
The Role of Copper in the Hydrogenation of Furfural and Levulinic Acid

 ,
,  , and
, and 
Abstract
1. Introduction
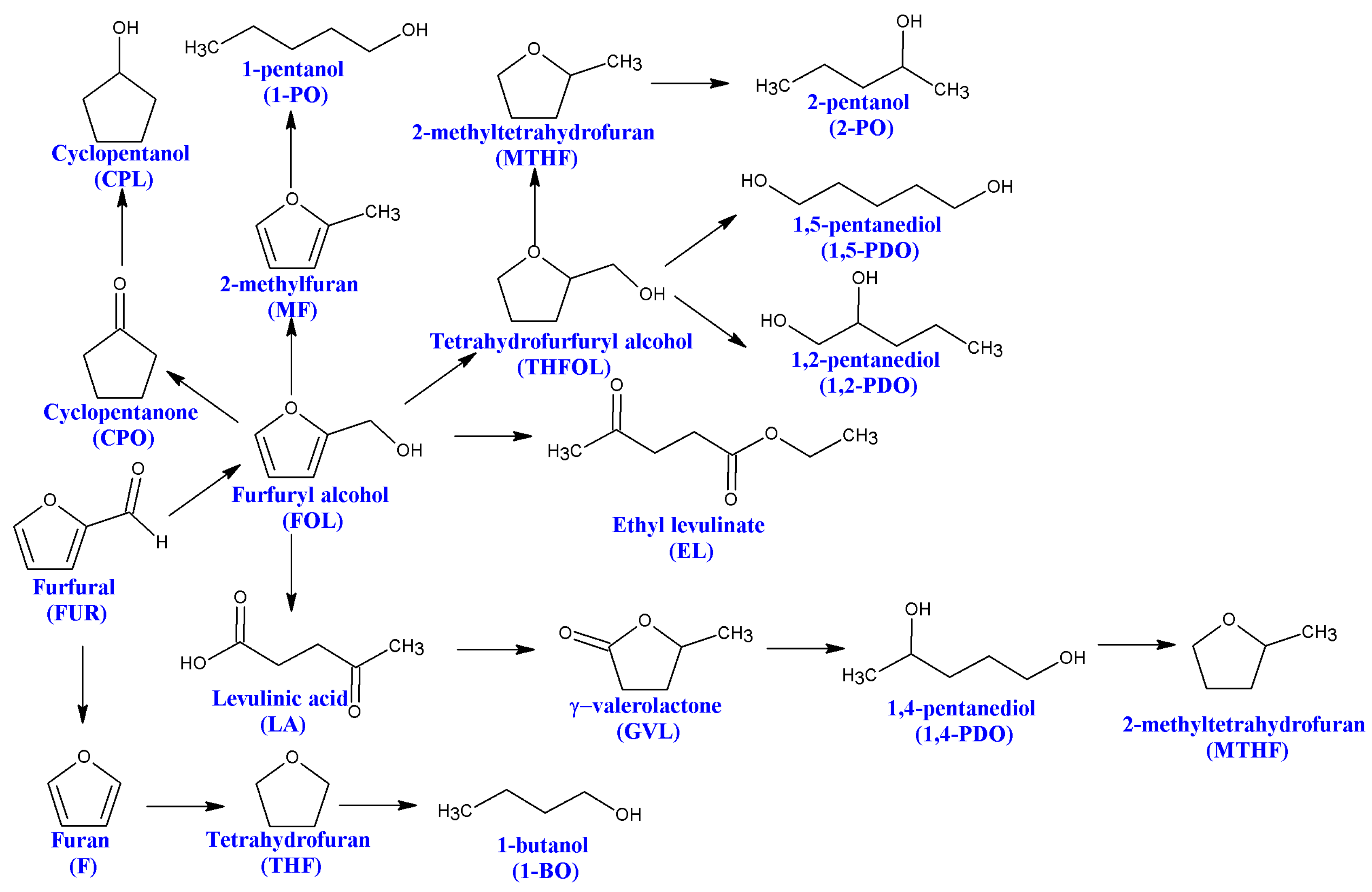
2. Hydrogenation of Furfural
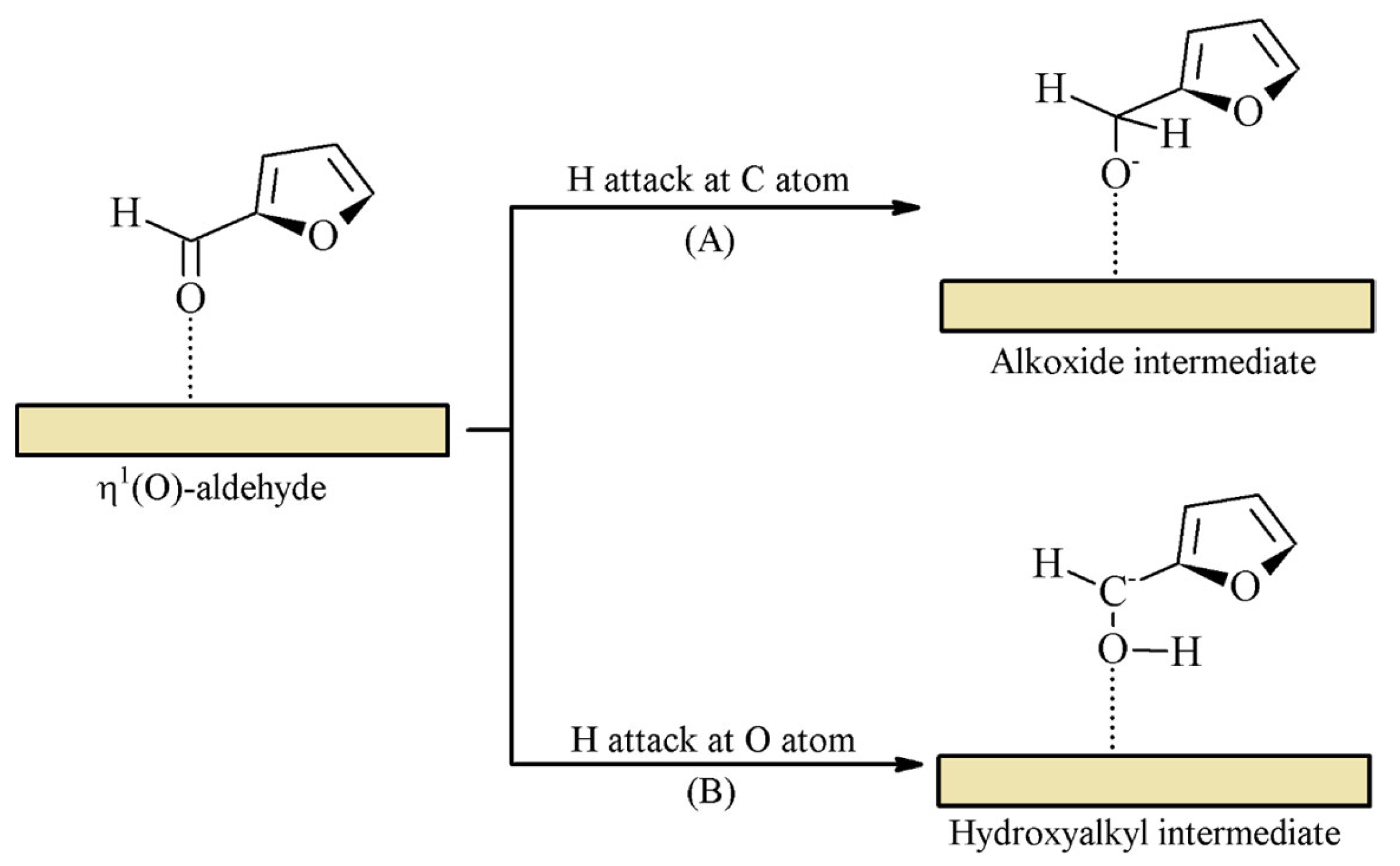
2.1. Hydrogenation of Furfural to Furfuryl Alcohol
2.2. Hydrogenation of Furfural through Catalytic Transfer Hydrogenation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Hydrogen Source | T (°C) | P (MPa) | t (h) | C a (%) | YFOL b (%) | Ref. |
|---|---|---|---|---|---|---|---|
| Cu/Fe2O3 | Isopropanol | 180 | 0.1 | 7.5 | 37.0 | 28.0 | [87] |
| Cu/Ac-SO3H | Isopropanol | 105 | 0.4 | 2 | >99.9 | >99.9 | [92] |
| Co–Cu/SBA-15 | Isopropanol | 170 | 2 | 4 | 80.1 | 79.6 | [93] |
| Co–Cu/SBA-15 | Isopropanol | 130 | 4 | 3 | - | 94.8 | [94] |
| Cu1Co5 | Isopropanol | 180 | 2 | 5 | 100 | 38.1 | [95] |
| Cu/MgO–Al2O3 | Isopropanol | 210 | - | 1 | >99.9 | 89.3 | [96] |
| Cu/SiO2 | Isopropanol | 110 | 1 | 4 | 66.3 | 66.3 | [97] |
| CuZnAl | Isopropanol | 110 | 1 | 4 | 48.1 | 48.1 | [97] |
| CuMgAl | Isopropanol | 110 | 1 | 4 | 100 | 100 | [97] |
| CuCr | Isopropanol | 110 | 1 | 4 | 93.2 | 93.2 | [97] |
| CuMgAl | Isopropanol | 150 | 1 | 6 | 100 | 100 | [98] |
| Cu/ZnO–Cr2O3-ZrO2 | Isopropanol | 170 | 2 | 3.5 | 100 | 96 | [99] |
| CuNi2.5@C | H2O | 130 | 5 | 5 | 71.6 | 16.7 | [100] |
| Cu/ZrO2 | Isopropanol | 220 | 0.1 | 4 | 98.9 | 38.7 | [101] |
| Cu–Ru/ZrO2 | Isopropanol | 220 | 0.1 | 4 | 100 | 36.3 | [101] |
| Cu–Ni/ZrO2 | Isopropanol | 220 | 0.1 | 4 | 100 | 33.7 | [101] |
| Cu@Pt | Isopropanol | 250 | 0.69 | 1.5 | 15.3 | 13.8 | [102] |
| Cu–Ni/Al2O3 | Isopropanol | 190 | 0.1 | 4 | 90 | 54 | [103] |
| Cu–Ni (bulk) | Decanol | 130 | 5 | 6 | 39 | 39 | [104] |
| Cu/Al2O3 | Methanol | 245 | 1 | 1.5 | >99 | 540 | [105] |
| Cu–Pd/C | 1,4-dioxane | 170 | 0.1 | 3 | 100 | 98.1 | [106] |
| Cu/MgAl2O4 | Formic acid | 210 | - | 1 | 90.0 | 89.1 | [107] |
| Cu–Ni/γ-Al2O3 c | Isopropanol | 130 | 4 | 4 | 92.6 | 86.7 | [108] |
| NiCu/Al2O3 | Isopropanol | 200 | 0.5 | 2 | >99.9 | 50.0 | [109] |
| CoCu/Al2O3 | Isopropanol | 200 | 0.5 | 2 | >99.9 | 63.0 | [109] |
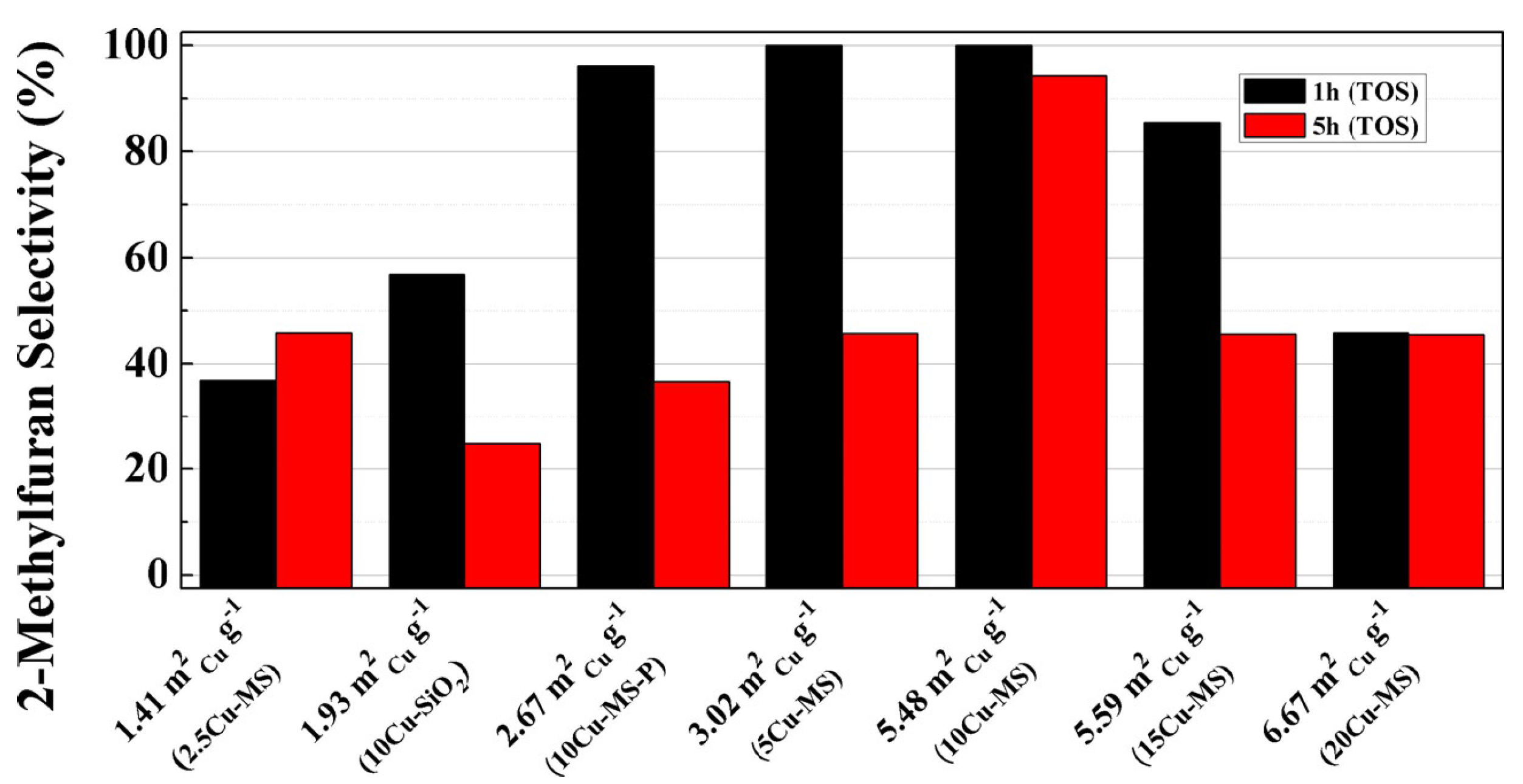
2.3. Hydrogenolysis of Furfural to 2-Methylfuran
2.3.1. Gas-Phase Hydrogenolysis of Furfural to 2-Methylfurane
2.3.2. Liquid-Phase Hydrogenolysis of Furfural to 2-Methylfurane
2.3.3. Conversion of Furfural to 2-Methylfurane through Catalytic Transfer Hydrogenation
| Catalyst | Hydrogen Source | T (°C) | t (h) | C a (%) | YMF b (%) | Ref. |
|---|---|---|---|---|---|---|
| CuPd/ZrO2 | Isopropanol | 220 | 4 | 100 | 61.9 | [101] |
| Cu–Zn–Al | 1,4-butanediol | 225 | 10 | 99.9 | 93.0 | [117] |
| Cu–Zn–Al | Cyclohexanol | 270 | 8 | 99.2 | 93.0 | [152] |
| Cu–Mn–Si | Cyclohexanol | 290 | 8 | 99.8 | 94.0 | [153] |
| Copper–chromite | 1,4-butanediol | 205 | 10 | 99.7 | 94.3 | [154] |
| Cu–FeOx | Isopropanol | 190 | 4 | 98.0 | 82.2 | [155] |
| NiCu/Al2O3 | Formic acid | 210 | 4 | 97.4 | 75.6 | [156] |
| CuRe/Al2O3 | Isopropanol | 220 | 4 | 100 | 94.0 | [157] |
| Cu–Zn–Al | Isopropanol | 180 | 4 | 96.0 | 72.0 | [158] |
| Cu/γAl2O3 | Isopropanol | 200 | 2 | 100 | 20.7 | [109] |
| CoCu/γ-Al2O3 | Isopropanol | 200 | 2 | 100 | 29.0 | [109] |
| NiCu/γ-Al2O3 | Isopropanol | 200 | 2 | 100 | 41.1 | [109] |
| Cuº/Cu2O·SiO2 | Methanol | 220 | 2 | 100 | 90.0 | [159] |
| CuO | Methanol | 240 | 4 | 95.0 | 60.0 | [160] |
| CuFe2O4 | Isopropanol | 200 | 1.5 | 99.4 | 97.0 | [161] |
2.4. Hydrogenation of Furfural to Cyclopentanone
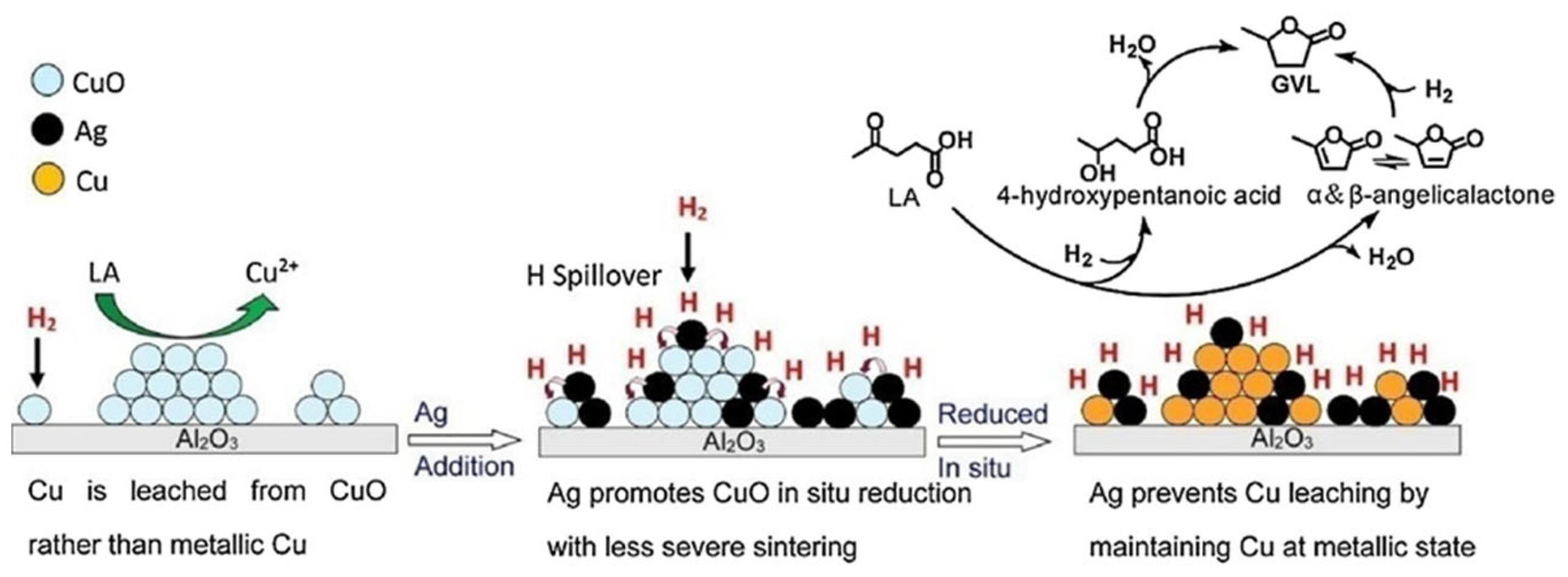
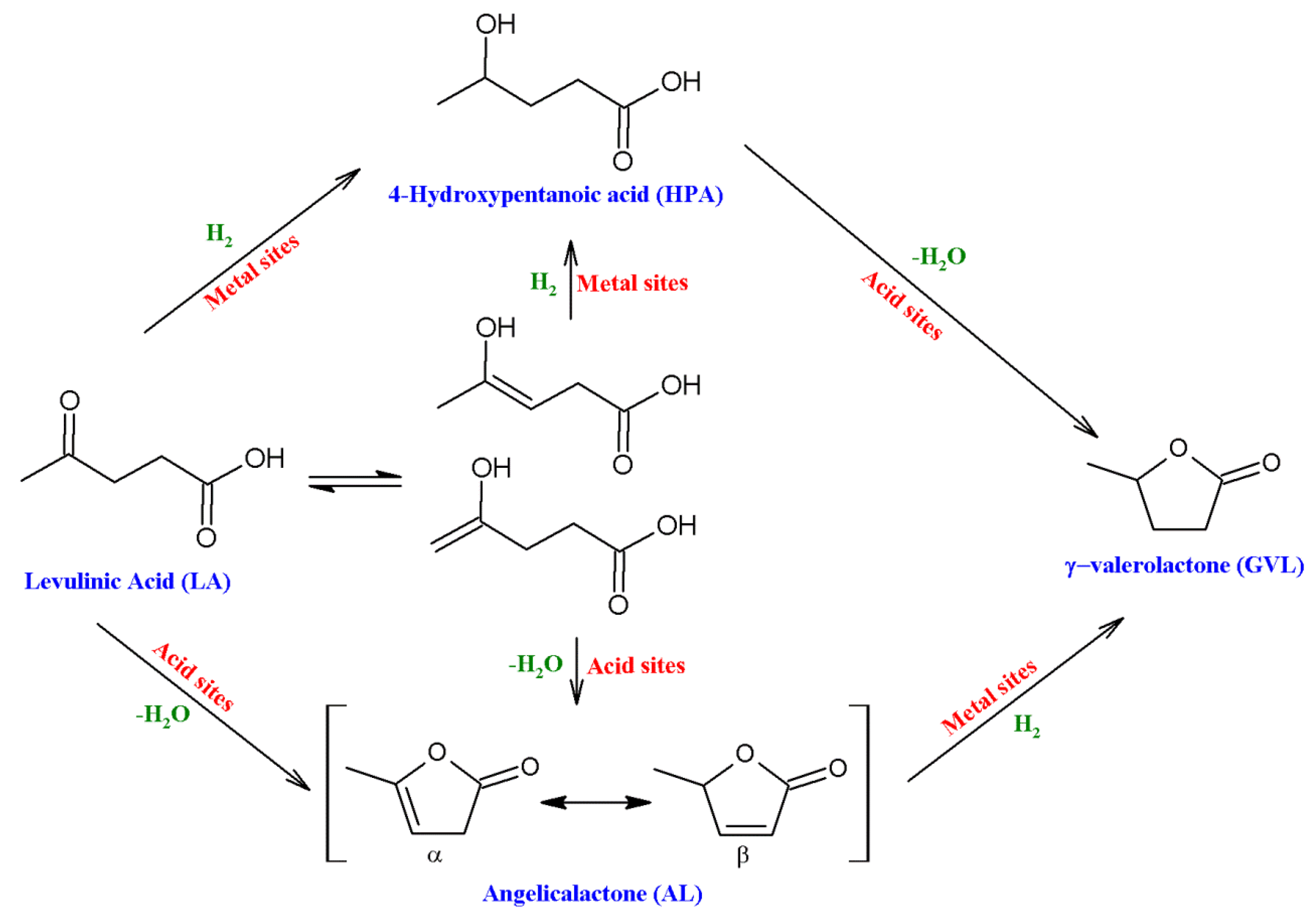
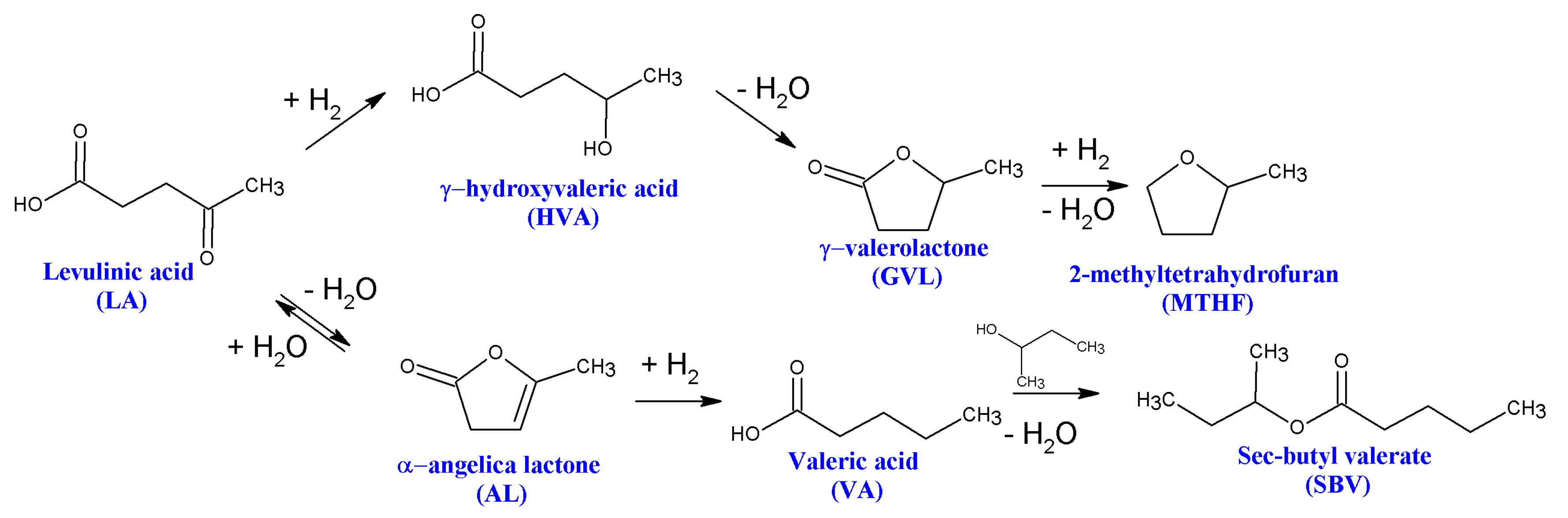
3. Hydrogenation of Levulinic Acid to γ-Valerolactone
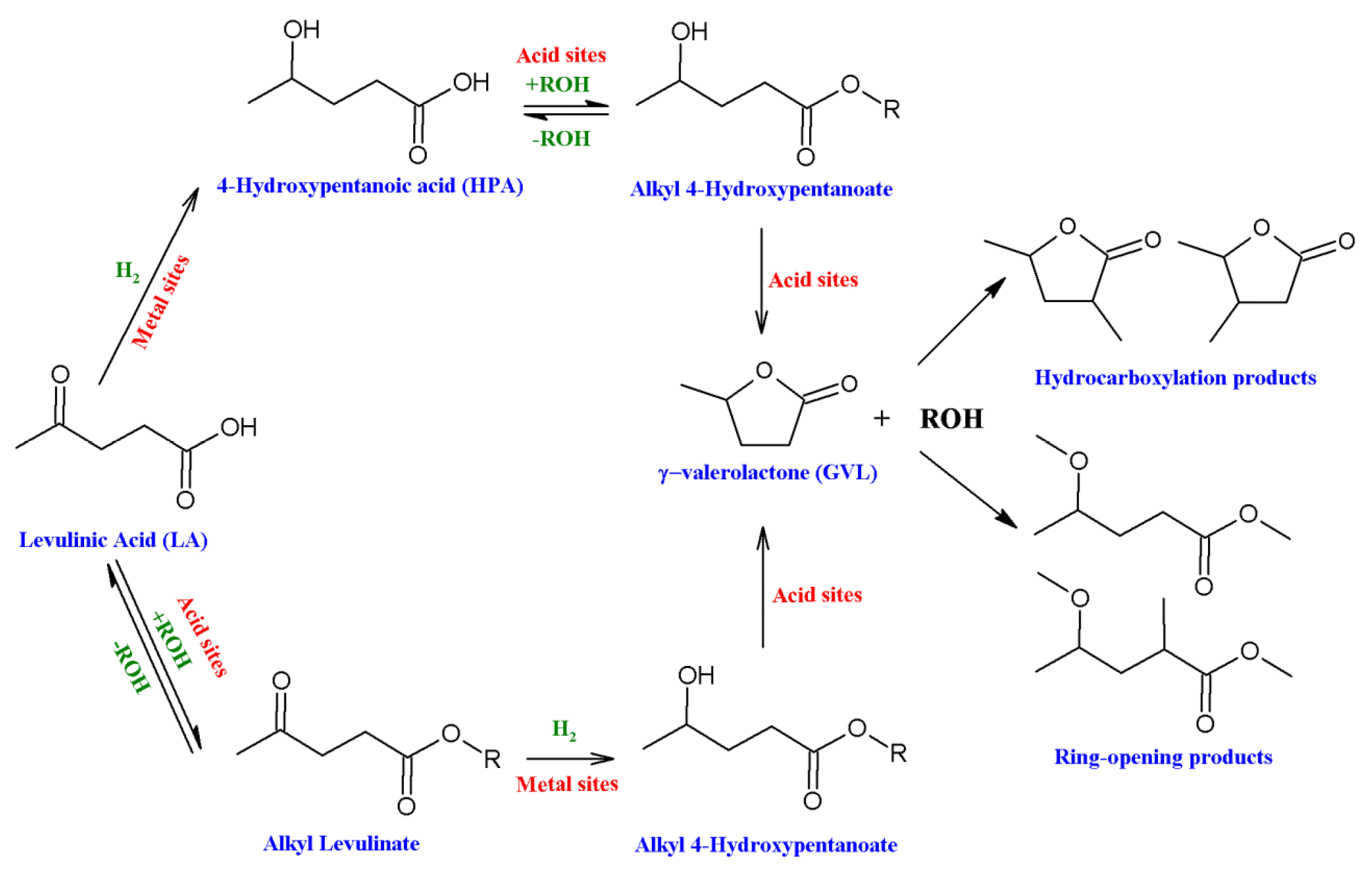
3.1. Hydrogenation of Alkyl Levulinate to γ-Valerolactone
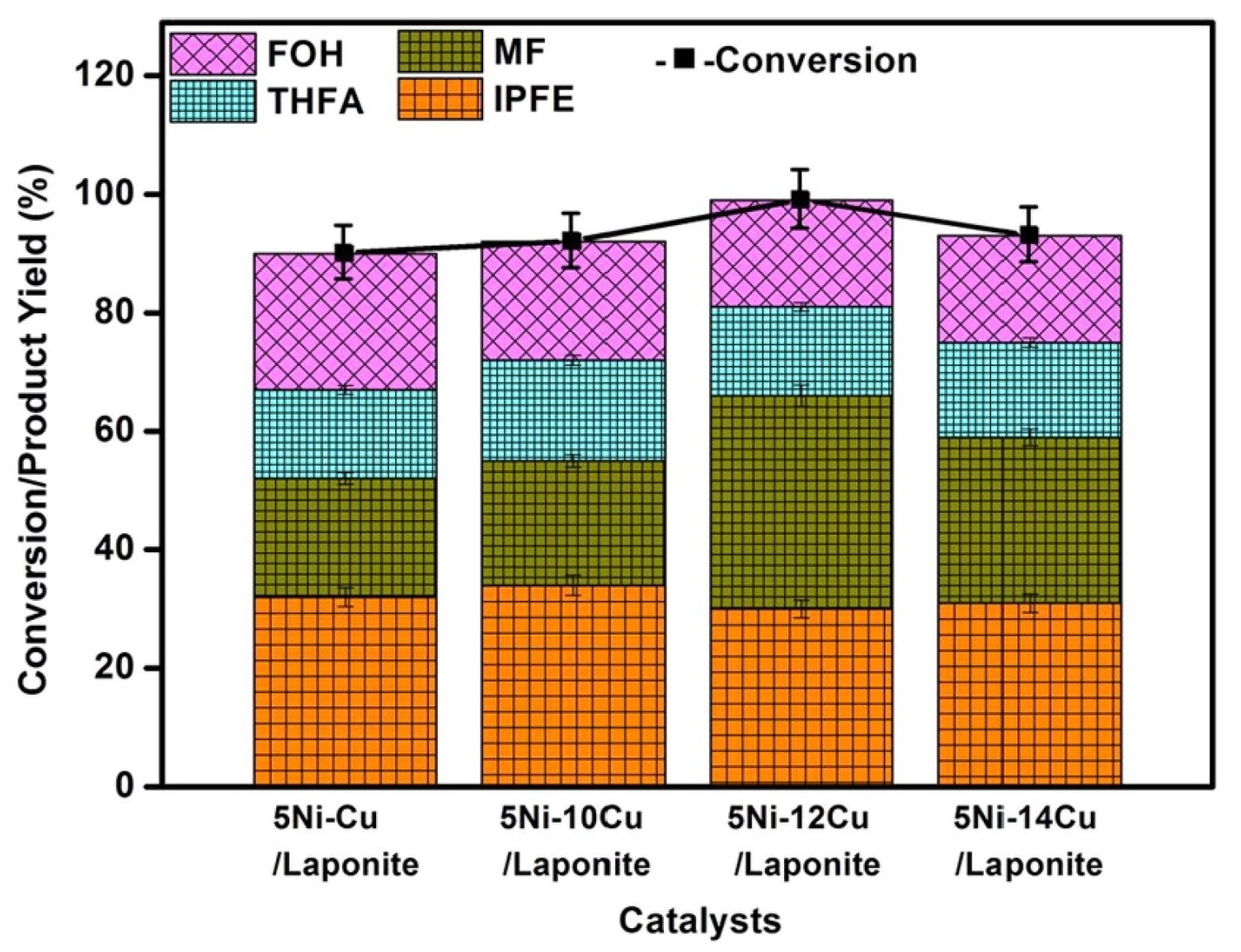
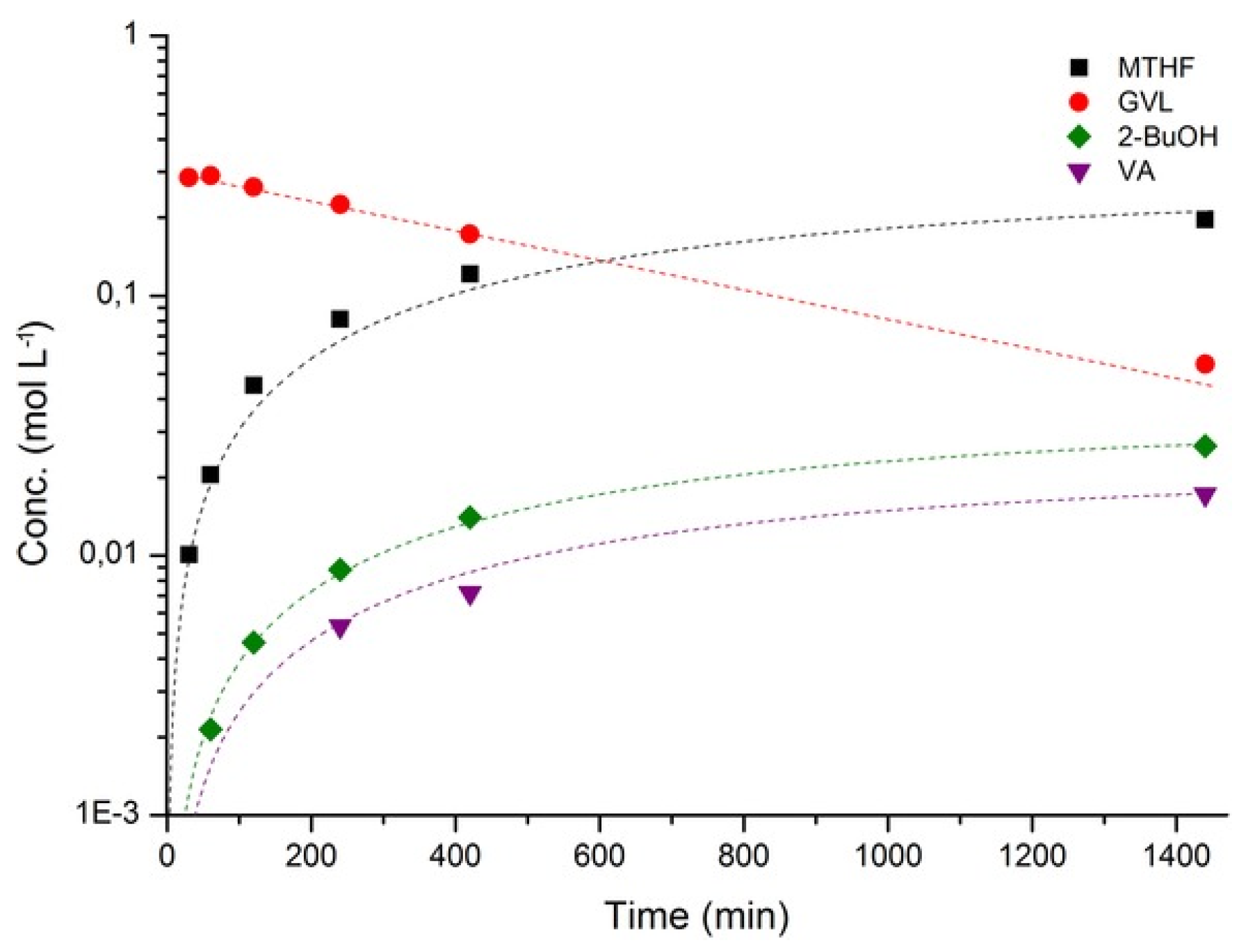
3.2. Hydrogenation of Levulinic Acid to 2-Methyltetrahydrofuran
4. Future Outlooks
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass; U.S. Department of Energy: Richland, WA, USA, 2004; Volume 1, p. 76. [CrossRef]
- Yan, K.; Wu, G.; Lafleur, T.; Jarvis, C. Production, Properties and Catalytic Hydrogenation of Furfural to Fuel Additives and Value-Added Chemicals. Renew. Sustain. Energy Rev. 2014, 38, 663–676. [Google Scholar] [CrossRef]
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; López Granados, M. Furfural: A Renewable and Versatile Platform Molecule for the Synthesis of Chemicals and Fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Gilkey, M.J.; Xu, B. Heterogeneous Catalytic Transfer Hydrogenation as an Effective Pathway in Biomass Upgrading. ACS Catal. 2016, 6, 1420–1436. [Google Scholar] [CrossRef]
- Chen, S.; Wojcieszak, R.; Dumeignil, F.; Marceau, E.; Royer, S. How Catalysts and Experimental Conditions Determine the Selective Hydroconversion of Furfural and 5-Hydroxymethylfurfural. Chem. Rev. 2018, 118, 11023–11117. [Google Scholar] [CrossRef] [PubMed]
- Bayan, S.; Beati, E. Furfural and its derivatives as motor fuels. Chim. Ind. 1941, 23, 432–434. [Google Scholar]
- Wang, C.; Xu, H.; Daniel, R.; Ghafourian, A.; Herreros, J.M.; Shuai, S.; Ma, X. Combustion Characteristics and Emissions of 2-Methylfuran Compared to 2,5-Dimethylfuran, Gasoline and Ethanol in a DISI Engine. Fuel 2013, 103, 200–211. [Google Scholar] [CrossRef]
- Scognamiglio, J.; Jones, L.; Letizia, C.S.; Api, A.M. Fragrance Material Review on Cyclopentanone. Food Chem. Toxicol. 2012, 50, S608–S612. [Google Scholar] [CrossRef]
- Siegel, H.; Eggersdorfer, M. Ketones. In Ullman’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Gong, W.; Chen, C.; Fan, R.; Zhang, H.; Wang, G.; Zhao, H. Transfer-Hydrogenation of Furfural and Levulinic Acid over Supported Copper Catalyst. Fuel 2018, 231, 165–171. [Google Scholar] [CrossRef]
- Hengne, A.M.; Rode, C.V. Cu-ZrO2 nanocomposite Catalyst for Selective Hydrogenation of Levulinic Acid and Its Ester to γ-Valerolactone. Green Chem. 2012, 14, 1064–1072. [Google Scholar] [CrossRef]
- Luo, W.; Sankar, M.; Beale, A.M.; He, Q.; Kiely, C.J.; Bruijnincx, P.C.A.; Weckhuysen, B.M. High Performing and Stable Supported Nano-Alloys for the Catalytic Hydrogenation of Levulinic Acid to γ-Valerolactone. Nat. Commun. 2015, 6, 6540. [Google Scholar] [CrossRef]
- Cen, Y.; Zhu, S.; Guo, J.; Chai, J.; Jiao, W.; Wang, J.; Fan, W. Supported Cobalt Catalysts for the Selective Hydrogenation of Ethyl Levulinate to Various Chemicals. RSC Adv. 2018, 8, 9152–9160. [Google Scholar] [CrossRef]
- Upare, P.P.; Lee, J.M.; Hwang, Y.K.; Hwang, D.W.; Lee, J.-H.; Halligudi, S.B.; Hwang, J.S. Direct Hydrocyclization of Biomass-Derived Levulinic Acid to 2-Methyltetrahydrofuran over Nanocomposite Copper/Silica Catalysts. ChemSusChem 2011, 4, 1749–1752. [Google Scholar] [CrossRef]
- Fernandes, D.R.; Rocha, A.S.; Mai, E.F.; Mota, C.J.A.; Teixeira Da Silva, V. Levulinic Acid Esterification with Ethanol to Ethyl Levulinate Production over Solid Acid Catalysts. Appl. Catal. A Gen. 2012, 425–426, 199–204. [Google Scholar] [CrossRef]
