

Hyperpolarized 13C-Pyruvate to Assess Response to Anti-PD1 Immune Checkpoint Inhibition in YUMMER 1.7 Melanoma Xenografts
 ,
,  ,
,  , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
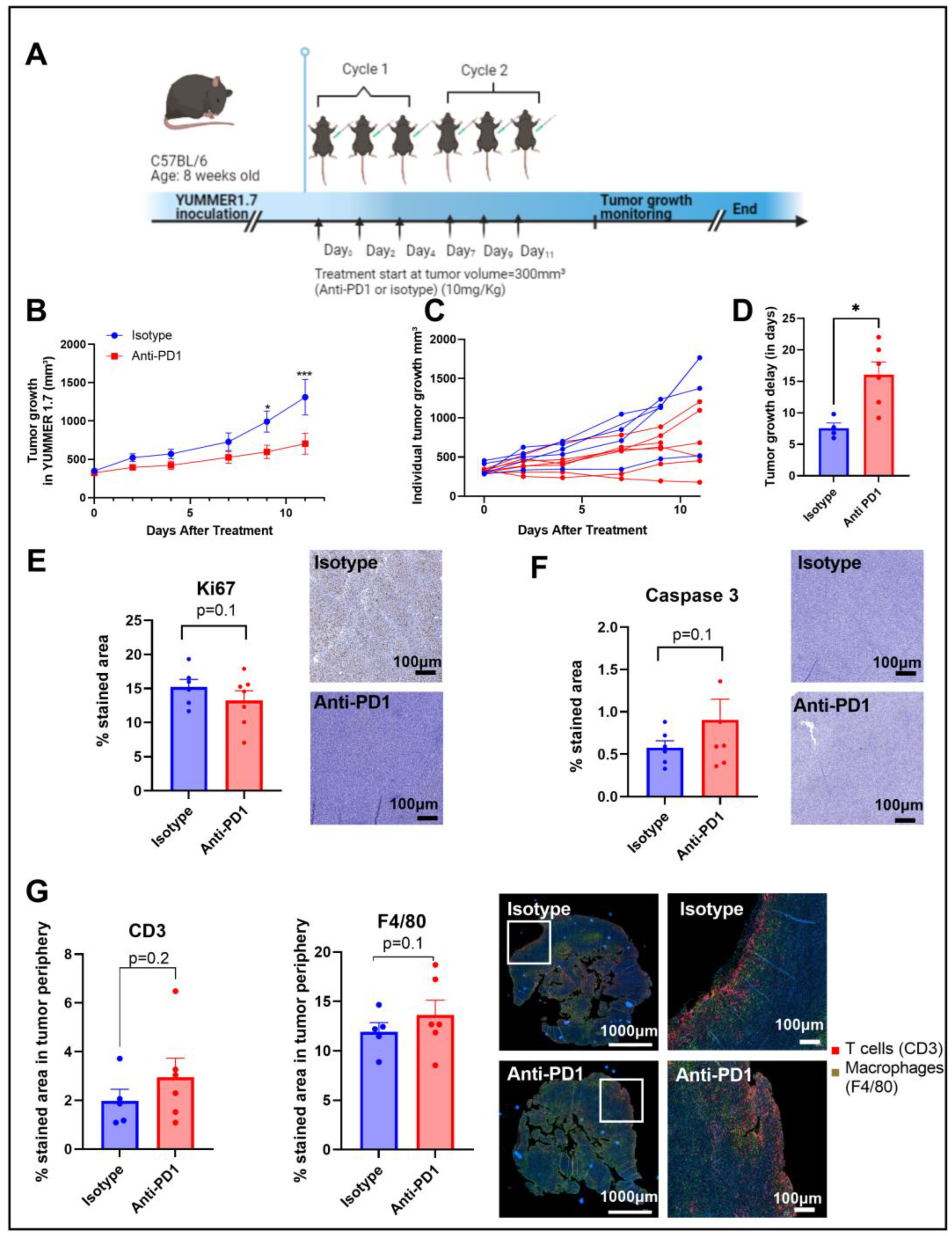
2.1. Anti-PD1 Monoclonal Antibody Delays Tumor Growth in Syngeneic YUMMER1.7 Melanoma Xenografts
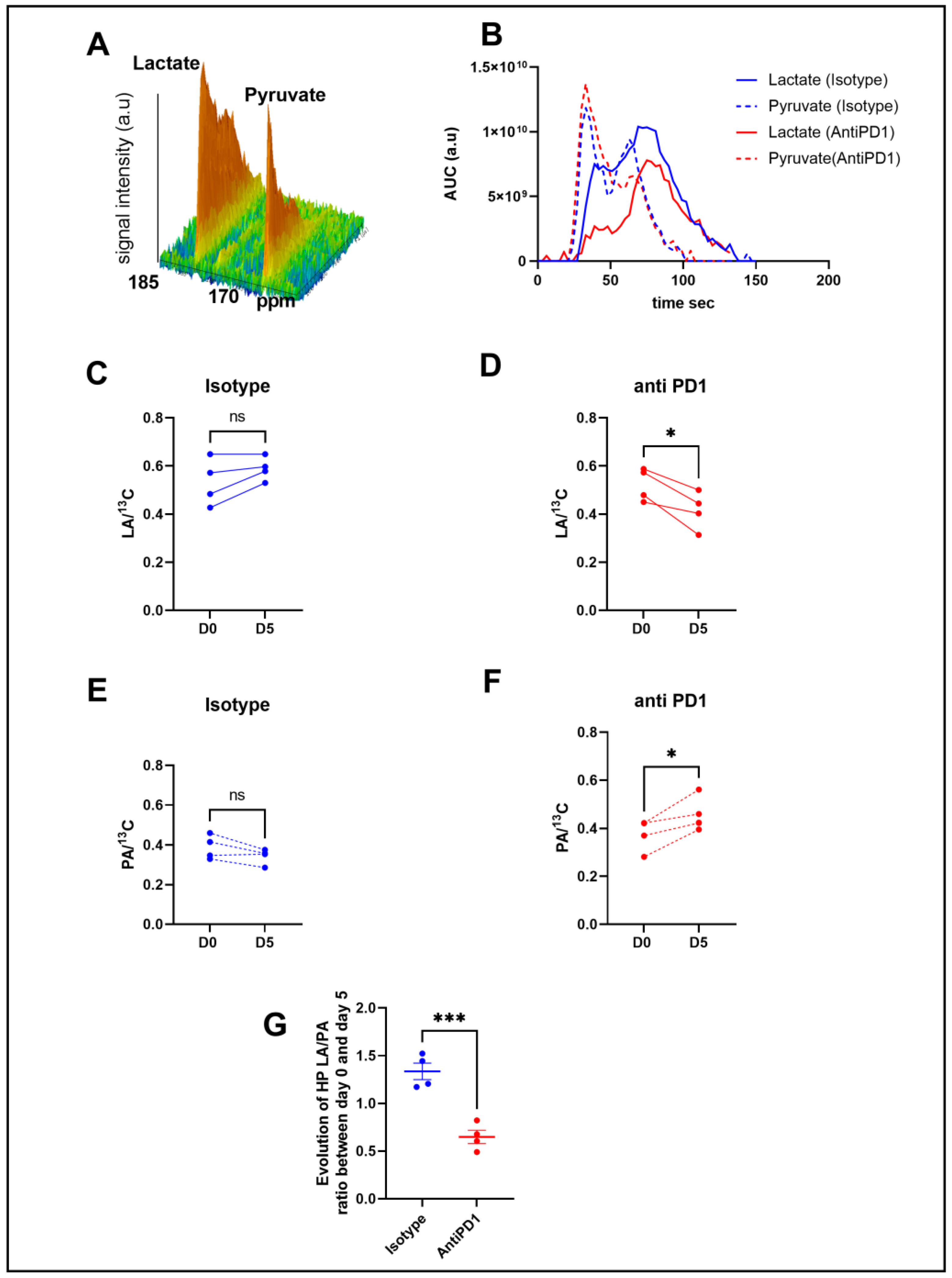
2.2. Hyperpolarized 13C-Pyruvate Detects Real-Time Metabolic Changes Induced by Anti-PD1 In Vivo
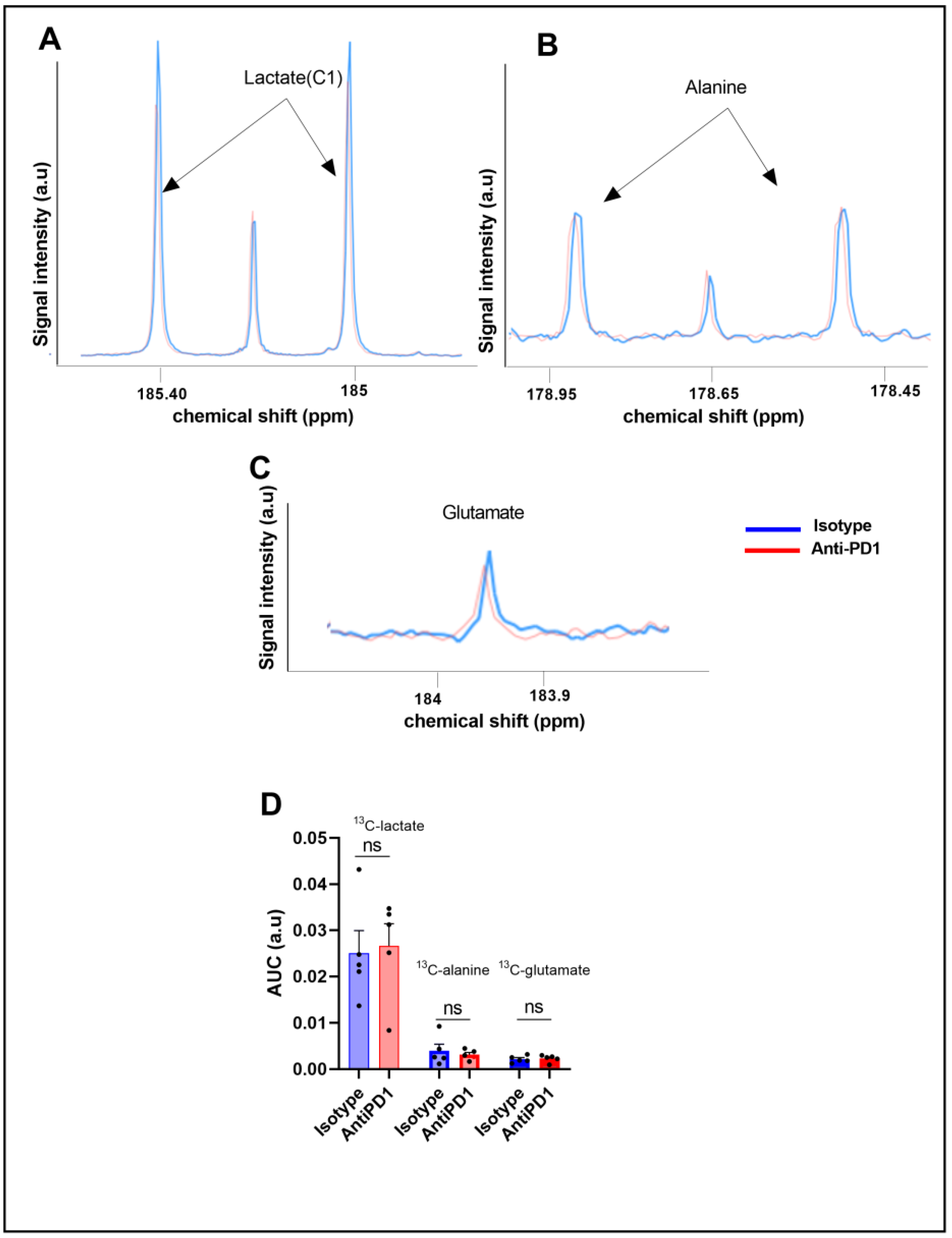
2.3. Ex Vivo Metabolic Profiling Using 13C-Glucose Tracing Is Not Modified in Response to Anti-PD1 Treatment
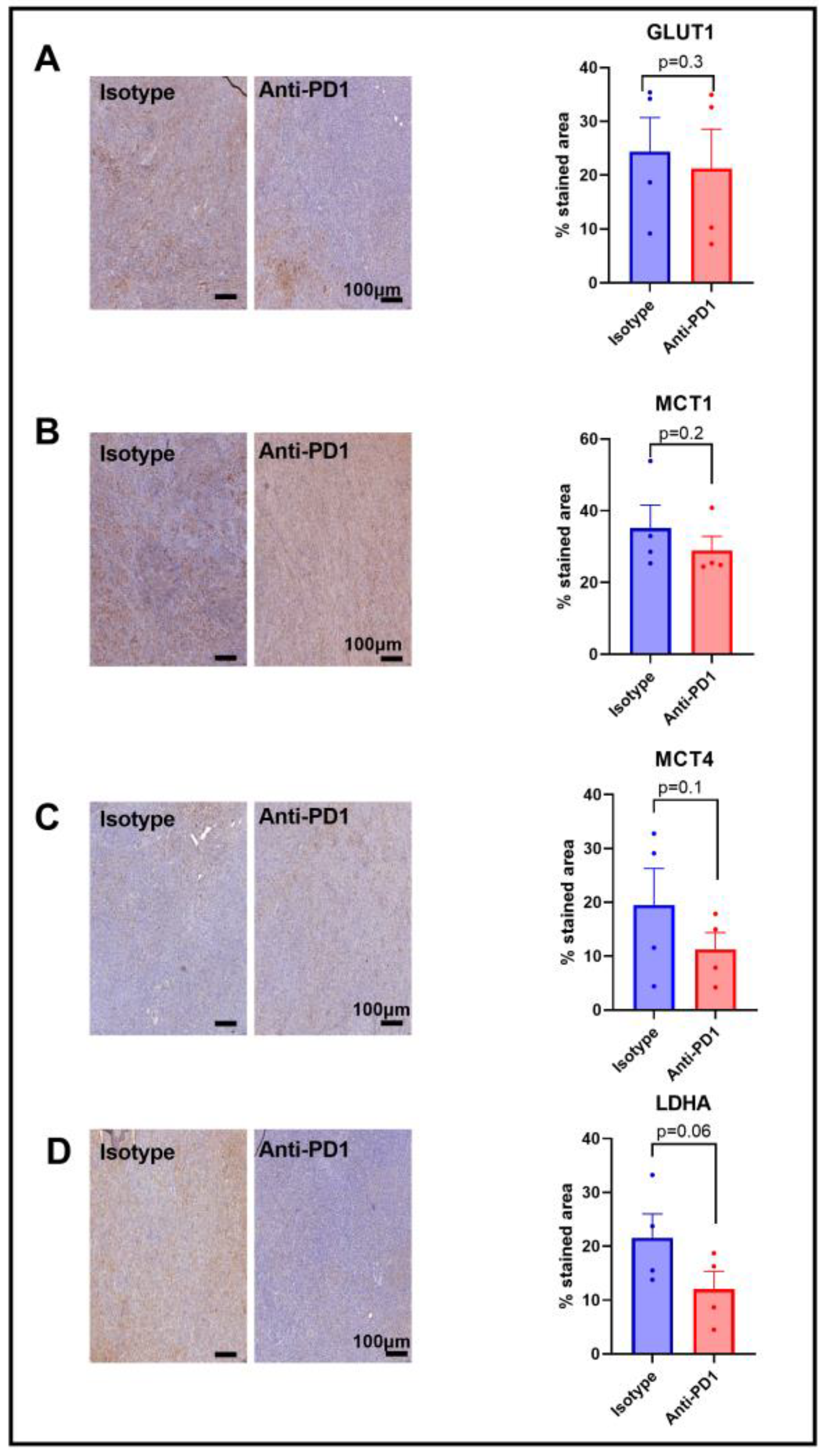
2.4. Expression of Metabolic Transporters and LDH-A in Response to Anti-PD1
3. Discussion
4. Materials and Methods
4.1. Mice and Tumors
4.2. In Vivo Checkpoint Blockade Treatment
4.3. Tissue Fixation, Freezing and IHC Stainings
4.4. Multiplex Immunofluorescence (mIF)
4.5. Computer-Assisted Quantitative Evaluation of Immunostaining in Whole Tissue Sections
4.6. Hyperpolarized 13C-MRS
4.7. U-13Cglucose Administration
4.8. Metabolite Extraction and 13C-MRS
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthews, N.H.; Li, W.-Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-4-0. [Google Scholar]
- Pasquali, S.; Hadjinicolaou, A.V.; Chiarion Sileni, V.; Rossi, C.R.; Mocellin, S. Systemic Treatments for Metastatic Cutaneous Melanoma. Cochrane Database Syst. Rev. 2018, 2, CD011123. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Prieto, P.A.; Reuben, A.; Cooper, Z.A.; Wargo, J.A. Targeted Therapies Combined with Immune Checkpoint Therapy. Cancer J. Sudbury Mass 2016, 22, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted Agents and Immunotherapies: Optimizing Outcomes in Melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A Review of Cancer Immunotherapy: From the Past, to the Present, to the Future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef]
