
The SMN Complex at the Crossroad between RNA Metabolism and Neurodegeneration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. SMN Complex Function in RNA Metabolism
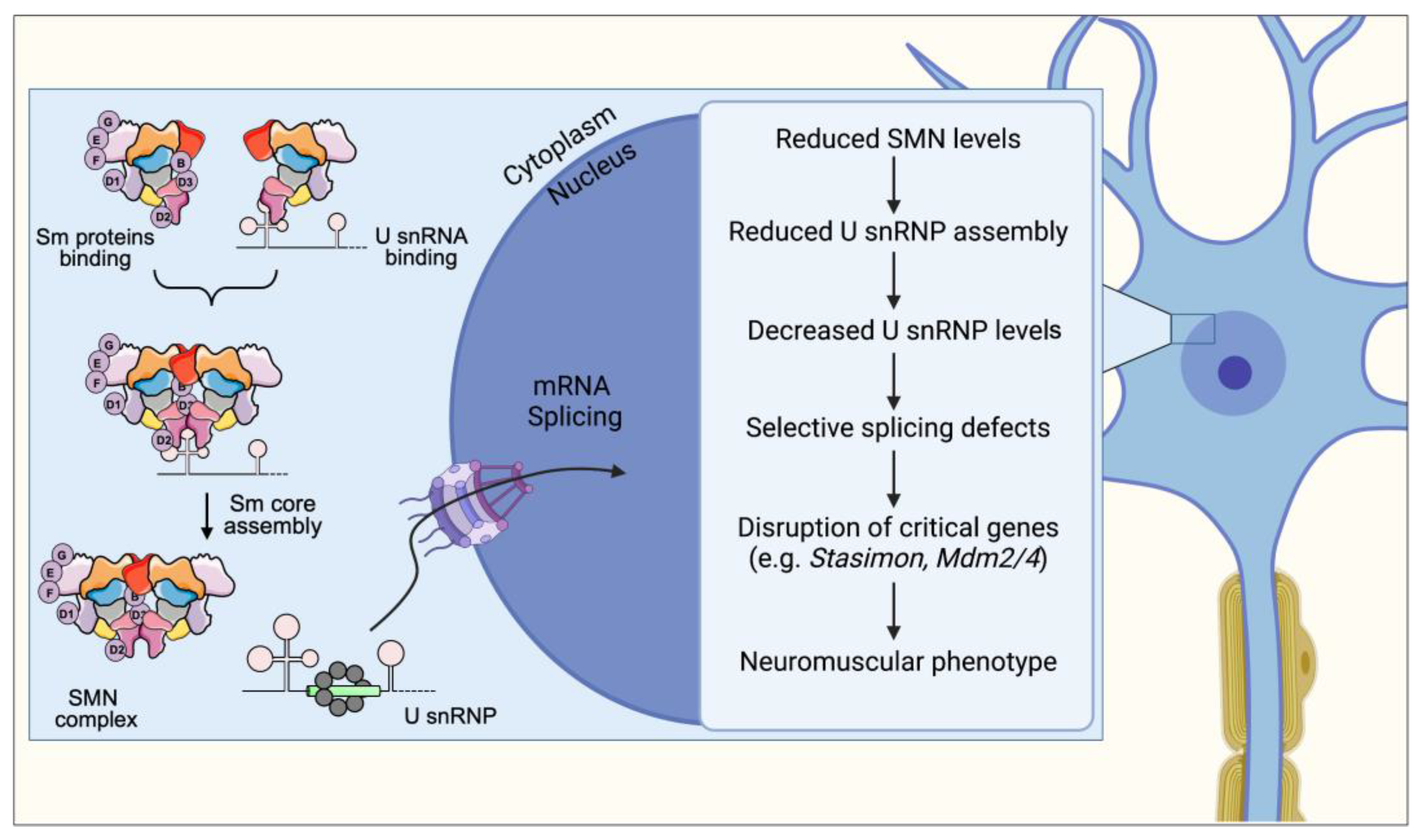
3. Spliceosomal snRNP Assembly
4. U7 snRNP Assembly
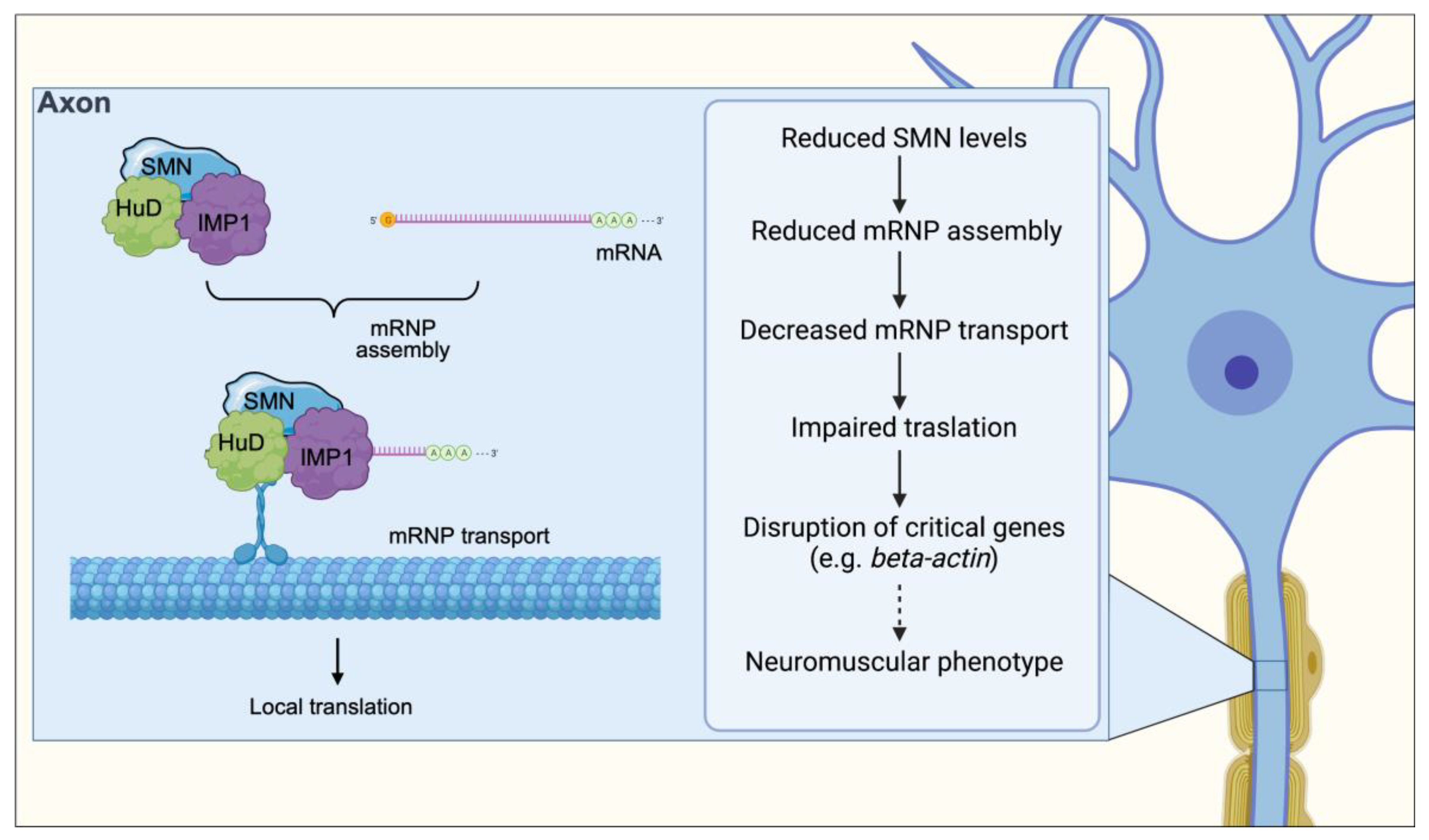
5. Messenger RNP Assembly and Axonal Transport
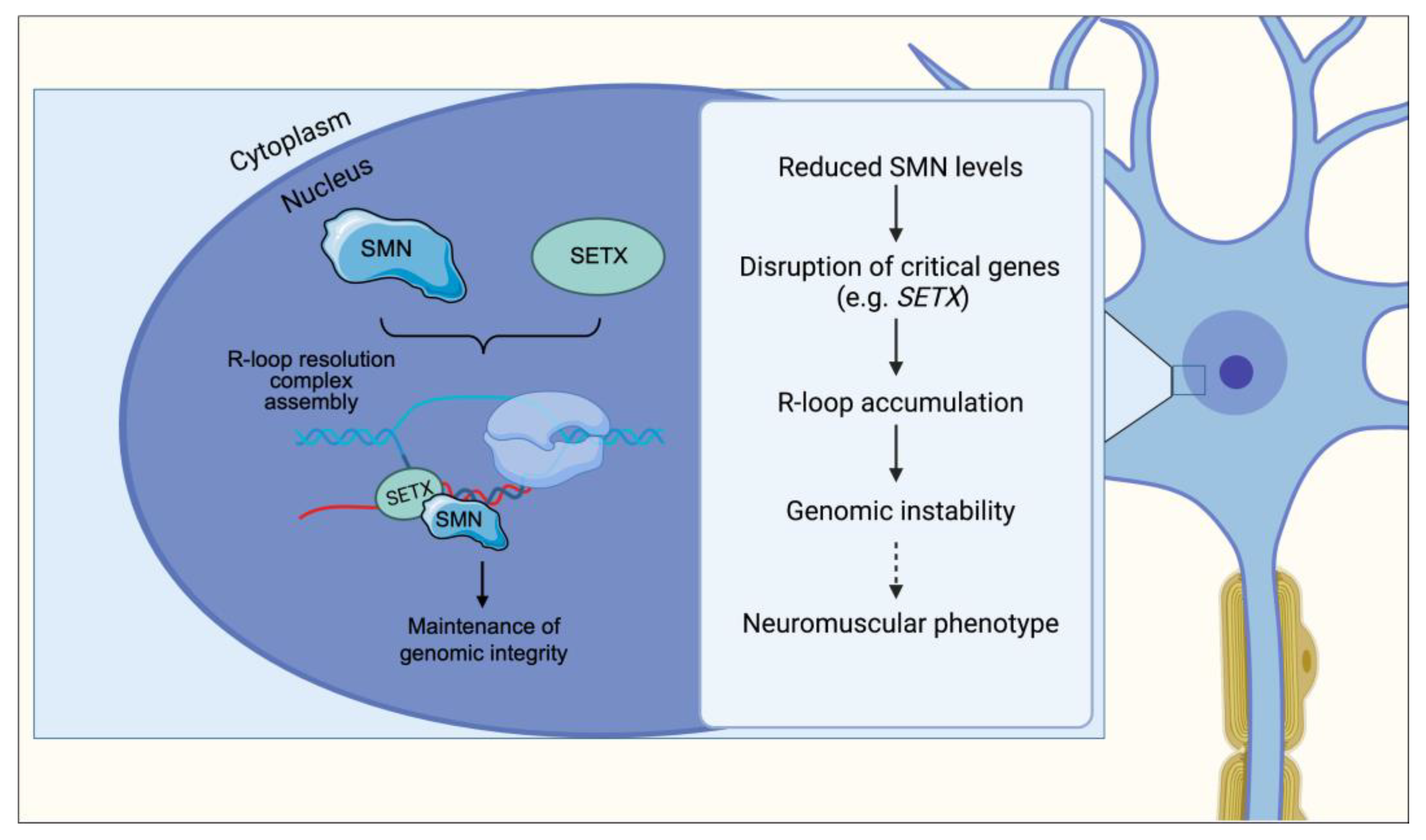
6. R-Loop Resolution and Transcription Termination
7. RNA Metabolism Defects in SMA
8. Spliceosomal snRNP Assembly Defects and Its Consequences on mRNA Splicing
9. U7 snRNP Assembly and Histone mRNA Processing Impairment
10. Altered mRNP Assembly and Axonal Transport
11. R-Loop Accumulation and Genomic Instability
12. Alterations in Non-Coding RNAs
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Battle, D.J.; Kasim, M.; Yong, J.; Lotti, F.; Lau, C.-K.; Mouaikel, J.; Zhang, Z.; Han, K.; Wan, L.; Dreyfuss, G. The SMN Complex: An Assembly Machine for RNPs. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Sumner, C.J.; Muntoni, F.; Darras, B.T.; Finkel, R.S. Spinal Muscular Atrophy. Nat. Rev. Dis. Primer 2022, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Pellizzoni, L. Chaperoning Ribonucleoprotein Biogenesis in Health and Disease. EMBO Rep. 2007, 8, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Li, D.K.; Tisdale, S.; Lotti, F.; Pellizzoni, L. SMN Control of RNP Assembly: From Post-Transcriptional Gene Regulation to Motor Neuron Disease. Semin. Cell Dev. Biol. 2014, 32, 22–29. [Google Scholar] [CrossRef]
- Tisdale, S.; Pellizzoni, L. Disease Mechanisms and Therapeutic Approaches in Spinal Muscular Atrophy. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 8691–8700. [Google Scholar] [CrossRef]
- Costa, C.J.; Willis, D.E. To the End of the Line: Axonal MRNA Transport and Local Translation in Health and Neurodegenerative Disease. Dev. Neurobiol. 2017, 78, 209–220. [Google Scholar] [CrossRef]