- Kang, S.; Fu, J.; Zhang, G. From Lignocellulosic Biomass to Levulinic Acid: A Review on Acid-Catalyzed Hydrolysis. Renew. Sustain. Energy Rev. 2018, 94, 340–362. [Google Scholar] [CrossRef]
- Lange, J.P.; Price, R.; Ayoub, P.M.; Louis, J.; Petrus, L.; Clarke, L.; Gosselink, H. Valeric Biofuels: A Platform of Cellulosic Transportation Fuels. Angew. Chem. Int. Ed. 2010, 49, 4479–4483. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Gamma-Valerolactone, a Sustainable Platform Molecule Derived from Lignocellulosic Biomass. Green Chem. 2013, 15, 584–595. [Google Scholar] [CrossRef]
- Wright, W.R.H.; Palkovits, R. Development of Heterogeneous Catalysts for the Conversion of Levulinic Acid to γ-Valerolactone. ChemSusChem 2012, 5, 1657–1667. [Google Scholar] [CrossRef]
- Yoshida, R.; Sun, D.; Yamada, Y.; Sato, S. Stable Cu-Ni/SiO2 Catalysts Prepared by Using Citric Acid-Assisted Impregnation for Vapor-Phase Hydrogenation of Levulinic Acid. Mol. Catal. 2018, 454, 70–76. [Google Scholar] [CrossRef]
- Tang, X.; Li, Z.; Zeng, X.; Jiang, Y.; Liu, S.; Lei, T.; Sun, Y.; Lin, L. In Situ Catalytic Hydrogenation of Biomass-Derived Methyl Levulinate to γ-Valerolactone in Methanol. ChemSusChem 2015, 8, 1601–1607. [Google Scholar] [CrossRef]
- Fábos, V.; Koczó, G.; Mehdi, H.; Boda, L.; Horváth, I.T. Bio-Oxygenates and the Peroxide Number: A Safety Issue Alert. Energy Environ. Sci. 2009, 2, 767–769. [Google Scholar] [CrossRef]
- Xu, Q.; Li, X.; Pan, T.; Yu, C.; Deng, J.; Guo, Q.; Fu, Y. Supported Copper Catalysts for Highly Efficient Hydrogenation of Biomass-Derived Levulinic Acid and γ-Valerolactone. Green Chem. 2016, 18, 1287–1294. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Converting Carbohydrates to Bulk Chemicals and Fine Chemicals over Heterogeneous Catalysts. Green Chem. 2011, 13, 520. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Conversion of Biomass Platform Molecules into Fuel Additives and Liquid Hydrocarbon Fuels. Green Chem. 2014, 16, 516–547. [Google Scholar] [CrossRef]
- Upare, P.P.; Lee, M.; Lee, S.-K.; Yoon, J.W.; Bae, J.; Hwang, D.W.; Lee, U.-H.; Chang, J.-S.; Hwang, Y.K. Ru Nanoparticles Supported Graphene Oxide Catalyst for Hydrogenation of Bio-Based Levulinic Acid to Cyclic Ethers. Catal. Today 2015, 265, 174–183. [Google Scholar] [CrossRef]
- Son, P.A.; Nishimura, S.; Ebitani, K. Production of γ-Valerolactone from Biomass-Derived Compounds Using Formic Acid as a Hydrogen Source over Supported Metal Catalysts in Water Solvent. RSC Adv. 2014, 4, 10525–10530. [Google Scholar] [CrossRef]
- Patankar, S.C.; Yadav, G.D. Cascade Engineered Synthesis of γ-Valerolactone, 1,4-Pentanediol, and 2-Methyltetrahydrofuran from Levulinic Acid Using Pd-Cu/ZrO2Catalyst in Water as Solvent. ACS Sustain. Chem. Eng. 2015, 3, 2619–2630. [Google Scholar] [CrossRef]
- Ishikawa, S.; Jones, D.R.; Iqbal, S.; Reece, C.; Morgan, D.J.; Willock, D.J.; Miedziak, P.J.; Bartley, J.K.; Edwards, J.K.; Murayama, T.; et al. Identification of the Catalytically Active Component of Cu-Zr-O Catalyst for the Hydrogenation of Levulinic Acid to γ-Valerolactone. Green Chem. 2017, 19, 225–236. [Google Scholar] [CrossRef]
- Levulinic Acid Market Size, Share, Price | Global Industry Report. 2020. Available online: https://www.grandviewresearch.com/industry-analysis/levulinic-acid-market (accessed on 22 January 2023).
- Pace, V.; Hoyos, P.; Castoldi, L.; Domínguez De María, P.; Alcántara, A.R. 2-Methyltetrahydrofuran (2-MeTHF): A Biomass-Derived Solvent with Broad Application in Organic Chemistry. ChemSusChem 2012, 5, 1369–1379. [Google Scholar] [CrossRef]
- Antonucci, V.; Coleman, J.; Ferry, J.B.; Johnson, N.; Mathe, M.; Scott, J.P.; Xu, J. Toxicological Assessment of 2-Methyltetrahydrofuran and Cyclopentyl Methyl Ether in Support of Their Use in Pharmaceutical Chemical Process Development. Org. Process Res. Dev. 2011, 15, 939–941. [Google Scholar] [CrossRef]
- Biddy, M.J.; Scarlata, C.; Kinchin, C. Chemicals from Biomass: A Market Assessment of Bioproducts with Near-Term Potential; National Renewable Energy Laboratory: Golden, CO, USA, 2016.
- Omoruyi, U.; Page, S.; Hallett, J.; Miller, P.W. Homogeneous Catalyzed Reactions of Levulinic Acid: To γ-Valerolactone and Beyond. ChemSusChem 2016, 9, 2037–2047. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef]
- Yu, I.K.M. Mechanistic Understanding of the Catalytic Hydrogenation of Bio-Derived Aromatics. Green Chem. 2021, 23, 9239–9253. [Google Scholar] [CrossRef]
- Ye, L.; Han, Y.; Feng, J.; Lu, X. A Review about GVL Production from Lignocellulose: Focusing on the Full Components Utilization. Ind. Crops Prod. 2020, 144, 112031. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, D.; Rodríguez-Padrón, D.; Len, C. Recent Advances in Catalytic Hydrogenation of Furfural. Catalysts 2019, 9, 796. [Google Scholar] [CrossRef]
- Fang, W.; Riisager, A. Recent Advances in Heterogeneous Catalytic Transfer Hydrogenation/Hydrogenolysis for Valorization of Biomass-Derived Furanic Compounds. Green Chem. 2021, 23, 670–688. [Google Scholar] [CrossRef]
- Wang, J.; Xiang, Z.; Huang, Z.; Xu, Q.; Yin, D. Recent Advances on Bifunctional Catalysts for One-Pot Conversion of Furfural to γ-Valerolactone. Front. Chem. 2022, 10, 959572. [Google Scholar] [CrossRef]
- Kwon, Y.; Schouten, K.J.P.; Van Der Waal, J.C.; De Jong, E.; Koper, M.T.M. Electrocatalytic Conversion of Furanic Compounds. ACS Catal. 2016, 6, 6704–6717. [Google Scholar] [CrossRef]
- Li, K.; Sun, Y. Electrocatalytic Upgrading of Biomass-Derived Intermediate Compounds to Value-Added Products. Chem. Eur. J. 2018, 24, 18258–18270. [Google Scholar] [CrossRef]
- Xu, C.; Paone, E.; Rodríguez-Padrón, D.; Luque, R.; Mauriello, F. Recent Catalytic Routes for the Preparation and the Upgrading of Biomass Derived Furfural and 5-Hydroxymethylfurfural. Chem. Soc. Rev. 2020, 49, 4273–4306. [Google Scholar] [CrossRef]
- Li, N.; Zong, M.H. (Chemo)Biocatalytic Upgrading of Biobased Furanic Platforms to Chemicals, Fuels, and Materials: A Comprehensive Review. ACS Catal. 2022, 12, 10080–10114. [Google Scholar] [CrossRef]
- Becerra, M.L.; Prieto, G.A.; Rendueles, M.; Diaz, M. Biological Transformations of Furanic Platform Molecules to Obtain Biomass-Derived Furans: A Review. Biomass Convers. Biorefin. 2022. [Google Scholar] [CrossRef]
- Ricard, E.; Guinot, H.M. Process for the Manufacture of Furfuryl Alcohol and Methylfurane. U.S. Patent 1,739,919, 17 December 1929. [Google Scholar]
- Lazier, W.A. Process for Hydrogenating Furfural. U.S. Patent 2,077,422, 20 April 1937. [Google Scholar]
- Swadesh, S. Catalytic Production of Furfuryl Alcohol and Catalyst Therefor. U.S. Patent 2,754,304, 10 July 1956. [Google Scholar]
- Hansen, J.B.; Nielsen, P.E.H. Handbook of Heterogeneous Catalysis; Ertl, G., Knozinger, H., Schuth, F., Weitkamp, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Rao, R.; Dandekar, A.; Baker, R.T.K.; Vannice, M.A.; Baker, T.K.; Vannice, M.A.; Baker, R.T.K.; Vannice, M.A. Properties of Copper Chromite Catalysts in Hydrogenation Reactions. J. Catal. 1997, 171, 406–419. [Google Scholar] [CrossRef]