- Haanen, J.B.A.G.; Robert, C. Immune Checkpoint Inhibitors. Prog. Tumor Res. 2015, 42, 55–66. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Robert, C. CTLA-4 and PD-1/PD-L1 Blockade: New Immunotherapeutic Modalities with Durable Clinical Benefit in Melanoma Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5300–5309. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Walunas, T.L.; Bluestone, J.A. CD28/B7 System of T Cell Costimulation. Annu. Rev. Immunol. 1996, 14, 233–258. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer Immunotherapy Comes of Age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L.A. Keytruda (Pembrolizumab): First PD-1 Inhibitor Approved for Previously Treated Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 96–100. [Google Scholar] [PubMed]
- Raedler, L.A. Opdivo (Nivolumab): Second PD-1 Inhibitor Receives FDA Approval for Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 180–183. [Google Scholar] [PubMed]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring Immune-Checkpoint Blockade: Response Evaluation and Biomarker Development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Hope, H.C.; Salmond, R.J. Targeting the Tumor Microenvironment and T Cell Metabolism for Effective Cancer Immunotherapy. Eur. J. Immunol. 2019, 49, 1147–1152. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Almuhaideb, A.; Papathanasiou, N.; Bomanji, J. 18F-FDG PET/CT Imaging in Oncology. Ann. Saudi Med. 2011, 31, 3–13. [Google Scholar] [CrossRef]