- Donlin-Asp, P.G.; Bassell, G.J.; Rossoll, W. A Role for the Survival of Motor Neuron Protein in MRNP Assembly and Transport. Curr. Opin. Neurobiol. 2016, 39, 53–61. [Google Scholar] [CrossRef]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A Single Nucleotide in the SMN Gene Regulates Splicing and Is Responsible for Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to SMN2 Pre-MRNA-Protein Complex Elicits Specificity for Small Molecule Splicing Modifiers. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Lally, C.; Jones, C.; Farwell, W.; Reyna, S.P.; Cook, S.F.; Flanders, W.D. Indirect Estimation of the Prevalence of Spinal Muscular Atrophy Type I, II, and III in the United States. Orphanet J. Rare Dis. 2017, 12, 175. [Google Scholar] [CrossRef]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, Incidence and Carrier Frequency of 5q–Linked Spinal Muscular Atrophy – a Literature Review. Orphanet J. Rare Dis. 2017, 12. [Google Scholar] [CrossRef]
- De Sanctis, R.; Coratti, G.; Pasternak, A.; Montes, J.; Pane, M.; Mazzone, E.S.; Young, S.D.; Salazar, R.; Quigley, J.; Pera, M.C.; et al. Developmental Milestones in Type I Spinal Muscular Atrophy. Neuromuscul. Disord. 2016, 26, 754–759. [Google Scholar] [CrossRef]
- Talbot, K.; Tizzano, E.F. The Clinical Landscape for SMA in a New Therapeutic Era. Gene Ther. 2017, 24, 529–533. [Google Scholar] [CrossRef]
- Harada, Y.; Sutomo, R.; Sadewa, A.H.; Akutsu, T.; Takeshima, Y.; Wada, H.; Matsuo, M.; Nishio, H. Correlation between SMN2 Copy Number and Clinical Phenotype of Spinal Muscular Atrophy: Three SMN2 Copies Fail to Rescue Some Patients from the Disease Severity. J. Neurol. 2002, 249, 1211–1219. [Google Scholar] [CrossRef]
- Liu, Q.; Dreyfuss, G. A Novel Nuclear Structure Containing the Survival of Motor Neurons Protein. EMBO J. 1996, 15, 3555–3565. [Google Scholar] [CrossRef]