- Liu, D.; Zemlyanov, D.; Wu, T.; Lobo-Lapidus, R.J.; Dumesic, J.A.; Miller, J.T.; Marshall, C.L. Deactivation Mechanistic Studies of Copper Chromite Catalyst for Selective Hydrogenation of 2-Furfuraldehyde. J. Catal. 2013, 299, 336–345. [Google Scholar] [CrossRef]
- Zhang, H.; Canlas, C.; Jeremy Kropf, A.; Elam, J.W.; Dumesic, J.A.; Marshall, C.L. Enhancing the Stability of Copper Chromite Catalysts for the Selective Hydrogenation of Furfural with ALD Overcoating (II)—Comparison between TiO2 and Al2O3 Overcoatings. J. Catal. 2015, 326, 172–181. [Google Scholar] [CrossRef]
- Zhang, H.; Lei, Y.; Kropf, A.J.; Zhang, G.; Elam, J.W.; Miller, J.T.; Sollberger, F.; Ribeiro, F.; Akatay, M.C.; Stach, E.A.; et al. Enhancing the Stability of Copper Chromite Catalysts for the Selective Hydrogenation of Furfural Using ALD Overcoating. J. Catal. 2014, 317, 284–292. [Google Scholar] [CrossRef]
- Boronat, M.; May, M.; Illas, F. Origin of Chemoselective Behavior of S-Covered Cu(1 1 1) towards Catalytic Hydrogenation of Unsaturated Aldehydes. Surf. Sci. 2008, 602, 3284–3290. [Google Scholar] [CrossRef]
- Sitthisa, S.; Sooknoi, T.; Ma, Y.G.; Balbuena, P.B.; Resasco, D.E. Kinetics and Mechanism of Hydrogenation of Furfural on Cu/SiO2 Catalysts. J. Catal. 2011, 277, 1–13. [Google Scholar] [CrossRef]
- Sitthisa, S.; Resasco, D.E. Hydrodeoxygenation of Furfural over Supported Metal Catalysts: A Comparative Study of Cu, Pd and Ni. Catal. Lett. 2011, 141, 784–791. [Google Scholar] [CrossRef]
- Rioux, R.M.; Vannice, M.A. Hydrogenation/Dehydrogenation Reactions: Isopropanol Dehydrogenation over Copper Catalysts. J. Catal. 2003, 216, 362–376. [Google Scholar] [CrossRef]
- Shi, Y.; Zhu, Y.; Yang, Y.; Li, Y.-W.; Jiao, H. Exploring Furfural Catalytic Conversion on Cu(111) from Computation. ACS Catal. 2015, 5, 4020–4032. [Google Scholar] [CrossRef]
- Liu, S.; Govindarajan, N.; Chan, K. Understanding Activity Trends in Furfural Hydrogenation on Transition Metal Surfaces. ACS Catal. 2022, 12, 12902–12910. [Google Scholar] [CrossRef]
- De Vrieze, J.E.; Thybaut, J.W.; Saeys, M. Role of Surface Hydroxyl Species in Copper-Catalyzed Hydrogenation of Ketones. ACS Catal. 2018, 8, 7539–7548. [Google Scholar] [CrossRef]
- Dong, F.; Zhu, Y.; Zheng, H.; Zhu, Y.; Li, X.; Li, Y. Cr-Free Cu-Catalysts for the Selective Hydrogenation of Biomass-Derived Furfural to 2-Methylfuran: The Synergistic Effect of Metal and Acid Sites. J. Mol. Catal. A Chem. 2015, 398, 140–148. [Google Scholar] [CrossRef]
- Jiménez-Gómez, C.P.; Cecilia, J.A.; Moreno-Tost, R.; Maireles-Torres, P. Selective Production of 2-Methylfuran by Gas-Phase Hydrogenation of Furfural on Copper Incorporated by Complexation in Mesoporous Silica Catalysts. ChemSusChem 2017, 10, 1448–1459. [Google Scholar] [CrossRef]
- Liu, J.; Liu, D.; Zhang, Y.; Wang, J.; Li, H.; Zhou, L.; Wu, S. Multiple Cores-Shell Structured Cu@SiO2 Ultrathin Leaf-Shaped Nanocomposite: Facile Fabrication and Excellent Selective Catalytic Hydrogenation Performance. ChemistrySelect 2018, 3, 4643–4652. [Google Scholar] [CrossRef]
- Ghashghaee, M.; Ghambarian, M.; Azizi, Z. Molecular-Level Insights into Furfural Hydrogenation Intermediates over Single-Atomic Cu Catalysts on Magnesia and Silica Nanoclusters. Mol. Simul. 2018, 7022, 154–163. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Siva Kumar, V.; Shasikala, V.; Padmasri, A.H.; Sreedhar, B.; David Raju, B.; Rama Rao, K.S. A Highly Efficient Cu/MgO Catalyst for Vapour Phase Hydrogenation of Furfural to Furfuryl Alcohol. Catal. Commun. 2003, 4, 287–293. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Padmasri, A.H.; Raju, B.D.; Rao, K.S.R.; David Raju, B.; Rama Rao, K.S.; Raju, B.D.; Rao, K.S.R. Vapor Phase Selective Hydrogenation of Furfural to Furfuryl Alcohol over Cu-MgO Coprecipitated Catalysts. J. Mol. Catal. A Chem. 2007, 265, 90–97. [Google Scholar] [CrossRef]
- Sadjadi, S.; Farzaneh, V.; Shirvani, S.; Ghashghaee, M. Preparation of Cu-MgO Catalysts with Different Copper Precursors and Precipitating Agents for the Vapor-Phase Hydrogenation of Furfural. Korean J. Chem. Eng. 2017, 34, 692–700. [Google Scholar] [CrossRef]
- Shirvani, S.; Ghashghaee, M.; Farzaneh, V.; Sadjadi, S. Influence of Catalyst Additives on Vapor-Phase Hydrogenation of Furfural to Furfuryl Alcohol on Impregnated Copper/Magnesia. Biomass Convers. Biorefin. 2018, 8, 79–86. [Google Scholar] [CrossRef]
- Ghashghaee, M.; Sadjadi, S.; Shirvani, S.; Farzaneh, V. A Novel Consecutive Approach for the Preparation of Cu–MgO Catalysts with High Activity for Hydrogenation of Furfural to Furfuryl Alcohol. Catal. Lett. 2017, 147, 318–327. [Google Scholar] [CrossRef]
- Jiménez-Gómez, C.P.; Cecilia, J.A.; Durán-Martín, D.; Moreno-Tost, R.; Santamaría-González, J.; Mérida-Robles, J.; Mariscal, R.; Maireles-Torres, P. Gas-Phase Hydrogenation of Furfural to Furfuryl Alcohol over Cu/ZnO Catalysts. J. Catal. 2016, 336, 107–115. [Google Scholar] [CrossRef]
- Yang, X.; Xiang, X.; Chen, H.; Zheng, H.; Li, Y.W.; Zhu, Y. Efficient Synthesis of Furfuryl Alcohol and 2-Methylfuran from Furfural over Mineral-Derived Cu/ZnO Catalysts. ChemCatChem 2017, 9, 3023–3030. [Google Scholar] [CrossRef]
- Kuld, S.; Conradsen, C.; Moses, P.G.; Chorkendorff, I.; Sehested, J. Quantification of Zinc Atoms in a Surface Alloy on Copper in an Industrial-Type Methanol Synthesis Catalyst. Angew. Chem. Int. Ed. 2014, 53, 5941–5945. [Google Scholar] [CrossRef]
- Yang, X.; Meng, Q.; Ding, G.; Wang, Y.; Chen, H.; Zhu, Y.L.; Li, Y.W. Construction of Novel Cu/ZnO-Al2O3 Composites for Furfural Hydrogenation: The Role of Al Components. Appl. Catal. A Gen. 2018, 561, 78–86. [Google Scholar] [CrossRef]
- Venkatesha, N.J.; Ramesh, S. Citric Acid-Assisted Synthesis of Nanoparticle Copper Catalyst Supported on an Oxide System for the Reduction of Furfural to Furfuryl Alcohol in the Vapor Phase. Ind. Eng. Chem. Res. 2018, 57, 1506–1515. [Google Scholar] [CrossRef]
- Jiménez-Gómez, C.P.; Cecilia, J.A.; Márquez-Rodríguez, I.; Moreno-Tost, R.; Santamaría-González, J.; Mérida-Robles, J.; Maireles-Torres, P. Gas-Phase Hydrogenation of Furfural over Cu/CeO2 Catalysts. Catal. Today 2017, 279, 327–338. [Google Scholar] [CrossRef]
- Jackson, M.A.; White, M.G.; Haasch, R.T.; Peterson, S.C.; Blackburn, J.A. Hydrogenation of Furfural at the Dynamic Cu Surface of CuOCeO2/Al2O3 in a Vapor Phase Packed Bed Reactor. Mol. Catal. 2018, 445, 124–132. [Google Scholar] [CrossRef]
- Vargas-Hernández, D.; Rubio-Caballero, J.M.; Santamaría-González, J.; Moreno-Tost, R.; Mérida-Robles, J.M.; Pérez-Cruz, M.A.; Jiménez-López, A.; Hernández-Huesca, R.; Maireles-Torres, P. Furfuryl Alcohol from Furfural Hydrogenation over Copper Supported on SBA-15 Silica Catalysts. J. Mol. Catal. A Chem. 2014, 383–384, 106–113. [Google Scholar] [CrossRef]
- Jiménez-Gómez, C.P.; Cecilia, J.A.; Moreno-Tost, R.; Maireles-Torres, P. Selective Furfural Hydrogenation to Furfuryl Alcohol Using Cu-Based Catalysts Supported on Clay Minerals. Top. Catal. 2017, 60, 1040–1053. [Google Scholar] [CrossRef]