- McArthur, G.A.; Puzanov, I.; Amaravadi, R.; Ribas, A.; Chapman, P.; Kim, K.B.; Sosman, J.A.; Lee, R.J.; Nolop, K.; Flaherty, K.T.; et al. Marked, Homogeneous, and Early [18F]Fluorodeoxyglucose-Positron Emission Tomography Responses to Vemurafenib in BRAF-Mutant Advanced Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 1628–1634. [Google Scholar] [CrossRef] [PubMed]
- Carlino, M.S.; Saunders, C.A.B.; Haydu, L.E.; Menzies, A.M.; Martin Curtis, C.; Lebowitz, P.F.; Kefford, R.F.; Long, G.V. 18F-Labelled Fluorodeoxyglucose–Positron Emission Tomography (FDG–PET) Heterogeneity of Response Is Prognostic in Dabrafenib Treated BRAF Mutant Metastatic Melanoma. Eur. J. Cancer 2013, 49, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Lipson, E.J.; Im, H.-J.; Rowe, S.P.; Gonzalez, E.M.; Blackford, A.; Chirindel, A.; Pardoll, D.M.; Topalian, S.L.; Wahl, R.L. Prediction of Response to Immune Checkpoint Inhibitor Therapy Using Early-Time-Point 18F-FDG PET/CT Imaging in Patients with Advanced Melanoma. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 1421–1428. [Google Scholar] [CrossRef]
- Kaira, K.; Higuchi, T.; Naruse, I.; Arisaka, Y.; Tokue, A.; Altan, B.; Suda, S.; Mogi, A.; Shimizu, K.; Sunaga, N.; et al. Metabolic Activity by 18F-FDG-PET/CT Is Predictive of Early Response after Nivolumab in Previously Treated NSCLC. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 56–66. [Google Scholar] [CrossRef]
- Sachpekidis, C.; Larribere, L.; Pan, L.; Haberkorn, U.; Dimitrakopoulou-Strauss, A.; Hassel, J.C. Predictive Value of Early 18F-FDG PET/CT Studies for Treatment Response Evaluation to Ipilimumab in Metastatic Melanoma: Preliminary Results of an Ongoing Study. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 386–396. [Google Scholar] [CrossRef]
