
Methyl Donors, Epigenetic Alterations, and Brain Health: Understanding the Connection

Abstract
1. Introduction
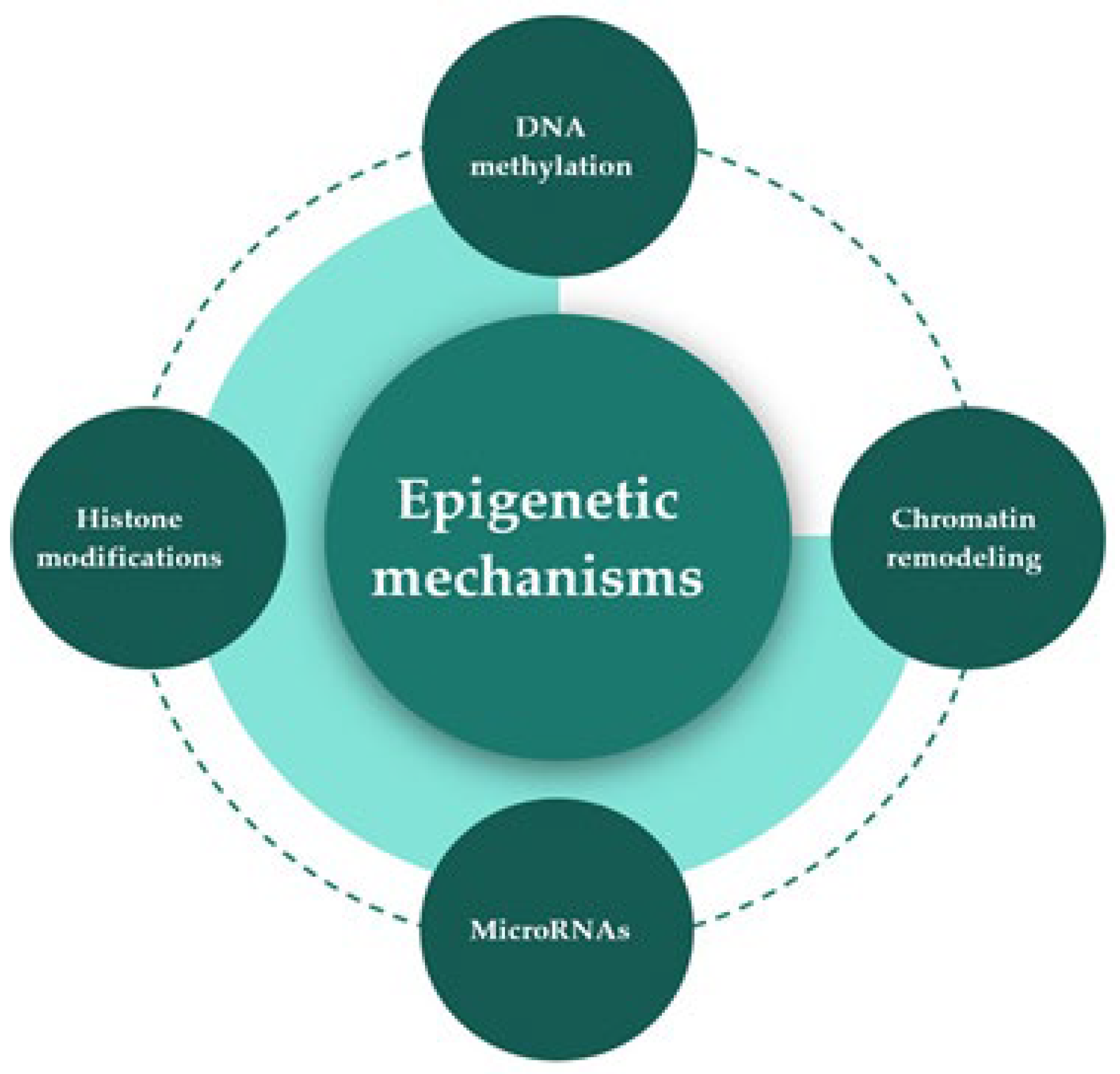
2. Epigenetic Mechanisms
3. One-Carbon Metabolism
4. Dietary Methyl Donors, Epigenetic Alterations, and Stress-Related Disorders
4.1. HPA Axis Programming by Early Life Stress and the Role of Methylation
4.2. Potential Neuroprotective Effects of Dietary Methyl Donors: More or Less?
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abdul, Q.A.; Yu, B.P.; Chung, H.Y.; Jung, H.A.; Choi, J.S. Epigenetic Modifications of Gene Expression by Lifestyle and Environment. Arch. Pharm. Res. 2017, 40, 1219–1237. [Google Scholar] [CrossRef]
- Zeisel, S.H. Choline: Needed for Normal Development of Memory. J. Am. Coll. Nutr. 2000, 19 (Suppl. S5), 528S–531S. [Google Scholar] [CrossRef]
- Niculescu, M.D.; Zeisel, S.H. Diet, Methyl Donors and DNA Methylation: Interactions between Dietary Folate, Methionine and Choline. J. Nutr. 2002, 132 (Suppl. S8), 2333S–2335S. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H. Nutritional Importance of Choline for Brain Development. J. Am. Coll. Nutr. 2004, 23 (Suppl. S6), 621S–626S. [Google Scholar] [CrossRef]
- Bekdash, R.A. Neuroprotective Effects of Choline and Other Methyl Donors. Nutrients 2019, 11, 2995. [Google Scholar] [CrossRef] [PubMed]
- Mentch, S.J.; Locasale, J.W. One-Carbon Metabolism and Epigenetics: Understanding the Specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.K.; Paal, M.C.; Donohue, T.M.; Ganesan, M.; Osna, N.A.; Kharbanda, K.K. Beneficial Effects of Betaine: A Comprehensive Review. Biology 2021, 10, 456. [Google Scholar] [CrossRef]
- Irvine, N.; England-Mason, G.; Field, C.J.; Dewey, D.; Aghajafari, F. Prenatal Folate and Choline Levels and Brain and Cognitive Development in Children: A Critical Narrative Review. Nutrients 2022, 14, 364. [Google Scholar] [CrossRef]
- Chmielewska, N.; Szyndler, J.; Maciejak, P.; Płaźnik, A. Epigenetic Mechanisms of Stress and Depression. Psychiatr. Pol. 2019, 53, 1413–1428. [Google Scholar] [CrossRef]
- Penner-Goeke, S.; Binder, E.B. Epigenetics and Depression. Dialogues Clin. Neurosci. 2019, 21, 397–405. [Google Scholar] [CrossRef]
- Klengel, T.; Pape, J.; Binder, E.B.; Mehta, D. The Role of DNA Methylation in Stress-Related Psychiatric Disorders. Neuropharmacology 2014, 80, 115–132. [Google Scholar] [CrossRef]
- Torres-Berrío, A.; Issler, O.; Parise, E.M.; Nestler, E.J. Unraveling the Epigenetic Landscape of Depression: Focus on Early Life Stress. Dialogues Clin. Neurosci. 2019, 21, 341–357. [Google Scholar] [CrossRef]
- Panariello, F.; Fanelli, G.; Fabbri, C.; Atti, A.R.; De Ronchi, D.; Serretti, A. Epigenetic Basis of Psychiatric Disorders: A Narrative Review. CNS Neurol. Disord. Drug Targets 2022, 21, 302–315. [Google Scholar] [CrossRef]
- Matrisciano, F.; Tueting, P.; Dalal, I.; Kadriu, B.; Grayson, D.R.; Davis, J.M.; Nicoletti, F.; Guidotti, A. Epigenetic Modifications of GABAergic Interneurons Are Associated with the Schizophrenia-like Phenotype Induced by Prenatal Stress in Mice. Neuropharmacology 2013, 68, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Anier, K.; Malinovskaja, K.; Pruus, K.; Aonurm-Helm, A.; Zharkovsky, A.; Kalda, A. Maternal Separation Is Associated with DNA Methylation and Behavioural Changes in Adult Rats. Eur. Neuropsychopharmacol. 2014, 24, 459–468. [Google Scholar] [CrossRef]
- Lux, V. Epigenetic Programming Effects of Early Life Stress: A Dual-Activation Hypothesis. Curr. Genom. 2018, 19, 638–652. [Google Scholar] [CrossRef]
- van Bodegom, M.; Homberg, J.R.; Henckens, M.J.A.G. Modulation of the Hypothalamic-Pituitary-Adrenal Axis by Early Life Stress Exposure. Front. Cell. Neurosci. 2017, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Henzel, K.S.; Pearson, B.L.; Siwek, M.E.; Papazoglou, A.; Guo, L.; Paesler, K.; Yu, M.; Müller, R.; Xie, K.; et al. A Paternal Methyl Donor-Rich Diet Altered Cognitive and Neural Functions in Offspring Mice. Mol. Psychiatry 2018, 23, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Ishii, D.; Matsuzawa, D.; Matsuda, S.; Tomizawa, H.; Sutoh, C.; Shimizu, E. Methyl Donor-Deficient Diet during Development Can Affect Fear and Anxiety in Adulthood in C57BL/6J Mice. PLoS ONE 2014, 9, e105750. [Google Scholar] [CrossRef]