- Jiménez-Gómez, C.P.; Cecilia, J.A.; Franco-Duro, F.I.; Pozo, M.; Moreno-Tost, R.; Maireles-Torres, P. Promotion Effect of Ce or Zn Oxides for Improving Furfuryl Alcohol Yield in the Furfural Hydrogenation Using Inexpensive Cu-Based Catalysts. Mol. Catal. 2018, 455, 121–131. [Google Scholar] [CrossRef]
- Reddy, B.M.; Reddy, G.K.; Rao, K.N.; Khan, A.; Ganesh, I. Silica Supported Transition Metal-Based Bimetallic Catalysts for Vapour Phase Selective Hydrogenation of Furfuraldehyde. J. Mol. Catal. A Chem. 2007, 265, 276–282. [Google Scholar] [CrossRef]
- Wu, J.; Shen, Y.M.; Liu, C.H.; Wang, H.B.; Geng, C.; Zhang, Z.X. Vapor Phase Hydrogenation of Furfural to Furfuryl Alcohol over Environmentally Friendly Cu-Ca/SiO2 Catalyst. Catal. Commun. 2005, 6, 633–637. [Google Scholar] [CrossRef]
- Seo, G.; Chon, H. Hydrogenation of Furfural over Copper-Containing Catalysts. J. Catal. 1981, 67, 424–429. [Google Scholar] [CrossRef]
- Guerrero-Torres, A.; Jiménez-Gómez, C.P.; Cecilia, J.A.; García-Sancho, C.; Quirante-Sánchez, J.J.; Mérida-Robles, J.M.; Maireles-Torres, P. Influence of the Incorporation of Basic or Amphoteric Oxides on the Performance of Cu-Based Catalysts Supported on Sepiolite in Furfural Hydrogenation. Catalysts 2019, 9, 315. [Google Scholar] [CrossRef]
- O’Neill, B.J.; Miller, J.T.; Dietrich, P.J.; Sollberger, F.G.; Ribeiro, F.H.; Dumesic, J.A. Operando X-ray Absorption Spectroscopy Studies of Sintering for Supported Copper Catalysts during Liquid-Phase Reaction. ChemCatChem 2014, 6, 2493–2496. [Google Scholar] [CrossRef]
- Xu, C.H.; Zheng, L.K.; Deng, D.F.; Liu, J.Y.; Liu, S.Y. Effect of Activation Temperature on the Surface Copper Particles and Catalytic Properties of Cu-Ni-Mg-Al Oxides from Hydrotalcite-like Precursors. Catal. Commun. 2011, 12, 996–999. [Google Scholar] [CrossRef]
- Lesiak, M.; Binczarski, M.; Karski, S.; Maniukiewicz, W.; Rogowski, J.; Szubiakiewicz, E.; Berlowska, J.; Dziugan, P.; Witońska, I. Hydrogenation of Furfural over Pd-Cu/Al2O3 Catalysts. The Role of Interaction between Palladium and Copper on Determining Catalytic Properties. J. Mol. Catal. A Chem. 2014, 395, 337–348. [Google Scholar] [CrossRef]
- Scholz, D.; Aellig, C.; Hermans, I. Catalytic Transfer Hydrogenation/Hydrogenolysis for Reductive Upgrading of Furfural and 5-(Hydroxymethyl)Furfural. ChemSusChem 2014, 7, 268–275. [Google Scholar] [CrossRef]
- Huang, L.; Zhu, Y.; Huo, C.; Zheng, H.; Feng, G.; Zhang, C.; Li, Y. Mechanistic Insight into the Heterogeneous Catalytic Transfer Hydrogenation over Cu/Al2O3: Direct Evidence for the Assistant Role of Support. J. Mol. Catal. A Chem. 2008, 288, 109–115. [Google Scholar] [CrossRef]
- Pellet, R.J. Hydrogen Transfer Catalysis by Platinum on Zeolites. J. Catal. 1998, 177, 40–52. [Google Scholar] [CrossRef]
- Sawadjoon, S.; Lundstedt, A.; Samec, J.S.M. Pd-Catalyzed Transfer Hydrogenolysis of Primary, Secondary, and Tertiary Benzylic Alcohols by Formic Acid: A Mechanistic Study. ACS Catal. 2013, 3, 635–642. [Google Scholar] [CrossRef]
- Rajagopal, S.; Spatola, A.F. Palladium-Catalyzed Transfer Hydrogenolysis of Benzyl Acetate with Ammonium Formate. Appl. Catal. A Gen. 1997, 152, 69–81. [Google Scholar] [CrossRef]
- Gong, W.; Chen, C.; Zhang, Y.; Zhou, H.; Wang, H.; Zhang, H.; Zhang, Y.; Wang, G.; Zhao, H. Efficient Synthesis of Furfuryl Alcohol from H2-Hydrogenation/Transfer Hydrogenation of Furfural Using Sulfonate Group Modified Cu Catalyst. ACS Sustain. Chem. Eng. 2017, 5, 2172–2180. [Google Scholar] [CrossRef]
- Srivastava, S.; Mohanty, P.; Parikh, J.K.; Dalai, A.K.; Amritphale, S.S.; Khare, A.K. Cr-Free Co-Cu/SBA-15 Catalysts for Hydrogenation of Biomass-Derived α-, β-Unsaturated Aldehyde to Alcohol. Cuihua Xuebao Chin. J. Catal. 2015, 36, 933–942. [Google Scholar] [CrossRef]
- Srivastava, S.; Solanki, N.; Mohanty, P.; Shah, K.A.; Parikh, J.K.J.; Dalai, A. Optimization and Kinetic Studies on Hydrogenation of Furfural to Furfuryl Alcohol over SBA-15 Supported Bimetallic Copper-Cobalt Catalyst. Catal. Lett. 2015, 145, 816–823. [Google Scholar] [CrossRef]
- Nguyen-Huy, C.; Lee, H.; Lee, J.; Kwak, J.H.; An, K. Mesoporous Mixed CuCo Oxides as Robust Catalysts for Liquid-Phase Furfural Hydrogenation. Appl. Catal. A Gen. 2019, 571, 118–126. [Google Scholar] [CrossRef]
- Chen, H.; Ruan, H.; Lu, X.; Fu, J.; Langrish, T.; Lu, X. Efficient Catalytic Transfer Hydrogenation of Furfural to Furfuryl Alcohol in Near-Critical Isopropanol over Cu/MgO-Al2O3 Catalyst. Mol. Catal. 2018, 445, 94–101. [Google Scholar] [CrossRef]
- Villaverde, M.M.; Bertero, N.M.; Garetto, T.F.; Marchi, A.J. Selective Liquid-Phase Hydrogenation of Furfural to Furfuryl Alcohol over Cu-Based Catalysts. Catal. Today 2013, 213, 87–92. [Google Scholar] [CrossRef]
- Villaverde, M.M.M.M.; Garetto, T.F.T.F.; Marchi, A.J.A.J. Liquid-Phase Transfer Hydrogenation of Furfural to Furfuryl Alcohol on Cu-Mg-Al Catalysts. Catal. Commun. 2014, 58, 6–10. [Google Scholar] [CrossRef]
- Sharma, R.V.; Das, U.; Sammynaiken, R.; Dalai, A.K. Liquid Phase Chemo-Selective Catalytic Hydrogenation of Furfural to Furfuryl Alcohol. Appl. Catal. A Gen. 2013, 454, 127–136. [Google Scholar] [CrossRef]
- Wang, Y.; Sang, S.; Zhu, W.; Gao, L.; Xiao, G. CuNi@C Catalysts with High Activity Derived from Metal-Organic Frameworks Precursor for Conversion of Furfural to Cyclopentanone. Chem. Eng. J. 2016, 299, 104–111. [Google Scholar] [CrossRef]
- Chang, X.; Liu, A.-F.; Cai, B.; Luo, J.-Y.; Pan, H.; Huang, Y.-B.; Chang, X.; Huang, Y.-B.; Cai, B. Catalytic Transfer Hydrogenation of Furfural to 2-Methylfuran and 2-Methyltetrahydrofuran over Bimetallic Copper-Palladium Catalysts. ChemSusChem 2016, 9, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lai, Q.; Holles, J.H. Bimetallic Overlayer Catalysts with High Selectivity and Reactivity for Furfural Hydrogenation. Catal. Commun. 2017, 89, 77–80. [Google Scholar] [CrossRef]
- Zhang, Z.; Pei, Z.; Chen, H.; Chen, K.; Hou, Z.; Lu, X.; Ouyang, P.; Fu, J. Catalytic In-Situ Hydrogenation of Furfural over Bimetallic Cu-Ni Alloy Catalysts in Isopropanol. Ind. Eng. Chem. Res. 2018, 57, 4225–4230. [Google Scholar] [CrossRef]
- Khromova, S.A.; Bykova, M.V.; Bulavchenko, O.A.; Ermakov, D.Y.; Saraev, A.A.; Kaichev, V.V.; Venderbosch, R.H.; Yakovlev, V.A. Furfural Hydrogenation to Furfuryl Alcohol over Bimetallic Ni–Cu Sol–Gel Catalyst: A Model Reaction for Conversion of Oxygenates in Pyrolysis Liquids. Top. Catal. 2016, 59, 1413–1423. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, J. Selective Transfer Hydrogenation of Biomass-Based Furfural and 5-Hydroxymethylfurfural over Hydrotalcite-Derived Copper Catalysts Using Methanol as a Hydrogen Donor. ACS Sustain. Chem. Eng. 2017, 5, 5982–5993. [Google Scholar] [CrossRef]
- Du, J.; Zhang, J.; Sun, Y.; Jia, W.; Si, Z.; Gao, H.; Tang, X.; Zeng, X.; Lei, T.; Liu, S.; et al. Catalytic Transfer Hydrogenation of Biomass-Derived Furfural to Furfuryl Alcohol over In-Situ Prepared Nano Cu-Pd/C Catalyst Using Formic Acid as Hydrogen Source. J. Catal. 2018, 368, 69–78. [Google Scholar] [CrossRef]
- Nagaiah, P.; Gidyonu, P.; Ashokraju, M.; Rao, M.V.; Challa, P.; Burri, D.R.; Kamaraju, S.R.R. Magnesium Aluminate Supported Cu Catalyst for Selective Transfer Hydrogenation of Biomass Derived Furfural to Furfuryl Alcohol with Formic Acid as Hydrogen Donor. ChemistrySelect 2019, 4, 145–151. [Google Scholar] [CrossRef]