- Liu, Q.; Fischer, U.; Wang, F.; Dreyfuss, G. The Spinal Muscular Atrophy Disease Gene Product, SMN, and Its Associated Protein SIP1 Are in a Complex with Spliceosomal SnRNP Proteins. Cell 1997, 90, 1013–1021. [Google Scholar] [CrossRef]
- Brahms, H.; Meheus, L.; de Brabandere, V.; Fischer, U.; Lührmann, R. Symmetrical Dimethylation of Arginine Residues in Spliceosomal Sm Protein B/B’ and the Sm-like Protein LSm4, and Their Interaction with the SMN Protein. RNA 2001, 7, 1531–1542. [Google Scholar] [CrossRef]
- Tripsianes, K.; Madl, T.; Machyna, M.; Fessas, D.; Englbrecht, C.; Fischer, U.; Neugebauer, K.M.; Sattler, M. Structural Basis for Dimethylarginine Recognition by the Tudor Domains of Human SMN and SPF30 Proteins. Nat. Struct. Mol. Biol. 2011, 18, 1414–1420. [Google Scholar] [CrossRef]
- Lorson, C.L.; Strasswimmer, J.; Yao, J.M.; Baleja, J.D.; Hahnen, E.; Wirth, B.; Le, T.; Burghes, A.H.; Androphy, E.J. SMN Oligomerization Defect Correlates with Spinal Muscular Atrophy Severity. Nat. Genet. 1998, 19, 63–66. [Google Scholar] [CrossRef]
- Pellizzoni, L.; Charroux, B.; Dreyfuss, G. SMN Mutants of Spinal Muscular Atrophy Patients Are Defective in Binding to SnRNP Proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11167–11172. [Google Scholar] [CrossRef]
- Martin, R.; Gupta, K.; Ninan, N.S.; Perry, K.; Van Duyne, G.D. The Survival Motor Neuron Protein Forms Soluble Glycine Zipper Oligomers. Struct. Lond. Engl. 1993 2012, 20, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Battle, D.J.; Kasim, M.; Wang, J.; Dreyfuss, G. SMN-Independent Subunits of the SMN Complex. Identification of a Small Nuclear Ribonucleoprotein Assembly Intermediate. J. Biol. Chem. 2007, 282, 27953–27959. [Google Scholar] [CrossRef] [PubMed]