- Paternain, L.; Martisova, E.; Campión, J.; Martínez, J.A.; Ramírez, M.J.; Milagro, F.I. Methyl Donor Supplementation in Rats Reverses the Deleterious Effect of Maternal Separation on Depression-like Behaviour. Behav. Brain Res. 2016, 299, 51–58. [Google Scholar] [CrossRef]
- Sahara, Y.; Matsuzawa, D.; Ishii, D.; Fuchida, T.; Goto, T.; Sutoh, C.; Shimizu, E. Paternal Methyl Donor Deficient Diets during Development Affect Male Offspring Behavior and Memory-Related Gene Expression in Mice. Dev. Psychobiol. 2019, 61, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Robertson, K.D.; Wolffe, A.P. DNA Methylation in Health and Disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P.; Wolffe, A.P. Methylation-Induced Repression—Belts, Braces, and Chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Esteller, M. DNA Methylomes, Histone Codes and MiRNAs: Tying It All Together. Int. J. Biochem. Cell Biol. 2009, 41, 87–95. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA Methylation in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG Islands and the Regulation of Transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Klose, R.J.; Bird, A.P. Genomic DNA Methylation: The Mark and Its Mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Chen, Z.; Riggs, A.D. DNA Methylation and Demethylation in Mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef]
- Jeltsch, A. Beyond Watson and Crick: DNA Methylation and Molecular Enzymology of DNA Methyltransferases. Chembiochem 2002, 3, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and Biology of Mammalian DNA Methyltransferases. Cell. Mol. Life Sci. 2004, 61, 2571–2587. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Christensen, J.; Helin, K. DNA Methylation: TET Proteins—Guardians of CpG Islands? EMBO Rep. 2012, 13, 28–35. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Clark, E.; Mulcahey, M.; Huang, S.; Shi, Y.G. TET1 Is a DNA-Binding Protein That Modulates DNA Methylation and Gene Transcription via Hydroxylation of 5-Methylcytosine. Cell Res. 2010, 20, 1390–1393. [Google Scholar] [CrossRef]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and Epigenetics: An Interplay of Dietary Methyl Donors, One-Carbon Metabolism and DNA Methylation. J. Nutr. Biochem. 2012, 23, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Zhang, Y.; Reinberg, D. Transcription Regulation by Histone Methylation: Interplay between Different Covalent Modifications of the Core Histone Tails. Genes Dev. 2001, 15, 2343–2360. [Google Scholar] [CrossRef] [PubMed]
- Lachner, M.; Jenuwein, T. The Many Faces of Histone Lysine Methylation. Curr. Opin. Cell Biol. 2002, 14, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Gregory, P.D.; Wagner, K.; Hörz, W. Histone Acetylation and Chromatin Remodeling. Exp. Cell Res. 2001, 265, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Schratt, G. Fine-Tuning Neural Gene Expression with MicroRNAs. Curr. Opin. Neurobiol. 2009, 19, 213–219. [Google Scholar] [CrossRef]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The Impact of MicroRNAs on Protein Output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef]
- Kapsimali, M.; Kloosterman, W.P.; de Bruijn, E.; Rosa, F.; Plasterk, R.H.A.; Wilson, S.W. MicroRNAs Show a Wide Diversity of Expression Profiles in the Developing and Mature Central Nervous System. Genome Biol. 2007, 8, R173. [Google Scholar] [CrossRef] [PubMed]
- Barbato, C.; Giorgi, C.; Catalanotto, C.; Cogoni, C. Thinking about RNA? MicroRNAs in the Brain. Mamm. Genome 2008, 19, 541–551. [Google Scholar] [CrossRef]