- Kong, B.Y.; Menzies, A.M.; Saunders, C.A.B.; Liniker, E.; Ramanujam, S.; Guminski, A.; Kefford, R.F.; Long, G.V.; Carlino, M.S. Residual FDG-PET Metabolic Activity in Metastatic Melanoma Patients with Prolonged Response to Anti-PD-1 Therapy. Pigment Cell Melanoma Res. 2016, 29, 572–577. [Google Scholar] [CrossRef]
- Bier, G.; Hoffmann, V.; Kloth, C.; Othman, A.E.; Eigentler, T.; Garbe, C.; La Fougère, C.; Pfannenberg, C.; Nikolaou, K.; Klumpp, B. CT Imaging of Bone and Bone Marrow Infiltration in Malignant Melanoma--Challenges and Limitations for Clinical Staging in Comparison to 18FDG-PET/CT. Eur. J. Radiol. 2016, 85, 732–738. [Google Scholar] [CrossRef]
- Nelson, S.J.; Vigneron, D.; Kurhanewicz, J.; Chen, A.; Bok, R.; Hurd, R. DNP-Hyperpolarized 13C Magnetic Resonance Metabolic Imaging for Cancer Applications. Appl. Magn. Reson. 2008, 34, 533–544. [Google Scholar] [CrossRef]
- Mäurer, J.; Tosi, C.; Hierholzer, J.; Bier, J.; Felix, J.; Lagalla, R.; Midiri, M.; Finazzo, M. The assessment of skin tumors with magnetic resonance with high resolution and a paramagnetic contrast medium. Radiol. Med. 1998, 95, 563–566. [Google Scholar]
- Shulman, R.G.; Brown, T.R.; Ugurbil, K.; Ogawa, S.; Cohen, S.M.; den Hollander, J.A. Cellular Applications of 31P and 13C Nuclear Magnetic Resonance. Science 1979, 205, 160–166. [Google Scholar] [CrossRef]
- Golman, K.; Zandt, R.I.; Lerche, M.; Pehrson, R.; Ardenkjaer-Larsen, J.H. Metabolic Imaging by Hyperpolarized 13C Magnetic Resonance Imaging for in Vivo Tumor Diagnosis. Cancer Res. 2006, 66, 10855–10860. [Google Scholar] [CrossRef] [PubMed]