- Srivastava, S.; Jadeja, G.C.; Parikh, J. Copper-Cobalt Catalyzed Liquid Phase Hydrogenation of Furfural to 2-Methylfuran: An Optimization, Kinetics and Reaction Mechanism Study. Chem. Eng. Res. Des. 2018, 132, 313–324. [Google Scholar] [CrossRef]
- Kalong, M.; Hongmanorom, P.; Ratchahat, S.; Koo-amornpattana, W.; Faungnawakij, K.; Assabumrungrat, S.; Srifa, A.; Kawi, S. Hydrogen-Free Hydrogenation of Furfural to Furfuryl Alcohol and 2-Methylfuran over Ni and Co-Promoted Cu/γ-Al2O3 Catalysts. Fuel Process. Technol. 2021, 214, 106721. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Martin, N.; Vlachos, D.G. Effect of Hydrogen Donor on Liquid Phase Catalytic Transfer Hydrogenation of Furfural over a Ru/RuO2/C Catalyst. J. Mol. Catal. A Chem. 2014, 392, 223–228. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, Y.B.; Guo, Q.X.; Fu, Y. RANEY® Ni Catalyzed Transfer Hydrogenation of Levulinate Esters to γ-Valerolactone at Room Temperature. Chem. Commun. 2013, 49, 5328–5330. [Google Scholar] [CrossRef]
- Bonrath, W.; Castelijns, A.M.C.F.; De Vries, J.G.; Guit, R.P.M.; Schütz, J.; Sereinig, N.; Vaessen, H.W.L.M. Gas Phase Hydrogenation of Levulinic Acid to γ-Valerolactone. Catal. Letters 2016, 146, 28–34. [Google Scholar] [CrossRef]
- Putrakumar, B.; Nagaraju, N.; Kumar, V.P.; Chary, K.V.R. Hydrogenation of Levulinic Acid to γ-Valerolactone over Copper Catalysts Supported on γ-Al2O3. Catal. Today 2015, 250, 209–217. [Google Scholar] [CrossRef]
- Zeitsch, K.J. Furfuryl Alcohol. In The Chemistry and Technology of Furfural and Its Many By-Products; Elsevier: Amsterdam, The Netherlands, 2000; pp. 150–155. [Google Scholar]
- Corma, A.; De La Torre, O.; Renz, M.; Villandier, N. Production of High-Quality Diesel from Biomass Waste Products. Angew. Chem. Int. Ed. 2011, 50, 2375–2378. [Google Scholar] [CrossRef]
- Zhu, Y.L.; Xiang, H.W.; Li, Y.W.; Jiao, H.J.; Wu, G.S.; Zhong, B.; Guo, G.Q. A New Strategy for the Efficient Synthesis of 2-Methylfuran and Gamma-Butyrolactone. New J. Chem. 2003, 27, 208–210. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, H.Y.; Zhu, Y.L.; Zhao, G.W.; Zhang, C.H.; Teng, B.T.; Xiang, H.W.; Li, Y.W. Effects of Calcination Temperature on Performance of Cu-Zn-Al Catalyst for Synthesizing Gamma-Butyrolactone and 2-Methylfuran through the Coupling of Dehydrogenation and Hydrogenation. Catal. Commun. 2004, 5, 505–510. [Google Scholar] [CrossRef]
- Xiu, S.; Shahbazi, A. Bio-Oil Production and Upgrading Research: A Review. Renew. Sustain. Energy Rev. 2012, 16, 4406–4414. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Tamura, M.; Tomishige, K. Catalytic Reduction of Biomass-Derived Furanic Compounds with Hydrogen. ACS Catal. 2013, 3, 2655–2668. [Google Scholar] [CrossRef]
- Zheng, H.Y.; Zhu, Y.L.; Teng, B.T.; Bai, Z.Q.; Zhang, C.H.; Xiang, H.W.; Li, Y.W. Towards Understanding the Reaction Pathway in Vapour Phase Hydrogenation of Furfural to 2-Methylfuran. J. Mol. Catal. A Chem. 2006, 246, 18–23. [Google Scholar] [CrossRef]
- Holdren, R.F. Manufacture of Methylfuran. U.S. Patent 2,445,714, 20 July 1948. [Google Scholar]
- Deutsch, K.L.; Shanks, B.H. Active Species of Copper Chromite Catalyst in C-O Hydrogenolysis of 5-Methylfurfuryl Alcohol. J. Catal. 2012, 285, 235–241. [Google Scholar] [CrossRef]
- Yan, K.; Liao, J.; Wu, X.; Xie, X. A Noble-Metal Free Cu-Catalyst Derived from Hydrotalcite for Highly Efficient Hydrogenation of Biomass-Derived Furfural and Levulinic Acid. RSC Adv. 2013, 3, 3853–3856. [Google Scholar] [CrossRef]
- Biradar, N.S.; Hengne, A.A.; Birajdar, S.N.; Swami, R.; Rode, C.V. Tailoring the Product Distribution with Batch and Continuous Process Options in Catalytic Hydrogenation of Furfural. Org. Process Res. Dev. 2014, 18, 1434–1442. [Google Scholar] [CrossRef]
- Chen, B.; Li, F.; Huang, Z.; Yuan, G. Tuning Catalytic Selectivity of Liquid-Phase Hydrogenation of Furfural via Synergistic Effects of Supported Bimetallic Catalysts. Appl. Catal. A Gen. 2015, 500, 23–29. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Vlachos, D.G. Liquid Phase Catalytic Transfer Hydrogenation of Furfural over a Ru/C Catalyst. Appl. Catal. A Gen. 2014, 480, 17–24. [Google Scholar] [CrossRef]
- Ren, G.; Wang, G.; Mei, H.; Xu, Y.; Huang, L. Reaction Mechanism Investigation of Furfural Conversion to 2-Methylfuran on Cu(1 1 1) Surface. Chem. Phys. Lett. 2018, 703, 1–7. [Google Scholar] [CrossRef]
- Fu, X.; Liu, Y.; Liu, Q.; Liu, Z.; Peng, Z. Preparation of Highly Active Cu/SiO2 Catalysts for Furfural to 2-Methylfuran by Ammonia Evaporation Method. Catalysts 2022, 12, 276. [Google Scholar] [CrossRef]
- Jiménez-Gómez, C.P.; Cecilia, J.A.; Alba-Rubio, A.C.; Cassidy, A.; Moreno-Tost, R.; García-Sancho, C.; Maireles-Torres, P. Tailoring the Selectivity of Cu-Based Catalysts in the Furfural Hydrogenation Reaction: Influence of the Morphology of the Silica Support. Fuel 2022, 319, 123827. [Google Scholar] [CrossRef]
- Park, S.; Kannapu, H.P.R.; Jeong, C.; Kim, J.; Suh, Y.W. Highly Active Mesoporous Cu−Al2O3 Catalyst for the Hydrodeoxygenation of Furfural to 2-Methylfuran. ChemCatChem 2020, 12, 105–111. [Google Scholar] [CrossRef]
- Lessard, J.; Morin, J.F.; Wehrung, J.F.; Magnin, D.; Chornet, E. High Yield Conversion of Residual Pentoses into Furfural via Zeolite Catalysis and Catalytic Hydrogenation of Furfural to 2-Methylfuran. Top. Catal. 2010, 53, 1231–1234. [Google Scholar] [CrossRef]
- Sheng, H.; Lobo, R.F. Iron-Promotion of Silica-Supported Copper Catalysts for Furfural Hydrodeoxygenation. ChemCatChem 2016, 8, 3402–3408. [Google Scholar] [CrossRef]
- Xiong, K.; Wan, W.; Chen, J.G. Reaction Pathways of Furfural, Furfuryl Alcohol and 2-Methylfuran on Cu(111) and NiCu Bimetallic Surfaces. Surf. Sci. 2016, 652, 91–97. [Google Scholar] [CrossRef]
- Golub, K.W.; Sulmonetti, T.P.; Darunte, L.A.; Shealy, M.S.; Jones, C.W. Metal-Organic-Framework-Derived Co/Cu-Carbon Nanoparticle Catalysts for Furfural Hydrogenation. ACS Appl. Nano Mater. 2019, 2, 6040–6056. [Google Scholar] [CrossRef]
- Hutchings, G.S.; Luc, W.; Lu, Q.; Zhou, Y.; Vlachos, D.G.; Jiao, F. Nanoporous Cu-Al-Co Alloys for Selective Furfural Hydrodeoxygenation to 2-Methylfuran. Ind. Eng. Chem. Res. 2017, 56, 3866–3872. [Google Scholar] [CrossRef]
- Srivastava, S.; Jadeja, G.C.C.; Parikh, J. A Versatile Bi-Metallic Copper-Cobalt Catalyst for Liquid Phase Hydrogenation of Furfural to 2-Methylfuran. RSC Adv. 2016, 6, 1649–1658. [Google Scholar] [CrossRef]
- Gilkey, M.J.M.J.; Panagiotopoulou, P.; Mironenko, A.V.A.V.; Jenness, G.R.G.R.; Vlachos, D.G.D.G.; Xu, B. Mechanistic Insights into Metal Lewis Acid-Mediated Catalytic Transfer Hydrogenation of Furfural to 2-Methylfuran. ACS Catal. 2015, 5, 3988–3994. [Google Scholar] [CrossRef]
- Bhogeswararao, S.; Srinivas, D. Catalytic Conversion of Furfural to Industrial Chemicals over Supported Pt and Pd Catalysts. J. Catal. 2015, 327, 65–77. [Google Scholar] [CrossRef]