- Wahid, F.; Khan, T.; Kim, Y.Y. MicroRNA and Diseases: Therapeutic Potential as New Generation of Drugs. Biochimie 2014, 104, 12–26. [Google Scholar] [CrossRef]
- Juźwik, C.A.; Drake, S.S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. MicroRNA Dysregulation in Neurodegenerative Diseases: A Systematic Review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef]
- Wang, W.; Kwon, E.J.; Tsai, L.-H. MicroRNAs in Learning, Memory, and Neurological Diseases. Learn. Mem. 2012, 19, 359–368. [Google Scholar] [CrossRef]
- Mehta, S.L.; Chokkalla, A.K.; Vemuganti, R. Noncoding RNA Crosstalk in Brain Health and Diseases. Neurochem. Int. 2021, 149, 105139. [Google Scholar] [CrossRef] [PubMed]
- Friso, S.; Udali, S.; De Santis, D.; Choi, S.-W. One-Carbon Metabolism and Epigenetics. Mol. Asp. Med. 2017, 54, 28–36. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Shuvalov, O.; Petukhov, A.; Daks, A.; Fedorova, O.; Vasileva, E.; Barlev, N.A. One-Carbon Metabolism and Nucleotide Biosynthesis as Attractive Targets for Anticancer Therapy. Oncotarget 2017, 8, 23955–23977. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. One-Carbon Epigenetics and Redox Biology of Neurodegeneration. Free Radic. Biol. Med. 2021, 170, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Kalhan, S.C.; Marczewski, S.E. Methionine, Homocysteine, One Carbon Metabolism and Fetal Growth. Rev. Endocr. Metab. Disord. 2012, 13, 109–119. [Google Scholar] [CrossRef]
- Zeisel, S.H. Choline: Essential for Brain Development and Function. Adv. Pediatr. 1997, 44, 263–295. [Google Scholar]
- Fisher, M.C.; Zeisel, S.H.; Mar, M.-H.; Sadler, T.W. Perturbations in Choline Metabolism Cause Neural Tube Defects in Mouse Embryos in Vitro. FASEB J. 2002, 16, 619–621. [Google Scholar] [CrossRef]
- Zeisel, S.H. A Brief History of Choline. Ann. Nutr. Metab. 2012, 61, 254–258. [Google Scholar] [CrossRef]
- Tayebati, S.K.; Marucci, G.; Santinelli, C.; Buccioni, M.; Amenta, F. Choline-Containing Phospholipids: Structure-Activity Relationships Versus Therapeutic Applications. Curr. Med. Chem. 2015, 22, 4328–4340. [Google Scholar] [CrossRef]
- Ueland, P.M. Choline and Betaine in Health and Disease. J. Inherit. Metab. Dis. 2011, 34, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; He, F.; Wu, C.; Li, P.; Li, N.; Deng, J.; Zhu, G.; Ren, W.; Peng, Y. Betaine in Inflammation: Mechanistic Aspects and Applications. Front. Immunol. 2018, 9, 1070. [Google Scholar] [CrossRef] [PubMed]
- Slow, S.; Elmslie, J.; Lever, M. Dietary Betaine and Inflammation. Am. J. Clin. Nutr. 2008, 88, 247–248, author reply 248. [Google Scholar] [CrossRef]
- van Gool, J.D.; Hirche, H.; Lax, H.; De Schaepdrijver, L. Folic Acid and Primary Prevention of Neural Tube Defects: A Review. Reprod. Toxicol. 2018, 80, 73–84. [Google Scholar] [CrossRef]
- Bottiglieri, T. Folate, Vitamin B₁₂, and S-Adenosylmethionine. Psychiatr. Clin. N. Am. 2013, 36, 1–13. [Google Scholar] [CrossRef]
- Mattson, M.P.; Shea, T.B. Folate and Homocysteine Metabolism in Neural Plasticity and Neurodegenerative Disorders. Trends Neurosci. 2003, 26, 137–146. [Google Scholar] [CrossRef]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, Folate, and the Methionine Remethylation Cycle-Biochemistry, Pathways, and Regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef]
- Smith, A.D.; Warren, M.J.; Refsum, H. Vitamin B12. Adv. Food Nutr. Res. 2018, 83, 215–279. [Google Scholar] [CrossRef] [PubMed]
- van de Lagemaat, E.E.; de Groot, L.C.P.G.M.; van den Heuvel, E.G.H.M. Vitamin B12 in Relation to Oxidative Stress: A Systematic Review. Nutrients 2019, 11, 482. [Google Scholar] [CrossRef]
- di Salvo, M.L.; Contestabile, R.; Safo, M.K. Vitamin B(6) Salvage Enzymes: Mechanism, Structure and Regulation. Biochim. Biophys. Acta 2011, 1814, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Spinneker, A.; Sola, R.; Lemmen, V.; Castillo, M.J.; Pietrzik, K.; González-Gross, M. Vitamin B6 Status, Deficiency and Its Consequences—An Overview. Nutr. Hosp. 2007, 22, 7–24. [Google Scholar] [PubMed]