- Kurhanewicz, J.; Vigneron, D.B.; Brindle, K.; Chekmenev, E.Y.; Comment, A.; Cunningham, C.H.; Deberardinis, R.J.; Green, G.G.; Leach, M.O.; Rajan, S.S.; et al. Analysis of Cancer Metabolism by Imaging Hyperpolarized Nuclei: Prospects for Translation to Clinical Research. Neoplasia 2011, 13, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Ros, S.; Wright, A.J.; D’Santos, P.; Hu, D.-E.; Hesketh, R.L.; Lubling, Y.; Georgopoulou, D.; Lerda, G.; Couturier, D.-L.; Razavi, P.; et al. Metabolic Imaging Detects Resistance to PI3Kα Inhibition Mediated by Persistent FOXM1 Expression in ER+ Breast Cancer. Cancer Cell 2020, 38, 516–533.e9. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Eskandari, R.; Ray, C.; Granlund, K.L.; Santos-Cunha, L.D.; Miloushev, V.Z.; Tee, S.S.; Jeong, S.; Aras, O.; Chen, Y.-B.; et al. Hyperpolarized MRI Visualizes Warburg Effects and Predicts Treatment Response to MTOR Inhibitors in Patient-Derived CcRCC Xenograft Models. Cancer Res. 2019, 79, 242–250. [Google Scholar] [CrossRef]
- Woitek, R.; McLean, M.A.; Ursprung, S.; Rueda, O.M.; Manzano Garcia, R.; Locke, M.J.; Beer, L.; Baxter, G.; Rundo, L.; Provenzano, E.; et al. Hyperpolarized Carbon-13 MRI for Early Response Assessment of Neoadjuvant Chemotherapy in Breast Cancer Patients. Cancer Res. 2021, 81, 6004–6017. [Google Scholar] [CrossRef]
- Wang, J.; Perry, C.J.; Meeth, K.; Thakral, D.; Damsky, W.; Micevic, G.; Kaech, S.; Blenman, K.; Bosenberg, M. UV-Induced Somatic Mutations Elicit a Functional T Cell Response in the YUMMER1.7 Mouse Melanoma Model. Pigment Cell Melanoma Res. 2017, 30, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Blenman, K.R.M.; Wang, J.; Cowper, S.; Bosenberg, M. Pathology of Spontaneous and Immunotherapy-Induced