- Carissimi, C.; Saieva, L.; Gabanella, F.; Pellizzoni, L. Gemin8 Is Required for the Architecture and Function of the Survival Motor Neuron Complex. J. Biol. Chem. 2006, 281, 37009–37016. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Zhao, S.; Francisco-Velilla, R.; Zhang, J.; Embarc-Buh, A.; Abellan, S.; Lv, M.; Tang, P.; Gong, Q.; Shen, H.; et al. Structural Basis for Gemin5 Decamer-Mediated MRNA Binding. Nat. Commun. 2022, 13, 5166. [Google Scholar] [CrossRef]
- Pacheco, A.; López de Quinto, S.; Ramajo, J.; Fernández, N.; Martínez-Salas, E. A Novel Role for Gemin5 in MRNA Translation. Nucleic Acids Res. 2009, 37, 582–590. [Google Scholar] [CrossRef]
- Workman, E.; Kalda, C.; Patel, A.; Battle, D.J. Gemin5 Binds to the Survival Motor Neuron MRNA to Regulate SMN Expression. J. Biol. Chem. 2015, 290, 15662–15669. [Google Scholar] [CrossRef]
- Yong, J.; Kasim, M.; Bachorik, J.L.; Wan, L.; Dreyfuss, G. Gemin5 Delivers SnRNA Precursors to the SMN Complex for SnRNP Biogenesis. Mol. Cell 2010, 38, 551–562. [Google Scholar] [CrossRef]
- Kroiss, M.; Schultz, J.; Wiesner, J.; Chari, A.; Sickmann, A.; Fischer, U. Evolution of an RNP Assembly System: A Minimal SMN Complex Facilitates Formation of UsnRNPs in Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 2008, 105, 10045–10050. [Google Scholar] [CrossRef]
- Burghes, A.H.M.; Beattie, C.E. Spinal Muscular Atrophy: Why Do Low Levels of Survival Motor Neuron Protein Make Motor Neurons Sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef]
- Bradrick, S.S.; Gromeier, M. Identification of Gemin5 as a Novel 7-Methylguanosine Cap-Binding Protein. PloS ONE 2009, 4, e7030. [Google Scholar] [CrossRef]
- Francisco-Velilla, R.; Fernandez-Chamorro, J.; Dotu, I.; Martinez-Salas, E. The Landscape of the Non-Canonical RNA-Binding Site of Gemin5 Unveils a Feedback Loop Counteracting the Negative Effect on Translation. Nucleic Acids Res. 2018, 46, 7339–7353. [Google Scholar] [CrossRef]
