; Li, J.; Mu, H.; Liu, X.; Qin, L.; Zhu, X.; Zheng, M. The effects of TRAF6 on proliferation, apoptosis and invasion in osteosarcoma are regulated by miR-124. Int. J. Mol. Med. 2018, 41, 2968–2976. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Lei, L.; Hui, H.; Tao, Z. MicroRNA-124 regulates TRAF6 expression and functions as an independent prognostic factor in colorectal cancer. Oncol Lett. 2019, 18, 856–863. [Google Scholar] [CrossRef]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell. 2013, 51, 283–296. [Google Scholar] [CrossRef]
- Hongming, H.; Jian, H. Bortezomib inhibits maturation and function of osteoclasts from PBMCs of patients with multiple myeloma by downregulating TRAF6. Leuk. Res. 2009, 33, 115–122. [Google Scholar] [CrossRef]
- Chiu, H.W.; Lin, S.W.; Lin, L.C.; Hsu, Y.H.; Lin, Y.F.; Ho, S.Y.; Wu, Y.H.; Wang, Y.J. Synergistic antitumor effects of radiation and proteasome inhibitor treatment in pancreatic cancer through the induction of autophagy and the downregulation of TRAF6. Cancer Lett. 2015, 365, 229–239. [Google Scholar] [CrossRef]
- Brenke, J.K.; Popowicz, G.M.; Schorpp, K.; Rothenaigner, I.; Roesner, M.; Meininger, I.; Kalinski, C.; Ringelstetter, L.; R’kyek, O.; Jürjens, G.; et al. Targeting TRAF6 E3 ligase activity with a small-molecule inhibitor combats autoimmunity. J. Biol. Chem. 2018, 293, 13191–13203. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, Z.; Huang, Z.; Zhang, X.; Zhou, Y.; Luo, Z.; Zeng, W.; Su, J.; Peng, C.; Chen, X. Epigallocatechin-3-gallate(EGCG) suppresses melanoma cell growth and metastasis by targeting TRAF6 activity. Oncotarget 2016, 7, 79557–79571. [Google Scholar] [CrossRef]
- Khusbu, F.Y.; Zhou, X.; Roy, M.; Chen, F.Z.; Cao, Q.; Chen, H.C. Resveratrol induces depletion of TRAF6 and suppresses prostate cancer cell proliferation and migration. Int. J. Biochem. Cell Biol. 2020, 118, 105644. [Google Scholar] [CrossRef]
- Qi, Z.; Xu, Y.; Wang, X.; Wang, S.; Zhang, Q.; Wang, Z.; Gao, Q. TLR13, TLR22, TRAF6, and TAK1 in the soiny mullet (Liza haematocheila): Molecular characterization and expression profiling analysis. Dev. Comp. Immunol. 2020, 112, 103774. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Chen, Z.; Wang, G.; Nardella, C.; Lee, S.W.; Chan, C.H.; Yang, W.L.; Wang, J.; Egia, A.; Nakayama, K.I.; et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 2010, 464, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Iqbal, N.J.; Sukrithan, V.; Nicholas, C.; Xue, Y.; Yu, C.; Locker, J.; Zou, J.; Schwartz, E.L.; Zhu, L. Targeted Inhibition of the E3 Ligase SCFSkp2/Cks1 Has Antitumor Activity in RB1-Deficient Human and Mouse Small-Cell Lung Cancer. Cancer Res. 2020, 80, 2355–2367. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Bauzon, F.; Fu, H.; Lu, Z.; Cui, J.; Nakayama, K.; Nakayama, K.I.; Locker, J.; Zhu, L. Skp2 deletion unmasks a p27 safeguard that blocks tumorigenesis in the absence of pRb and p53 tumor suppressors. Cancer Cell. 2013, 24, 645–659. [Google Scholar] [CrossRef]
- Chan, C.H.; Morrow, J.K.; Li, C.F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.E.; Cai, Z.; Xu, D.; et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef]
- Jiang, W.; Lin, M.; Wang, Z. Dioscin: A new potential inhibitor of Skp2 for cancer therapy. EBioMedicine 2020, 51, 102593. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- You, I.; Erickson, E.C.; Donovan, K.A.; Eleuteri, N.A.; Fischer, E.S.; Gray, N.S.; Toker, A. Discovery of an Akt Degrader with Prolonged Inhibition of Downstream Signaling. Cell Chem Biol. 2020, 27, 66–73. [Google Scholar] [CrossRef]
- Yu, X.; Xu, J.; Shen, Y.; Cahuzac, K.M.; Park, K.S.; Dale, B.; Liu, J.; Parsons, R.E.; Jin, J. Discovery of Potent, Selective, and In Vivo Efficacious Akt Kinase Protein Degraders via Structure-Activity Relationship Studies. J. Med. Chem. 2022, 65, 3644–3666. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paccosi, E.; Balzerano, A.; Proietti-De-Santis, L. Interfering with the Ubiquitin-Mediated Regulation of Akt as a Strategy for Cancer Treatment. Int. J. Mol. Sci. 2023, 24, 2809. https://doi.org/10.3390/ijms24032809
Paccosi E, Balzerano A, Proietti-De-Santis L. Interfering with the Ubiquitin-Mediated Regulation of Akt as a Strategy for Cancer Treatment. International Journal of Molecular Sciences. 2023; 24(3):2809. https://doi.org/10.3390/ijms24032809
Chicago/Turabian StylePaccosi, Elena, Alessio Balzerano, and Luca Proietti-De-Santis. 2023. "Interfering with the Ubiquitin-Mediated Regulation of Akt as a Strategy for Cancer Treatment" International Journal of Molecular Sciences 24, no. 3: 2809. https://doi.org/10.3390/ijms24032809
APA StylePaccosi, E., Balzerano, A., & Proietti-De-Santis, L. (2023). Interfering with the Ubiquitin-Mediated Regulation of Akt as a Strategy for Cancer Treatment. International Journal of Molecular Sciences, 24(3), 2809. https://doi.org/10.3390/ijms24032809