Tumor Regression in a Murine Model of Melanoma. Pigment Cell Melanoma Res. 2019, 32, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Larson, P.E.Z.; Harzstark, A.L.; Ferrone, M.; van Criekinge, M.; Chang, J.W.; Bok, R.; Park, I.; et al. Metabolic Imaging of Patients with Prostate Cancer Using Hyperpolarized [1-13C]Pyruvate. Sci. Transl. Med. 2013, 5, 198ra108. [Google Scholar] [CrossRef]
- Boland, G.M.; Flaherty, K.T. Tracking Early Response to Immunotherapy. Nat. Cancer 2020, 1, 160–162. [Google Scholar] [CrossRef]
- Palmer, C.S.; Ostrowski, M.; Balderson, B.; Christian, N.; Crowe, S.M. Glucose Metabolism Regulates T Cell Activation, Differentiation, and Functions. Front. Immunol. 2015, 6, 1. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Available online: https://pubmed.ncbi.nlm.nih.gov/13298683/ (accessed on 2 November 2022).
- Doherty, J.R.; Cleveland, J.L. Targeting Lactate Metabolism for Cancer Therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Kettunen, M.I.; Hu, D.; Witney, T.H.; McLaughlin, R.; Gallagher, F.A.; Bohndiek, S.E.; Day, S.E.; Brindle, K.M. Magnetization Transfer Measurements of Exchange between Hyperpolarized [1-13C]Pyruvate and [1-13C]Lactate in a Murine Lymphoma. Magn. Reson. Med. 2010, 63, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Gammon, S.; Zacharias, N.M.; Liu, T.; Salzillo, T.; Xi, Y.; Wang, J.; Bhattacharya, P.; Piwnica-Worms, D. Hyperpolarized [1- 13 C]Pyruvate-to-[1- 13 C]Lactate Conversion Is Rate-Limited by Monocarboxylate Transporter-1 in the Plasma Membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 22378–22389. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed]