- Francisco-Velilla, R.; Fernandez-Chamorro, J.; Ramajo, J.; Martinez-Salas, E. The RNA-Binding Protein Gemin5 Binds Directly to the Ribosome and Regulates Global Translation. Nucleic Acids Res. 2016, 44, 8335–8351. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-MRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef]
- Meister, G.; Hannus, S.; Plöttner, O.; Baars, T.; Hartmann, E.; Fakan, S.; Laggerbauer, B.; Fischer, U. SMNrp Is an Essential Pre-MRNA Splicing Factor Required for the Formation of the Mature Spliceosome. EMBO J. 2001, 20, 2304–2314. [Google Scholar] [CrossRef]
- Pellizzoni, L.; Charroux, B.; Rappsilber, J.; Mann, M.; Dreyfuss, G. A Functional Interaction between the Survival Motor Neuron Complex and RNA Polymerase II. J. Cell Biol. 2001, 152, 75–85. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Patel, A.A.; Steitz, J.A. Splicing Double: Insights from the Second Spliceosome. Nat. Rev. Mol. Cell Biol. 2003, 4, 960–970. [Google Scholar] [CrossRef]
- Chari, A.; Golas, M.M.; Klingenhäger, M.; Neuenkirchen, N.; Sander, B.; Englbrecht, C.; Sickmann, A.; Stark, H.; Fischer, U. An Assembly Chaperone Collaborates with the SMN Complex to Generate Spliceosomal SnRNPs. Cell 2008, 135, 497–509. [Google Scholar] [CrossRef]
- Neuenkirchen, N.; Englbrecht, C.; Ohmer, J.; Ziegenhals, T.; Chari, A.; Fischer, U. Reconstitution of the Human U SnRNP Assembly Machinery Reveals Stepwise Sm Protein Organization. EMBO J. 2015, 34, 1925–1941. [Google Scholar] [CrossRef]
- Friesen, W.J.; Paushkin, S.; Wyce, A.; Massenet, S.; Pesiridis, G.S.; Van Duyne, G.; Rappsilber, J.; Mann, M.; Dreyfuss, G. The Methylosome, a 20S Complex Containing JBP1 and PICln, Produces Dimethylarginine-Modified Sm Proteins. Mol. Cell. Biol. 2001, 21, 8289–8300. [Google Scholar] [CrossRef]