- Hellmann, H.; Mooney, S. Vitamin B6: A Molecule for Human Health? Molecules 2010, 15, 442–459. [Google Scholar] [CrossRef]
- Mandaviya, P.R.; Stolk, L.; Heil, S.G. Homocysteine and DNA Methylation: A Review of Animal and Human Literature. Mol. Genet. Metab. 2014, 113, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D. Metabolic Regulatory Properties of S-Adenosylmethionine and S-Adenosylhomocysteine. Clin. Chem. Lab. Med. 2007, 45, 1694–1699. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Zhou, X.; Yin, W.; Wan, K.; Zhang, W.; Li, C.; Li, M.; Zhu, W.; Zhu, X.; Sun, Z. The Influence of MTHFR Polymorphism on Gray Matter Volume in Patients with Amnestic Mild Cognitive Impairment. Front. Neurosci. 2021, 15, 778123. [Google Scholar] [CrossRef]
- Faravelli, C.; Lo Sauro, C.; Lelli, L.; Pietrini, F.; Lazzeretti, L.; Godini, L.; Benni, L.; Fioravanti, G.; Talamba, G.A.; Castellini, G.; et al. The Role of Life Events and HPA Axis in Anxiety Disorders: A Review. Curr. Pharm. Des. 2012, 18, 5663–5674. [Google Scholar] [CrossRef]
- Koe, A.S.; Salzberg, M.R.; Morris, M.J.; O’Brien, T.J.; Jones, N.C. Early Life Maternal Separation Stress Augmentation of Limbic Epileptogenesis: The Role of Corticosterone and HPA Axis Programming. Psychoneuroendocrinology 2014, 42, 124–133. [Google Scholar] [CrossRef]
- Weaver, I.C.G.; Korgan, A.C.; Lee, K.; Wheeler, R.V.; Hundert, A.S.; Goguen, D. Stress and the Emerging Roles of Chromatin Remodeling in Signal Integration and Stable Transmission of Reversible Phenotypes. Front. Behav. Neurosci. 2017, 11, 41. [Google Scholar] [CrossRef]
- Saunderson, E.A.; Spiers, H.; Mifsud, K.R.; Gutierrez-Mecinas, M.; Trollope, A.F.; Shaikh, A.; Mill, J.; Reul, J.M.H.M. Stress-Induced Gene Expression and Behavior Are Controlled by DNA Methylation and Methyl Donor Availability in the Dentate Gyrus. Proc. Natl. Acad. Sci. USA 2016, 113, 4830–4835. [Google Scholar] [CrossRef]
- Szyf, M. The Early Life Social Environment and DNA Methylation: DNA Methylation Mediating the Long-Term Impact of Social Environments Early in Life. Epigenetics 2011, 6, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Babenko, O.; Kovalchuk, I.; Metz, G.A.S. Stress-Induced Perinatal and Transgenerational Epigenetic Programming of Brain Development and Mental Health. Neurosci. Biobehav. Rev. 2015, 48, 70–91. [Google Scholar] [CrossRef] [PubMed]
- McKee, S.E.; Reyes, T.M. Effect of Supplementation with Methyl-Donor Nutrients on Neurodevelopment and Cognition: Considerations for Future Research. Nutr. Rev. 2018, 76, 497–511. [Google Scholar] [CrossRef]
- Dauncey, M.J. Nutrition, the Brain and Cognitive Decline: Insights from Epigenetics. Eur. J. Clin. Nutr. 2014, 68, 1179–1185. [Google Scholar] [CrossRef]
- Vanhees, K.; Vonhögen, I.G.C.; van Schooten, F.J.; Godschalk, R.W.L. You Are What You Eat, and so Are Your Children: The Impact of Micronutrients on the Epigenetic Programming of Offspring. Cell. Mol. Life Sci. 2014, 71, 271–285. [Google Scholar] [CrossRef]
- Lucassen, P.J.; Naninck, E.F.G.; van Goudoever, J.B.; Fitzsimons, C.; Joels, M.; Korosi, A. Perinatal Programming of Adult Hippocampal Structure and Function; Emerging Roles of Stress, Nutrition and Epigenetics. Trends Neurosci. 2013, 36, 621–631. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Physiology and Neurobiology of Stress and Adaptation: Central Role of the Brain. Physiol. Rev. 2007, 87, 873–904. [Google Scholar] [CrossRef]
- McEwen, B.S. Protective and Damaging Effects of Stress Mediators: Central Role of the Brain. Dialogues Clin. Neurosci. 2006, 8, 367–381. [Google Scholar] [CrossRef]
- de Kloet, E.R.; Joëls, M.; Holsboer, F. Stress and the Brain: From Adaptation to Disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef]
- Shirazi, S.N.; Friedman, A.R.; Kaufer, D.; Sakhai, S.A. Glucocorticoids and the Brain: Neural Mechanisms Regulating the Stress Response. Adv. Exp. Med. Biol. 2015, 872, 235–252. [Google Scholar] [CrossRef]