- Oz, G.; Alger, J.R.; Barker, P.B.; Bartha, R.; Bizzi, A.; Boesch, C.; Bolan, P.J.; Brindle, K.M.; Cudalbu, C.; Dinçer, A.; et al. Clinical Proton MR Spectroscopy in Central Nervous System Disorders. Radiology 2014, 270, 658–679. [Google Scholar] [CrossRef] [PubMed]
- Aboubakar Nana, F.; Hoton, D.; Ambroise, J.; Lecocq, M.; Vanderputten, M.; Sibille, Y.; Vanaudenaerde, B.; Pilette, C.; Bouzin, C.; Ocak, S. Increased Expression and Activation of FAK in Small-Cell Lung Cancer Compared to Non-Small-Cell Lung Cancer. Cancers 2019, 11, 1526. [Google Scholar] [CrossRef]
- Acciardo, S.; Mignion, L.; Lacomblez, E.; Schoonjans, C.; Joudiou, N.; Gourgue, F.; Bouzin, C.; Baurain, J.-F.; Gallez, B.; Jordan, B.F. Metabolic Imaging Using Hyperpolarized 13 C-Pyruvate to Assess Sensitivity to the B-Raf Inhibitor Vemurafenib in Melanoma Cells and Xenografts. J. Cell. Mol. Med. 2020, 24, 1934–1944. [Google Scholar] [CrossRef]
- Farah, C.; Neveu, M.-A.; Yelek, C.; Bouzin, C.; Gallez, B.; Baurain, J.-F.; Mignion, L.; Jordan, B.F. Combined HP 13C Pyruvate and 13C-Glucose Fluxomic as a Potential Marker of Response to Targeted Therapies in YUMM1.7 Melanoma Xenografts. Biomedicines 2022, 10, 717. [Google Scholar] [CrossRef]
- Yuan, M.; Kremer, D.M.; Huang, H.; Breitkopf, S.B.; Ben-Sahra, I.; Manning, B.D.; Lyssiotis, C.A.; Asara, J.M. Ex Vivo and in Vivo Stable Isotope Labelling of Central Carbon Metabolism and Related Pathways with Analysis by LC–MS/MS. Nat. Protoc. 2019, 14, 313–330. [Google Scholar] [CrossRef]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.D.