- Grimm, C.; Chari, A.; Pelz, J.-P.; Kuper, J.; Kisker, C.; Diederichs, K.; Stark, H.; Schindelin, H.; Fischer, U. Structural Basis of Assembly Chaperone- Mediated SnRNP Formation. Mol. Cell 2013, 49, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Mu, L.; Shen, C.; Kong, X.; Wang, Y.; Hou, Y.; Zhang, R. Negative Cooperativity between Gemin2 and RNA Provides Insights into RNA Selection and the SMN Complex’s Release in SnRNP Assembly. Nucleic Acids Res. 2020, 48, 895–911. [Google Scholar] [CrossRef] [PubMed]
- Fornerod, M.; Ohno, M. Exportin-Mediated Nuclear Export of Proteins and Ribonucleoproteins. Results Probl. Cell Differ. 2002, 35, 67–91. [Google Scholar] [PubMed]
- Fischer, U.; Lührmann, R. An Essential Signaling Role for the M3G Cap in the Transport of U1 SnRNP to the Nucleus. Science 1990, 249, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Mouaikel, J.; Narayanan, U.; Verheggen, C.; Matera, A.G.; Bertrand, E.; Tazi, J.; Bordonné, R. Interaction between the Small-Nuclear-RNA Cap Hypermethylase and the Spinal Muscular Atrophy Protein, Survival of Motor Neuron. EMBO Rep. 2003, 4, 616–622. [Google Scholar] [CrossRef]
- Mouaikel, J.; Verheggen, C.; Bertrand, E.; Tazi, J.; Bordonné, R. Hypermethylation of the Cap Structure of Both Yeast SnRNAs and SnoRNAs Requires a Conserved Methyltransferase That Is Localized to the Nucleolus. Mol. Cell 2002, 9, 891–901. [Google Scholar] [CrossRef]
- Narayanan, U.; Achsel, T.; Lührmann, R.; Matera, A.G. Coupled in Vitro Import of U SnRNPs and SMN, the Spinal Muscular Atrophy Protein. Mol. Cell 2004, 16, 223–234. [Google Scholar] [CrossRef]
- Matera, A.G.; Terns, R.M.; Terns, M.P. Non-Coding RNAs: Lessons from the Small Nuclear and Small Nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 209–220. [Google Scholar] [CrossRef]
- Courchaine, E.M.; Barentine, A.E.S.; Straube, K.; Lee, D.-R.; Bewersdorf, J.; Neugebauer, K.M. DMA-Tudor Interaction Modules Control the Specificity of in Vivo Condensates. Cell 2021, 184, 3612–3625.e17. [Google Scholar] [CrossRef]