- Finsterwald, C.; Alberini, C.M. Stress and Glucocorticoid Receptor-Dependent Mechanisms in Long-Term Memory: From Adaptive Responses to Psychopathologies. Neurobiol. Learn. Mem. 2014, 112, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Schatzberg, A.F.; Keller, J.; Tennakoon, L.; Lembke, A.; Williams, G.; Kraemer, F.B.; Sarginson, J.E.; Lazzeroni, L.C.; Murphy, G.M. HPA Axis Genetic Variation, Cortisol and Psychosis in Major Depression. Mol. Psychiatry 2014, 19, 220–227. [Google Scholar] [CrossRef]
- Porter, R.J.; Gallagher, P. Abnormalities of the HPA Axis in Affective Disorders: Clinical Subtypes and Potential Treatments. Acta Neuropsychiatr. 2006, 18, 193–209. [Google Scholar] [CrossRef]
- Shah, J.L.; Malla, A.K. Much Ado about Much: Stress, Dynamic Biomarkers and HPA Axis Dysregulation along the Trajectory to Psychosis. Schizophr. Res. 2015, 162, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A.; Beedle, A.S. Early Life Events and Their Consequences for Later Disease: A Life History and Evolutionary Perspective. Am. J. Hum. Biol. 2007, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Mood Disorders and Allostatic Load. Biol. Psychiatry 2003, 54, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Tyborowska, A.; Volman, I.; Niermann, H.C.M.; Pouwels, J.L.; Smeekens, S.; Cillessen, A.H.N.; Toni, I.; Roelofs, K. Early-Life and Pubertal Stress Differentially Modulate Grey Matter Development in Human Adolescents. Sci. Rep. 2018, 8, 9201. [Google Scholar] [CrossRef]
- Saleh, A.; Potter, G.G.; McQuoid, D.R.; Boyd, B.; Turner, R.; MacFall, J.R.; Taylor, W.D. Effects of Early Life Stress on Depression, Cognitive Performance and Brain Morphology. Psychol. Med. 2017, 47, 171–181. [Google Scholar] [CrossRef]
- Vaiserman, A.M.; Koliada, A.K. Early-Life Adversity and Long-Term Neurobehavioral Outcomes: Epigenome as a Bridge? Hum. Genom. 2017, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Perez-Polo, J.R. Epigenetics: Stress and Disease. Int. J. Dev. Neurosci. 2017, 62, 54–55. [Google Scholar] [CrossRef]
- Labonté, B.; Suderman, M.; Maussion, G.; Lopez, J.P.; Navarro-Sánchez, L.; Yerko, V.; Mechawar, N.; Szyf, M.; Meaney, M.J.; Turecki, G. Genome-Wide Methylation Changes in the Brains of Suicide Completers. Am. J. Psychiatry 2013, 170, 511–520. [Google Scholar] [CrossRef]
- Bick, J.; Naumova, O.; Hunter, S.; Barbot, B.; Lee, M.; Luthar, S.S.; Raefski, A.; Grigorenko, E.L. Childhood Adversity and DNA Methylation of Genes Involved in the Hypothalamus-Pituitary-Adrenal Axis and Immune System: Whole-Genome and Candidate-Gene Associations. Dev. Psychopathol. 2012, 24, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Blanco Rodríguez, J.; Camprubí Sánchez, C. Epigenetic Transgenerational Inheritance. Adv. Exp. Med. Biol. 2019, 1166, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.G. Integrating Early Life Experience, Gene Expression, Brain Development, and Emergent Phenotypes: Unraveling the Thread of Nature via Nurture. Adv. Genet. 2014, 86, 277–307. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.G.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic Programming by Maternal Behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef]
- McGowan, P.O.; Sasaki, A.; D’Alessio, A.C.; Dymov, S.; Labonté, B.; Szyf, M.; Turecki, G.; Meaney, M.J. Epigenetic Regulation of the Glucocorticoid Receptor in Human Brain Associates with Childhood Abuse. Nat. Neurosci. 2009, 12, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Lubin, F.D.; Funk, A.J.; Sweatt, J.D. Lasting Epigenetic Influence of Early-Life Adversity on the BDNF Gene. Biol. Psychiatry 2009, 65, 760–769. [Google Scholar] [CrossRef]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmühl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.X.; Spengler, D. Dynamic DNA Methylation Programs Persistent Adverse Effects of Early-Life Stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- McCoy, C.R.; Rana, S.; Stringfellow, S.A.; Day, J.J.; Wyss, J.M.; Clinton, S.M.; Kerman, I.A. Neonatal Maternal Separation Stress Elicits Lasting DNA Methylation Changes in the Hippocampus of Stress-Reactive Wistar Kyoto Rats. Eur. J. Neurosci. 2016, 44, 2829–2845. [Google Scholar] [CrossRef] [PubMed]