- Yan, K.; Chen, A. Selective Hydrogenation of Furfural and Levulinic Acid to Biofuels on the Ecofriendly Cu-Fe Catalyst. Fuel 2014, 115, 101–108. [Google Scholar] [CrossRef]
- Srivastava, S.; Jadeja, G.C.C.; Parikh, J. Synergism Studies on Alumina-Supported Copper-Nickel Catalysts towards Furfural and 5-Hydroxymethylfurfural Hydrogenation. J. Mol. Catal. A Chem. 2017, 426, 244–256. [Google Scholar] [CrossRef]
- Akmaz, S.; Algorabi, S.; Koc, S.N. Furfural Hydrogenation to 2-Methylfuran over Efficient Sol-Gel Copper-Cobalt/Zirconia Catalyst. Can. J. Chem. Eng. 2021, 99, S562–S574. [Google Scholar] [CrossRef]
- Geng, W.; Li, W.; Liu, L.L.; Liu, J.; Liu, L.L.; Kong, X. Facile Assembly of Cu-Cu2O/N-Reduced Graphene Oxide Nanocomposites for Efficient Synthesis of 2-Methylfuran. Fuel 2020, 259, 116267. [Google Scholar] [CrossRef]
- Jaatinen, S.K.; Karinen, R.S.; Lehtonen, J.S. Liquid Phase Furfural Hydrotreatment to 2-Methylfuran with Carbon Supported Copper, Nickel, and Iron Catalysts. ChemistrySelect 2017, 2, 51–60. [Google Scholar] [CrossRef]
- Smirnov, A.A.; Shilov, I.N.; Alekseeva, M.V.; Selishcheva, S.A.; Yakovlev, V.A. Study of the Composition Effect of Molybdenum-Modified Nickel–Copper Catalysts on Their Activity and Selectivity in the Hydrogenation of Furfural to Different Valuable Chemicals. Catal. Ind. 2018, 10, 228–236. [Google Scholar] [CrossRef]
- Seemala, B.; Cai, C.M.; Kumar, R.; Wyman, C.E.; Christopher, P. Effects of Cu-Ni Bimetallic Catalyst Composition and Support on Activity, Selectivity, and Stability for Furfural Conversion to 2-Methyfuran. ACS Sustain. Chem. Eng. 2018, 6, 2152–2161. [Google Scholar] [CrossRef]
- Varila, T.; Mäkelä, E.; Kupila, R.; Romar, H.; Hu, T.; Karinen, R.; Puurunen, R.L.; Lassi, U. Conversion of Furfural to 2-Methylfuran over CuNi Catalysts Supported on Biobased Carbon Foams. Catal. Today 2021, 367, 16–27. [Google Scholar] [CrossRef]
- Umasankar, S.; Santhana Krishnan, P.; Sonia Theres, G.; Tamizhdurai, P.; Shanthi, K. Liquid Phase Hydrogenation of Furfural to Biofuel over Robust NiCu/Laponite Catalyst: A Study on the Role of Copper Loading. Adv. Powder Technol. 2021, 32, 3034–3045. [Google Scholar] [CrossRef]
- Chuseang, J.; Nakwachara, R.; Kalong, M.; Ratchahat, S.; Koo-Amornpattana, W.; Klysubun, W.; Khemthong, P.; Faungnawakij, K.; Assabumrungrat, S.; Itthibenchapong, V.; et al. Selective Hydrogenolysis of Furfural into Fuel-Additive 2-Methylfuran over a Rhenium-Promoted Copper Catalyst. Sustain. Energy Fuels 2021, 5, 1379–1393. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, X.; Zhu, Y.; Zhu, Y.; Zheng, H.; Li, Y. Promoting Effect of Boron Oxide on Cu/SiO2 Catalyst for Glycerol Hydrogenolysis to 1,2-Propanediol. J. Catal. 2013, 303, 70–79. [Google Scholar] [CrossRef]
- Zheng, H.-Y.; Zhu, Y.-L.; Bai, Z.-Q.; Huang, L.; Xiang, H.-W.; Li, Y.-W. An Environmentally Benign Process for the Efficient Synthesis of Cyclohexanone and 2-Methylfuran. Green Chem. 2006, 8, 107–109. [Google Scholar] [CrossRef]
- Zheng, H.Y.; Zhu, Y.L.; Huang, L.; Zeng, Z.Y.; Wan, H.J.; Li, Y.W. Study on Cu-Mn-Si Catalysts for Synthesis of Cyclohexanone and 2-Methylfuran through the Coupling Process. Catal. Commun. 2008, 9, 342–348. [Google Scholar] [CrossRef]
- Zheng, H.Y.; Yang, J.; Zhu, Y.L.; Zhao, G.W. Synthesis of γ-Butyrolactone and 2-Methylfuran through the Coupling of Dehydrogenation and Hydrogenation over Copper-Chromite Catalyst. React. Kinet. Catal. Lett. 2004, 82, 263–269. [Google Scholar] [CrossRef]
- Luo, J.; Cheng, Y.; Niu, H.; Wang, T.; Liang, C. Efficient Cu/FeOx Catalyst with Developed Structure for Catalytic Transfer Hydrogenation of Furfural. J. Catal. 2022, 413, 575–587. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, Z.; Lin, W.; Song, W.; Li, S. High Efficient Conversion of Furfural to 2-Methylfuran over Ni-Cu/Al2O3 Catalyst with Formic Acid as a Hydrogen Donor. Appl. Catal. A Gen. 2017, 547, 248–255. [Google Scholar] [CrossRef]
- Zhou, K.; Chen, J.; Cheng, Y.; Chen, Z.; Kang, S.; Cai, Z.; Xu, Y.; Wei, J. Enhanced Catalytic Transfer Hydrogenation of Biomass-Based Furfural into 2-Methylfuran over Multifunctional Cu-Re Bimetallic Catalysts. ACS Sustain. Chem. Eng. 2020, 8, 16624–16636. [Google Scholar] [CrossRef]
- Niu, H.; Luo, J.; Li, C.; Wang, B.; Liang, C. Transfer Hydrogenation of Biomass-Derived Furfural to 2-Methylfuran over CuZnAl Catalysts. Ind. Eng. Chem. Res. 2019, 58, 6298–6308. [Google Scholar] [CrossRef]
- Li, B.; Li, L.; Sun, H.; Zhao, C. Selective Deoxygenation of Aqueous Furfural to 2-Methylfuran over Cu0/Cu2O·SiO2 Sites via a Copper Phyllosilicate Precursor without Extraneous Gas. ACS Sustain. Chem. Eng. 2018, 6, 12096–12103. [Google Scholar] [CrossRef]
- Zhang, J.; Li, C.; Hu, S.; Gu, J.; Yuan, H.; Chen, Y. Mechanistic Insights into Copper Oxides Catalyzed Bio-Based Furfural Hydrogenation Using Methanol as In-Situ Hydrogen Donor. Renew. Energy 2022, 200, 88–97. [Google Scholar] [CrossRef]
- More, G.S.; Shivhare, A.; Kaur, S.P.; Dhilip Kumar, T.J.; Srivastava, R. Catalytic Interplay of Metal Ions (Cu2+, Ni2+, and Fe2+) in MFe2O4 Inverse Spinel Catalysts for Enhancing the Activity and Selectivity during Selective Transfer Hydrogenation of Furfural into 2-Methylfuran. Catal. Sci. Technol. 2022, 12, 4857–4870. [Google Scholar] [CrossRef]
- Cui, J.; Tan, J.; Cui, X.; Zhu, Y.; Deng, T.; Ding, G.; Li, Y. Conversion of Xylose to Furfuryl Alcohol and 2-Methylfuran in a Continuous Fixed-Bed Reactor. ChemSusChem 2016, 9, 1259–1262. [Google Scholar] [CrossRef]
- Gandarias, I.; García-Fernández, S.; Obregón, I.; Agirrezabal-Telleria, I.; Arias, P.L. Production of 2-Methylfuran from Biomass through an Integrated Biorefinery Approach. Fuel Process. Technol. 2018, 178, 336–343. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, G.; Yang, L.; Li, F. Efficient Conversion of Furfural into Cyclopentanone over High Performing and Stable Cu/ZrO2 Catalysts. Appl. Catal. A Gen. 2018, 561, 117–126. [Google Scholar] [CrossRef]
- Wang, Y.; Miao, Y.; Li, S.; Gao, L.; Xiao, G. Metal-Organic Frameworks Derived Bimetallic Cu-Co Catalyst for Efficient and Selective Hydrogenation of Biomass-Derived Furfural to Furfuryl Alcohol. Mol. Catal. 2017, 436, 128–137. [Google Scholar] [CrossRef]
- Gong, W.; Chen, C.; Zhang, H.; Wang, G.; Zhao, H. In Situ Synthesis of Highly Dispersed Cu-Co Bimetallic Nanoparticles for Tandem Hydrogenation/Rearrangement of Bioderived Furfural in Aqueous-Phase. ACS Sustain. Chem. Eng. 2018, 6, 14919–14925. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, M.; Wang, T.; Xiao, G. Conversion of Furfural to Cyclopentanol on Cu/Zn/Al Catalysts Derived from Hydrotalcite-Like Materials. Catal. Letters 2015, 145, 1557–1565. [Google Scholar] [CrossRef]
- Dunlop, A.P.; Maddels, J.W.; Forest, P.; Quaker, T.; Com, O. Process of Preparing Gamma-Valerolactone. U.S. Patent 2,786,852, 26 March 1957. [Google Scholar]
- Serrano-Ruiz, J.C.; Wang, D.; Dumesic, J.A. Catalytic Upgrading of Levulinic Acid to 5-Nonanone. Green Chem. 2010, 12, 574–577. [Google Scholar] [CrossRef]