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic Profiling, Metabolomic and Metabonomic Procedures for NMR Spectroscopy of Urine, Plasma, Serum and Tissue Extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Schoonjans, C.A.; Joudiou, N.; Brusa, D.; Corbet, C.; Feron, O.; Gallez, B. Acidosis-Induced Metabolic Reprogramming in Tumor Cells Enhances the Anti-Proliferative Activity of the PDK Inhibitor Dichloroacetate. Cancer Lett. 2020, 470, 18–28. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farah, C.; Neveu, M.-A.; Bouzin, C.; Knezevic, Z.; Gallez, B.; Leucci, E.; Baurain, J.-F.; Mignion, L.; Jordan, B.F. Hyperpolarized 13C-Pyruvate to Assess Response to Anti-PD1 Immune Checkpoint Inhibition in YUMMER 1.7 Melanoma Xenografts. Int. J. Mol. Sci. 2023, 24, 2499. https://doi.org/10.3390/ijms24032499
Farah C, Neveu M-A, Bouzin C, Knezevic Z, Gallez B, Leucci E, Baurain J-F, Mignion L, Jordan BF. Hyperpolarized 13C-Pyruvate to Assess Response to Anti-PD1 Immune Checkpoint Inhibition in YUMMER 1.7 Melanoma Xenografts. International Journal of Molecular Sciences. 2023; 24(3):2499. https://doi.org/10.3390/ijms24032499
Chicago/Turabian StyleFarah, Chantale, Marie-Aline Neveu, Caroline Bouzin, Zorica Knezevic, Bernard Gallez, Eleonora Leucci, Jean-François Baurain, Lionel Mignion, and Bénédicte F. Jordan. 2023. "Hyperpolarized 13C-Pyruvate to Assess Response to Anti-PD1 Immune Checkpoint Inhibition in YUMMER 1.7 Melanoma Xenografts" International Journal of Molecular Sciences 24, no. 3: 2499. https://doi.org/10.3390/ijms24032499
APA StyleFarah, C., Neveu, M.-A., Bouzin, C., Knezevic, Z., Gallez, B., Leucci, E., Baurain, J.-F., Mignion, L., & Jordan, B. F. (2023). Hyperpolarized 13C-Pyruvate to Assess Response to Anti-PD1 Immune Checkpoint Inhibition in YUMMER 1.7 Melanoma Xenografts. International Journal of Molecular Sciences, 24(3), 2499. https://doi.org/10.3390/ijms24032499