- McGill, B.E.; Bundle, S.F.; Yaylaoglu, M.B.; Carson, J.P.; Thaller, C.; Zoghbi, H.Y. Enhanced Anxiety and Stress-Induced Corticosterone Release Are Associated with Increased Crh Expression in a Mouse Model of Rett Syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 18267–18272. [Google Scholar] [CrossRef]
- Fyffe, S.L.; Neul, J.L.; Samaco, R.C.; Chao, H.-T.; Ben-Shachar, S.; Moretti, P.; McGill, B.E.; Goulding, E.H.; Sullivan, E.; Tecott, L.H.; et al. Deletion of Mecp2 in Sim1-Expressing Neurons Reveals a Critical Role for MeCP2 in Feeding Behavior, Aggression, and the Response to Stress. Neuron 2008, 59, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, L.; Bellia, F.; Pavoncello, N.; Vigli, D.; D’Addario, C.; De Filippis, B. Methyl-CpG Binding Protein 2 Dysfunction Provides Stress Vulnerability with Sex- and Zygosity-Dependent Outcomes. Eur. J. Neurosci. 2022, 55, 2766–2776. [Google Scholar] [CrossRef] [PubMed]
- Abellán-Álvaro, M.; Stork, O.; Agustín-Pavón, C.; Santos, M. MeCP2 Haplodeficiency and Early-Life Stress Interaction on Anxiety-like Behavior in Adolescent Female Mice. J. Neurodev. Disord. 2021, 13, 59. [Google Scholar] [CrossRef]
- Bockmühl, Y.; Patchev, A.V.; Madejska, A.; Hoffmann, A.; Sousa, J.C.; Sousa, N.; Holsboer, F.; Almeida, O.F.X.; Spengler, D. Methylation at the CpG Island Shore Region Upregulates Nr3c1 Promoter Activity after Early-Life Stress. Epigenetics 2015, 10, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Wu, Y.; Bockmühl, Y.; Spengler, D. Genes Learn from Stress: How Infantile Trauma Programs Us for Depression. Epigenetics 2010, 5, 194–199. [Google Scholar] [CrossRef]
- Mueller, B.R.; Bale, T.L. Sex-Specific Programming of Offspring Emotionality after Stress Early in Pregnancy. J. Neurosci. 2008, 28, 9055–9065. [Google Scholar] [CrossRef]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA Methylation-Related Chromatin Remodeling in Activity-Dependent BDNF Gene Regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef]
- Su, M.; Hong, J.; Zhao, Y.; Liu, S.; Xue, X. MeCP2 Controls Hippocampal Brain-Derived Neurotrophic Factor Expression via Homeostatic Interactions with MicroRNA-132 in Rats with Depression. Mol. Med. Rep. 2015, 12, 5399–5406. [Google Scholar] [CrossRef]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic Regulation of BDNF Gene Transcription in the Consolidation of Fear Memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Carretero, J.; Benvegnù, S.; Dotti, C.G.; Martin, M.G. Neuronal Activity Controls Bdnf Expression via Polycomb De-Repression and CREB/CBP/JMJD3 Activation in Mature Neurons. Nat. Commun. 2016, 7, 11081. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, A.; Rao, B.S.S.; Nair, D.; Trinh, M.; Mawjee, N.; Tonegawa, S.; Chattarji, S. Transgenic Brain-Derived Neurotrophic Factor Expression Causes Both Anxiogenic and Antidepressant Effects. Proc. Natl. Acad. Sci. USA 2006, 103, 13208–13213. [Google Scholar] [CrossRef] [PubMed]
- Boersma, G.J.; Lee, R.S.; Cordner, Z.A.; Ewald, E.R.; Purcell, R.H.; Moghadam, A.A.; Tamashiro, K.L. Prenatal Stress Decreases Bdnf Expression and Increases Methylation of Bdnf Exon IV in Rats. Epigenetics 2014, 9, 437–447. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic Modifications and Human Disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Bailey, L.B.; Rampersaud, G.C.; Kauwell, G.P.A. Folic Acid Supplements and Fortification Affect the Risk for Neural Tube Defects, Vascular Disease and Cancer: Evolving Science. J. Nutr. 2003, 133, 1961S–1968S. [Google Scholar] [CrossRef]