- Al-Shaal, M.G.; Wright, W.R.H.; Palkovits, R. Exploring the Ruthenium Catalysed Synthesis of γ-Valerolactone in Alcohols and Utilisation of Mild Solvent-Free Reaction Conditions. Green Chem. 2012, 14, 1260–1263. [Google Scholar] [CrossRef]
- Christian, R.V.J.; Brown, H.D.; Hixon, R.M. Derivatives of γ-Valerolactone, 1,4-Pentanediol and 1,4-Di-(β-Cyanoethoxy)-Pentane. J. Am. Chem. Soc. 1947, 69, 1961–1963. [Google Scholar] [CrossRef]
- Jones, D.R.; Iqbal, S.; Ishikawa, S.; Reece, C.; Thomas, L.M.; Miedziak, P.J.; Morgan, D.J.; Edwards, J.K.; Bartley, J.K.; Willock, D.J.; et al. The Conversion of Levulinic Acid into γ-Valerolactone Using Cu-ZrO2 Catalysts. Catal. Sci. Technol. 2016, 6, 6022–6030. [Google Scholar] [CrossRef]
- Hirayama, J.; Orlowski, I.; Iqbal, S.; Douthwaite, M.; Ishikawa, S.; Miedziak, P.J.; Bartley, J.K.; Edwards, J.; He, Q.; Jenkins, R.L.; et al. The Effects of Dopants on the Cu-ZrO2 Catalyzed Hydrogenation of Levulinic Acid. J. Phys. Chem. C 2018, 123, 7879–7888. [Google Scholar] [CrossRef]
- Obregón, I.; Corro, E.; Izquierdo, U.; Requies, J.; Arias, P.L. Levulinic Acid Hydrogenolysis on Al2O3-Based Ni-Cu Bimetallic Catalysts. Chinese J. Catal. 2014, 35, 656–662. [Google Scholar] [CrossRef]
- Gupta, S.S.R.; Kantam, M.L. Selective Hydrogenation of Levulinic Acid into γ-Valerolactone over Cu/Ni Hydrotalcite-Derived Catalyst. Catal. Today 2018, 309, 189–194. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, J.; Li, S.; Yin, J.; Sun, X.; Guo, X.; Song, C.; Zhou, J. Hydrogenation of Levulinic Acid into Gamma-Valerolactone over In Situ Reduced CuAg Bimetallic Catalyst: Strategy and Mechanism of Preventing Cu Leaching. Appl. Catal. B Environ. 2018, 232, 1–10. [Google Scholar] [CrossRef]
- Guo, H.; Hiraga, Y.; Qi, X.; Smith, R.L. Hydrogen Gas-Free Processes for Single-Step Preparation of Transition-Metal Bifunctional Catalysts and One-Pot γ-Valerolactone Synthesis in Supercritical CO2-Ionic Liquid Systems. J. Supercrit. Fluids 2019, 147, 263–270. [Google Scholar] [CrossRef]
- Balla, P.; Perupogu, V.; Vanama, P.K.; Komandur, V.R.C. Hydrogenation of Biomass-Derived Levulinic Acid to γ-Valerolactone over Copper Catalysts Supported on ZrO2. J. Chem. Technol. Biotechnol. 2016, 91, 769–776. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Li, J.; Pippel, E.; Yang, H.; Gao, Z.; Qin, Y. High Efficiency Cu-ZnO Hydrogenation Catalyst: The Tailoring of Cu-ZnO Interface Sites by Molecular Layer Deposition. ACS Catal. 2015, 5, 5567–5573. [Google Scholar] [CrossRef]
- Sun, D.; Ohkubo, A.; Asami, K.; Katori, T.; Yamada, Y.; Sato, S. Vapor-Phase Hydrogenation of Levulinic Acid and Methyl Levulinate to Γ-Valerolactone over Non-Noble Metal-Based Catalysts. Mol. Catal. 2017, 437, 105–113. [Google Scholar] [CrossRef]
- Upare, P.P.; Jeong, M.G.; Hwang, Y.K.; Kim, D.H.; Kim, Y.D.; Hwang, D.W.; Lee, U.H.; Chang, J.S. Nickel-Promoted Copper-Silica Nanocomposite Catalysts for Hydrogenation of Levulinic Acid to Lactones Using Formic Acid as a Hydrogen Feeder. Appl. Catal. A Gen. 2015, 491, 127–135. [Google Scholar] [CrossRef]
- Yoshida, R.; Sun, D.; Yamada, Y.; Sato, S.; Hutchings, G.J. Vapor-Phase Hydrogenation of Levulinic Acid to Γ-Valerolactone over Cu-Ni Bimetallic Catalysts. Catal. Commun. 2017, 97, 79–82. [Google Scholar] [CrossRef]
- Lomate, S.; Sultana, A.; Fujitani, T. Effect of SiO2 support Properties on the Performance of Cu-SiO2 catalysts for the Hydrogenation of Levulinic Acid to Gamma Valerolactone Using Formic Acid as a Hydrogen Source. Catal. Sci. Technol. 2017, 7, 3073–3083. [Google Scholar] [CrossRef]
- Lomate, S.; Sultana, A.; Fujitani, T. Vapor Phase Catalytic Transfer Hydrogenation (CTH) of Levulinic Acid to γ-Valerolactone over Copper Supported Catalysts Using Formic Acid as Hydrogen Source. Catal. Letters 2018, 148, 348–358. [Google Scholar] [CrossRef]
- Ashokraju, M.; Mohan, V.; Murali, K.; Rao, M.V.; Raju, B.D.; Rao, K.S.R. Formic Acid Assisted Hydrogenation of Levulinic Acid to γ-Valerolactone over Ordered Mesoporous Cu/Fe2O3 catalyst Prepared by Hard Template Method. J. Chem. Sci. 2018, 130, 16. [Google Scholar] [CrossRef]
- Orlowski, I.; Douthwaite, M.; Iqbal, S.; Hayward, J.S.; Davies, T.E.; Bartley, J.K.; Miedziak, P.J.; Hirayama, J.; Morgan, D.J.; Willock, D.J.; et al. The Hydrogenation of Levulinic Acid to γ-Valerolactone over Cu–ZrO2 Catalysts Prepared by a PH-Gradient Methodology. J. Energy Chem. 2019, 36, 15–24. [Google Scholar] [CrossRef]
- Obregón, I.; Gandarias, I.; Miletić, N.; Ocio, A.; Arias, P.L. One-Pot 2-Methyltetrahydrofuran Production from Levulinic Acid in Green Solvents Using Ni-Cu/Al2O3 Catalysts. ChemSusChem 2015, 8, 3483–3488. [Google Scholar] [CrossRef]
- Tang, X.; Sun, Y.; Zeng, X.; Hao, W.; Lin, L.; Liu, S. Novel Process for the Extraction of Ethyl Levulinate by Toluene with Less Humins from the Ethanolysis Products of Carbohydrates. Energy and Fuels 2014, 28, 4251–4255. [Google Scholar] [CrossRef]
- Hu, X.; Lievens, C.; Larcher, A.; Li, C.Z. Reaction Pathways of Glucose during Esterification: Effects of Reaction Parameters on the Formation of Humin Type Polymers. Bioresour. Technol. 2011, 102, 10104–10113. [Google Scholar] [CrossRef]
- Hu, X.; Li, C.Z. Levulinic Esters from the Acid-Catalysed Reactions of Sugars and Alcohols as Part of a Bio-Refinery. Green Chem. 2011, 13, 1676–1679. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, J.; Guo, Y.; Chen, L. Effective Upgrade of Levulinic Acid into γ-Valerolactone over an Inexpensive and Magnetic Catalyst Derived from Hydrotalcite Precursor. ACS Sustain. Chem. Eng. 2015, 3, 1708–1714. [Google Scholar] [CrossRef]
- Zheng, J.; Zhu, J.; Xu, X.; Wang, W.; Li, J.; Zhao, Y.; Tang, K.; Song, Q.; Qi, X.; Kong, D.; et al. Continuous Hydrogenation of Ethyl Levulinate to γ-Valerolactone and 2-Methyl Tetrahydrofuran over Alumina Doped Cu/SiO2 Catalyst: The Potential of Commercialization. Sci. Rep. 2016, 6, 2–10. [Google Scholar] [CrossRef]
- Chia, M.; Dumesic, J.A. Liquid-Phase Catalytic Transfer Hydrogenation and Cyclization of Levulinic Acid and Its Esters to γ-Valerolactone over Metal Oxide Catalysts. Chem. Commun. 2011, 47, 12233–12235. [Google Scholar] [CrossRef]
- Tang, X.; Chen, H.; Hu, L.; Hao, W.; Sun, Y.; Zeng, X.; Lin, L.; Liu, S. Conversion of Biomass to γ-Valerolactone by Catalytic Transfer Hydrogenation of Ethyl Levulinate over Metal Hydroxides. Appl. Catal. B Environ. 2014, 147, 827–834. [Google Scholar] [CrossRef]
- Cao, X.; Liu, H.; Wei, J.; Tang, X.; Zeng, X.; Sun, Y.; Lei, T.; Zhao, G.; Lin, L. Effective Production of γ-Valerolactone from Biomass-Derived Methyl Levulinate over CuOx-CaCO3 Catalyst. Cuihua Xuebao Chin. J. Catal. 2019, 40, 192–203. [Google Scholar] [CrossRef]
- Yong, S.T.; Ooi, C.W.; Chai, S.P.; Wu, X.S. Review of Methanol Reforming-Cu-Based Catalysts, Surface Reaction Mechanisms, and Reaction Schemes. Int. J. Hydrogen Energy 2013, 38, 9541–9552. [Google Scholar] [CrossRef]
- Li, Z.; Tang, X.; Jiang, Y.; Wang, Y.; Zuo, M.; Chen, W.; Zeng, X.; Sun, Y.; Lin, L. Atom-Economical Synthesis of γ-Valerolactone with Self-Supplied Hydrogen from Methanol. Chem. Commun. 2015, 51, 16320–16323. [Google Scholar] [CrossRef]