- Lafarga, V.; Tapia, O.; Sharma, S.; Bengoechea, R.; Stoecklin, G.; Lafarga, M.; Berciano, M.T. CBP-Mediated SMN Acetylation Modulates Cajal Body Biogenesis and the Cytoplasmic Targeting of SMN. Cell. Mol. Life Sci. CMLS 2018, 75, 527–546. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Faravelli, I.; Kuwajima, T.; Delestrée, N.; Dermentzaki, G.; De Planell-Saguer, M.; Rinchetti, P.; Hao, L.T.; Beattie, C.C.; Corti, S.; et al. Sumoylation Regulates the Assembly and Activity of the SMN Complex. Nat. Commun. 2021, 12, 5040. [Google Scholar] [CrossRef]
- Schilling, M.; Prusty, A.B.; Boysen, B.; Oppermann, F.S.; Riedel, Y.L.; Husedzinovic, A.; Rasouli, H.; König, A.; Ramanathan, P.; Reymann, J.; et al. TOR Signaling Regulates Liquid Phase Separation of the SMN Complex Governing SnRNP Biogenesis. Cell Rep. 2021, 35, 109277. [Google Scholar] [CrossRef]
- Pellizzoni, L.; Yong, J.; Dreyfuss, G. Essential Role for the SMN Complex in the Specificity of SnRNP Assembly. Science 2002, 298, 1775–1779. [Google Scholar] [CrossRef]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and Regulation of Canonical Histone MRNAs: Life without a Poly(A) Tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef]
- Pillai, R.S.; Grimmler, M.; Meister, G.; Will, C.L.; Lührmann, R.; Fischer, U.; Schümperli, D. Unique Sm Core Structure of U7 SnRNPs: Assembly by a Specialized SMN Complex and the Role of a New Component, Lsm11, in Histone RNA Processing. Genes Dev. 2003, 17, 2321–2333. [Google Scholar] [CrossRef]
- Marzluff, W.F.; Koreski, K.P. Birth and Death of Histone MRNAs. Trends Genet. TIG 2017, 33, 745–759. [Google Scholar] [CrossRef]
- Romeo, V.; Schümperli, D. Cycling in the Nucleus: Regulation of RNA 3’ Processing and Nuclear Organization of Replication-Dependent Histone Genes. Curr. Opin. Cell Biol. 2016, 40, 23–31. [Google Scholar] [CrossRef]