- Lam, N.S.K.; Long, X.X.; Li, X.; Saad, M.; Lim, F.; Doery, J.C.; Griffin, R.C.; Galletly, C. The Potential Use of Folate and Its Derivatives in Treating Psychiatric Disorders: A Systematic Review. Biomed. Pharmacother. 2022, 146, 112541. [Google Scholar] [CrossRef]
- Cortés-Albornoz, M.C.; García-Guáqueta, D.P.; Velez-van-Meerbeke, A.; Talero-Gutiérrez, C. Maternal Nutrition and Neurodevelopment: A Scoping Review. Nutrients 2021, 13, 3530. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, T.; Chen, L.; Dai, Y.; Wu, L.-J.; Jia, F.-Y.; Hao, Y.; Li, L.; Zhang, J.; Ke, X.-Y.; et al. Serum Folate Status Is Primarily Associated with Neurodevelopment in Children with Autism Spectrum Disorders Aged Three and Under-A Multi-Center Study in China. Front. Nutr. 2021, 8, 661223. [Google Scholar] [CrossRef] [PubMed]
- Villamor, E.; Rifas-Shiman, S.L.; Gillman, M.W.; Oken, E. Maternal Intake of Methyl-Donor Nutrients and Child Cognition at 3 Years of Age. Paediatr. Perinat. Epidemiol. 2012, 26, 328–335. [Google Scholar] [CrossRef]
- Roth, C.; Magnus, P.; Schjølberg, S.; Stoltenberg, C.; Surén, P.; McKeague, I.W.; Davey Smith, G.; Reichborn-Kjennerud, T.; Susser, E. Folic Acid Supplements in Pregnancy and Severe Language Delay in Children. JAMA 2011, 306, 1566–1573. [Google Scholar] [CrossRef]
- Nilsson, T.K.; Yngve, A.; Böttiger, A.K.; Hurtig-Wennlöf, A.; Sjöström, M. High Folate Intake Is Related to Better Academic Achievement in Swedish Adolescents. Pediatrics 2011, 128, e358–e365. [Google Scholar] [CrossRef]
- Craciunescu, C.N.; Brown, E.C.; Mar, M.-H.; Albright, C.D.; Nadeau, M.R.; Zeisel, S.H. Folic Acid Deficiency during Late Gestation Decreases Progenitor Cell Proliferation and Increases Apoptosis in Fetal Mouse Brain. J. Nutr. 2004, 134, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Blaise, S.A.; Nédélec, E.; Schroeder, H.; Alberto, J.-M.; Bossenmeyer-Pourié, C.; Guéant, J.-L.; Daval, J.-L. Gestational Vitamin B Deficiency Leads to Homocysteine-Associated Brain Apoptosis and Alters Neurobehavioral Development in Rats. Am. J. Pathol. 2007, 170, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, S.M.; Mesfioui, A.; Berkiks, I.; Ennaciri, A.; Chahirou, Y.; Diagana, Y.; Ouichou, A.; El Midaoui, A.; El Hessni, A. Effects of the Methyl Donors Supplementation on Hippocampal Oxidative Stress, Depression and Anxiety in Chronically High Fructose-Treated Rats. Neuroscience 2021, 476, 1–11. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Otaegui-Arrazola, A.; Amiano, P.; Elbusto, A.; Urdaneta, E.; Martínez-Lage, P. Diet, Cognition, and Alzheimer’s Disease: Food for Thought. Eur. J. Nutr. 2014, 53, 1–23. [Google Scholar] [CrossRef]
- Ravaglia, G.; Forti, P.; Maioli, F.; Martelli, M.; Servadei, L.; Brunetti, N.; Porcellini, E.; Licastro, F. Homocysteine and Folate as Risk Factors for Dementia and Alzheimer Disease. Am. J. Clin. Nutr. 2005, 82, 636–643. [Google Scholar] [CrossRef]
- Gröber, U.; Kisters, K.; Schmidt, J. Neuroenhancement with Vitamin B12-Underestimated Neurological Significance. Nutrients 2013, 5, 5031–5045. [Google Scholar] [CrossRef]
- Dauncey, M.J. Recent Advances in Nutrition, Genes and Brain Health. Proc. Nutr. Soc. 2012, 71, 581–591. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Tang, M.-X.; Miller, J.; Green, R.; Mayeux, R. Relation of Higher Folate Intake to Lower Risk of Alzheimer Disease in the Elderly. Arch. Neurol. 2007, 64, 86–92. [Google Scholar] [CrossRef]
- Kennedy, B.P.; Bottiglieri, T.; Arning, E.; Ziegler, M.G.; Hansen, L.A.; Masliah, E. Elevated S-Adenosylhomocysteine in Alzheimer Brain: Influence on Methyltransferases and Cognitive Function. J. Neural Transm. 2004, 111, 547–567. [Google Scholar] [CrossRef]