- Cao, X.; Wei, J.; Liu, H.; Lv, X.; Tang, X.; Zeng, X.; Sun, Y.; Lei, T.; Liu, S.; Lin, L. Hydrogenation of Methyl Levulinate to γ-Valerolactone over Cu─Mg Oxide Using MeOH as in Situ Hydrogen Source. J. Chem. Technol. Biotechnol. 2019, 94, 167–177. [Google Scholar] [CrossRef]
- Zhang, R.; Ma, Y.; You, F.; Peng, T.; He, Z.; Li, K. Exploring to Direct the Reaction Pathway for Hydrogenation of Levulinic Acid into Γ-Valerolactone for Future Clean-Energy Vehicles over a Magnetic Cu-Ni Catalyst. Int. J. Hydrogen Energy 2017, 42, 25185–25194. [Google Scholar] [CrossRef]
- Dong, F.; Zhu, Y.; Ding, G.; Cui, J.; Li, X.; Li, Y.; Ding, G.; Li, Y.; Dong, F.; Cui, J.; et al. One-Step Conversion of Furfural into 2-Methyltetrahydrofuran under Mild Conditions. ChemSusChem 2015, 8, 1534–1537. [Google Scholar] [CrossRef]
- Du, X.-L.; Bi, Q.-Y.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Tunable Copper-Catalyzed Chemoselective Hydrogenolysis of Biomass-Derived γ-Valerolactone into 1,4-Pentanediol or 2-Methyltetrahydrofuran. Green Chem. 2012, 14, 935. [Google Scholar] [CrossRef]
- Bermudez, J.M.; Menendez, J.A.; Romero, A.A.; Serrano, E.; Garcia-Martinez, J.; Luque, R.; Menéndez, J.A.; Romero, A.A.; Serrano, E.; Garcia-Martinez, J.; et al. Continuous Flow Nanocatalysis: Reaction Pathways in the Conversion of Levulinic Acid to Valuable Chemicals. Green Chem. 2013, 15, 2786–2792. [Google Scholar] [CrossRef]
- Xie, Z.; Chen, B.; Wu, H.; Liu, M.; Liu, H.; Zhang, J.; Yang, G.; Han, B. Highly Efficient Hydrogenation of Levulinic Acid into 2-Methyltetrahydrofuran over Ni-Cu/Al2O3-ZrO2 Bifunctional Catalysts. Green Chem. 2019, 21, 606–613. [Google Scholar] [CrossRef]











| Catalyst | Space Velocity (h−1) | H2/FUR Molar Ratio | T (°C) | TOS (h) | C a (%) | Y b (%) | Ref. |
|---|---|---|---|---|---|---|---|
| CuCr2O7 | 52 (WHSV) | 25 | 200 | 4 | 22 | 20 (FOL) | [51] |
| Cu/SiO2 | 2.3 (WHSV) | 25 | 290 | 0.25 | 77 | 63 (FOL) | [55] |
| Cu/SiO2 | 0.5 (WHSV) | 17 | 140 | 10 | 90 | 73 (FOL) | [61] |
| Cu/SiO2 | 0.5 (WHSV) | 17 | 220 | 210 | 100 | 89.5 (MF) | [61] |
| Cu/SiO2 | 1.5 (WHSV) | 11.5 | 170 | 1 | 91 | 85 (FOL) | [77] |
| Cu/SiO2 | 1.5(WHSV) | 11.5 | 210 | 14 | 95 | 80 (MF) | [62] |
| Cu/Sep c | 1.5 (WHSV) | 11.5 | 210 | 5 | 83 | 72 (FOL) | [78] |
| Cu/Ker d | 1.5 (WHSV) | 11.5 | 190 | 5 | 91 | 50 (MF) | [79] |
| CuCo/SiO2 | 3.1 (WHSV) | 6 | 200 | 12 | 65 | 64 (FOL) | [80] |
| CuCa/SiO2 | 0.33 (LHSV) | 5.1 | 130 | 80 | 100 | 98 (FOL) | [81] |
| CuPd/Zeo e | 7.7 (WHSV) | 0.08 | 300 | - | 58 | 58 (FOL) | [82] |
| Cu/MgO | 4.8 (WHSV) | 2.5 | 180 | 5 | 98 | 96 (FOL) | [65] |
| Cu/MgO/Sep c | 1.5 (WHSV) | 11.5 | 210 | 5 | 73 | 64 (FOL) | [83] |
| CuCa/MgO | 1.7 (WHSV) | 10 | 180 | 0.5 | 91 | 90 (FOL) | [69] |
| Cu/ZnO | 0.5 (WHSV) | 17 | 220 | 10 | 95 | 31 (FOL) | [61] |
| Cu/ZnO | 1.5 (WHSV) | 11.5 | 210 | 5 | 93 | 76 (FOL) | [70] |
| Cu/ZnO | 1.5 (LHSV) | 15 | 200 | 16 | 100 | 94 (MF) | [71] |
| Cu/ZnO/Al2O3 | 0.5 (LHSV) | 15 | 120 | 16 | 97 | 94 (FOL) | [73] |
| Cu/ZnO/Al2O3 | 3.6 (LHSV) | 15 | 200 | 30 | 76 | 73 (FOL) | [74] |
| Cu/ZnO/Ker d | 1.5 (WHSV) | 11.5 | 190 | 5 | 62 | 55 (FOL) | [79] |
| Cu/ZnO/Sep c | 1.5 (WHSV) | 11.5 | 210 | 5 | 81 | 58 (FOL) | [83] |
| Cu/CeO2 | 1.5 (WHSV) | 11.5 | 190 | 5 | 81 | 67 (FOL) | [75] |
| Cu/CeO2/Al2O3 | 60 (W/F) | - | 175 | 6 | 90 | 72 (FOL) | [76] |
| Cu/CeO2/Ker d | 1.5 (WHSV) | 11.5 | 190 | 5 | 64 | 61 (FOL) | [79] |
| Cu/CeO2/Sep c | 1.5 (WHSV) | 11.5 | 210 | 5 | 66 | 61 (FOL) | [83] |
| Catalyst | Hydrogen Source | T (°C) | P (MPa) | t (h) | C a (%) | yGVL b (%) | Ref. |
|---|---|---|---|---|---|---|---|
| CuO–Cr2O3 | H2 | 273 | 10.1 | - | - | 62 | [170] |
| Cu–ZrO2 | H2 | 200 | 3.4 | 5 | 100 | 100 | [11] |
| Cu–Al2O3 | H2 | 200 | 3.4 | 5 | 100 | 100 | [11] |
| Cu–ZrO2 | H2 | 200 | 3.5 | 2 | 100 | 80 | [171] |
| Cu–ZrO2 | H2 | 200 | 3.5 | 2 | 100 | 100 | [29] |
| Mn/Cu–ZrO2 | H2 | 200 | 2.6 | 0.5 | 82 | 82 | [172] |
| Ni–Cu/Al2O3 | H2 | 250 | 6.4 | 2 | 100 | 96 | [173] |
| CuAl2O4 | H2 | 200 | 6.9 | 10 | 98 | 87 | [124] |
| CuCr2O4 | H2 | 200 | 6.9 | 10 | >99 | 91 | [124] |
| CuFe2O4 | H2 | 200 | 6.9 | 10 | >99 | 82 | [124] |
| Cu/Al | H2 | 140 | 3.0 | 3 | 76 | 58 | [174] |
| Cu/Mg/Al | H2 | 140 | 3.0 | 3 | 100 | 82 | [174] |
| Cu/Ni/Mg/Al | H2 | 140 | 3.0 | 3 | 100 | 100 | [174] |
| CuAg/Al2O3 | H2 | 180 | 1.4 | 4 | 100 | >99 | [175] |
| Cu/CuO–FC | FA c | 170 | 8.4 | 6 | 99 | 50 | [176] |
| CuO–SiO2 | H2 | 290 | 0.5 | 67 | >99 | 93 | [112] |
| Cu/Al2O3 | H2 | 265 | 0.1 | 4 | 98 | 85 | [113] |
| Cu/ZrO2 | H2 | 265 | 0.1 | 4 | 81 | 67 | [177] |
| Cu–ZnO | H2 | 240 | 1.0 | 20 | - | 70 | [178] |
| Cu/SiO2 | H2 | 250 | 0.1 | 5 | 73 | 55 | [179] |
| Cu/Al2O3 | H2 | 250 | 0.1 | 5 | 98 | 95 | [179] |
| Cu–Ni/SiO2 | H2 | 250 | 0.1 | - | 98 | 96 | [20] |
| Cu/SiO2 | H2 | 265 | 1.0 | 100 | 100 | 99.9 | [14] |
| Ni–Cu/SiO2 | FA c | 265 | 0.1 | 100 | 98 | 90 | [180] |
| Cu–Ni/SiO2 | H2 | 250 | 0.1 | 50 | 100 | 99 | [181] |
| Cu/SiO2 | FA c | 270 | 0.1 | - | 66 | 53 | [182] |
| Cu/SiO2 | FA c | 250 | 0.1 | - | 56 | 49 | [183] |
| Cu/Fe2O3 | FA c | 250 | 0.1 | 3 | 100 | 100 | [184] |
| Catalyst | Solvent | Hydrogen Source | T (°C) | P (MPa) | T (h) | C a (%) | yGVL b (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Cu–ZrO2 | Methanol | H2 | 200 | 3.4 | 5 | 100 | 90 | [11] |
| Magnetic Ni/Cu/Mg/Al/Fe | Methanol | H2 | 142 | 2 | 3 | 100 | 98 | [190] |
| Cu/Al2O3 | Methanol | H2 | 24 | 0.1 | 5 | 94 | 86 | [179] |
| Cu/γ-Al2O3 | Ethanol | H2 | 200 | 4.9 | 6 | 100 | 93 | [23] |
| Cu–WO3/ZrO2 | Ethanol | H2 | 200 | 4.9 | 6 | 100 | 94 | [23] |
| Cu | Methanol | Methanol | 240 | — | 1 | 97 | 85 | [21] |
| CuCr2O4 | Methanol | Methanol | 250 | 0.1 | 4 | 94 | 96 | [191] |
| Cu–MgO | Methanol | Methanol | 220 | — | 4 | 96 | 91 | [192] |
| CuOx–CaCO3 | Methanol | Methanol | 240 | — | 3 | >99 | 96 | [193] |
| Cu/C | 2-Propanol | 2-Propanol | 220 | 2.0 | 5 | >99 | 89 | [10] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Sancho, C.; Mérida-Robles, J.M.; Cecilia-Buenestado, J.A.; Moreno-Tost, R.; Maireles-Torres, P.J. The Role of Copper in the Hydrogenation of Furfural and Levulinic Acid. Int. J. Mol. Sci. 2023, 24, 2443. https://doi.org/10.3390/ijms24032443
García-Sancho C, Mérida-Robles JM, Cecilia-Buenestado JA, Moreno-Tost R, Maireles-Torres PJ. The Role of Copper in the Hydrogenation of Furfural and Levulinic Acid. International Journal of Molecular Sciences. 2023; 24(3):2443. https://doi.org/10.3390/ijms24032443
Chicago/Turabian StyleGarcía-Sancho, Cristina, Josefa María Mérida-Robles, Juan Antonio Cecilia-Buenestado, Ramón Moreno-Tost, and Pedro Jesús Maireles-Torres. 2023. "The Role of Copper in the Hydrogenation of Furfural and Levulinic Acid" International Journal of Molecular Sciences 24, no. 3: 2443. https://doi.org/10.3390/ijms24032443
APA StyleGarcía-Sancho, C., Mérida-Robles, J. M., Cecilia-Buenestado, J. A., Moreno-Tost, R., & Maireles-Torres, P. J. (2023). The Role of Copper in the Hydrogenation of Furfural and Levulinic Acid. International Journal of Molecular Sciences, 24(3), 2443. https://doi.org/10.3390/ijms24032443