- Fuso, A.; Nicolia, V.; Pasqualato, A.; Fiorenza, M.T.; Cavallaro, R.A.; Scarpa, S. Changes in Presenilin 1 Gene Methylation Pattern in Diet-Induced B Vitamin Deficiency. Neurobiol. Aging 2011, 32, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Ricceri, L.; Cavallaro, R.A.; Isopi, E.; Mangia, F.; Fiorenza, M.T.; Scarpa, S. S-Adenosylmethionine Reduces the Progress of the Alzheimer-like Features Induced by B-Vitamin Deficiency in Mice. Neurobiol. Aging 2012, 33, e1–e1482. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, H.; Yu, M.; Zhang, X.; Zhang, M.; Wilson, J.X.; Huang, G. Folic Acid Administration Inhibits Amyloid β-Peptide Accumulation in APP/PS1 Transgenic Mice. J. Nutr. Biochem. 2015, 26, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain Glutathione Levels—A Novel Biomarker for Mild Cognitive Impairment and Alzheimer’s Disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef]
- Saharan, S.; Mandal, P.K. The Emerging Role of Glutathione in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 40, 519–529. [Google Scholar] [CrossRef]
- Sun, J.; Wen, S.; Zhou, J.; Ding, S. Association between Malnutrition and Hyperhomocysteine in Alzheimer’s Disease Patients and Diet Intervention of Betaine. J. Clin. Lab. Anal. 2016, 31, e22090. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Slack, B.E.; Mellott, T.J. Neuroprotective Actions of Dietary Choline. Nutrients 2017, 9, 815. [Google Scholar] [CrossRef]
- Velazquez, R.; Ferreira, E.; Knowles, S.; Fux, C.; Rodin, A.; Winslow, W.; Oddo, S. Lifelong Choline Supplementation Ameliorates Alzheimer’s Disease Pathology and Associated Cognitive Deficits by Attenuating Microglia Activation. Aging Cell 2019, 18, e13037. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase Inhibitors as Alzheimer’s Therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef]
- Mellott, T.J.; Huleatt, O.M.; Shade, B.N.; Pender, S.M.; Liu, Y.B.; Slack, B.E.; Blusztajn, J.K. Perinatal Choline Supplementation Reduces Amyloidosis and Increases Choline Acetyltransferase Expression in the Hippocampus of the APPswePS1dE9 Alzheimer’s Disease Model Mice. PLoS ONE 2017, 12, e0170450. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, X.; Chen, X.; Cai, Y.; Ma, Y.; Ma, J.; Zhang, Q.; Dai, L.; Fan, X.; Bai, Y. Choline Supplementation Ameliorates Behavioral Deficits and Alzheimer’s Disease-Like Pathology in Transgenic APP/PS1 Mice. Mol. Nutr. Food Res. 2019, 63, 1801407. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Methyl Donors | Function | References |
|---|---|---|
| Methionine | Precursor for SAM formation, maintenance of the redox state, and brain health. | [57] |
| Choline | Regulation of cholinergic signaling, maintaining cellular membrane integrity, and contributing to the formation of SAM. | [58,59,60,61] |
| Betaine | Choline precursor, a methyl-donor in the BHMT pathway, and anti-inflammatory functions. | [62,63,64] |
| Folic acid | Normal brain development, nucleotide synthesis, and prevention of neural tube defects. | [65,66,67] |
| Vitamin B12 | Nucleotide synthesis, antioxidant properties, and maintaining brain health. | [68,69,70] |
| Vitamin B6 | Maintenance of the redox state and brain health. Role in transamination and decarboxylation reactions required for the metabolism of several neurotransmitters. Nucleotide synthesis and protein/lipid metabolism. | [71,72,73] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bekdash, R.A. Methyl Donors, Epigenetic Alterations, and Brain Health: Understanding the Connection. Int. J. Mol. Sci. 2023, 24, 2346. https://doi.org/10.3390/ijms24032346
Bekdash RA. Methyl Donors, Epigenetic Alterations, and Brain Health: Understanding the Connection. International Journal of Molecular Sciences. 2023; 24(3):2346. https://doi.org/10.3390/ijms24032346
Chicago/Turabian StyleBekdash, Rola A. 2023. "Methyl Donors, Epigenetic Alterations, and Brain Health: Understanding the Connection" International Journal of Molecular Sciences 24, no. 3: 2346. https://doi.org/10.3390/ijms24032346
APA StyleBekdash, R. A. (2023). Methyl Donors, Epigenetic Alterations, and Brain Health: Understanding the Connection. International Journal of Molecular Sciences, 24(3), 2346. https://doi.org/10.3390/ijms24032346