- Tatomer, D.C.; Terzo, E.; Curry, K.P.; Salzler, H.; Sabath, I.; Zapotoczny, G.; McKay, D.J.; Dominski, Z.; Marzluff, W.F.; Duronio, R.J. Concentrating Pre-MRNA Processing Factors in the Histone Locus Body Facilitates Efficient Histone MRNA Biogenesis. J. Cell Biol. 2016, 213, 557–570. [Google Scholar] [CrossRef]
- Azzouz, T.N.; Pillai, R.S.; Däpp, C.; Chari, A.; Meister, G.; Kambach, C.; Fischer, U.; Schümperli, D. Toward an Assembly Line for U7 SnRNPs: Interactions of U7-Specific Lsm Proteins with PRMT5 and SMN Complexes. J. Biol. Chem. 2005, 280, 34435–34440. [Google Scholar] [CrossRef]
- Pillai, R.S.; Will, C.L.; Lührmann, R.; Schümperli, D.; Müller, B. Purified U7 SnRNPs Lack the Sm Proteins D1 and D2 but Contain Lsm10, a New 14 KDa Sm D1-like Protein. EMBO J. 2001, 20, 5470–5479. [Google Scholar] [CrossRef]
- Tisdale, S.; Lotti, F.; Saieva, L.; Van Meerbeke, J.P.; Crawford, T.O.; Sumner, C.J.; Mentis, G.Z.; Pellizzoni, L. SMN Is Essential for the Biogenesis of U7 Small Nuclear Ribonucleoprotein and 3’-End Formation of Histone MRNAs. Cell Rep. 2013, 5, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Akten, B.; Kye, M.J.; Hao, L.T.; Wertz, M.H.; Singh, S.; Nie, D.; Huang, J.; Merianda, T.T.; Twiss, J.L.; Beattie, C.E.; et al. Interaction of Survival of Motor Neuron (SMN) and HuD Proteins with MRNA Cpg15 Rescues Motor Neuron Axonal Deficits. Proc. Natl. Acad. Sci. USA 2011, 108, 10337–10342. [Google Scholar] [CrossRef] [PubMed]
- Fallini, C.; Rouanet, J.P.; Donlin-Asp, P.G.; Guo, P.; Zhang, H.; Singer, R.H.; Rossoll, W.; Bassell, G.J. Dynamics of Survival of Motor Neuron (SMN) Protein Interaction with the MRNA-Binding Protein IMP1 Facilitates Its Trafficking into Motor Neuron Axons. Dev. Neurobiol. 2014, 74, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Fallini, C.; Zhang, H.; Su, Y.; Silani, V.; Singer, R.H.; Rossoll, W.; Bassell, G.J. The Survival of Motor Neuron (SMN) Protein Interacts with the MRNA-Binding Protein HuD and Regulates Localization of Poly(A) MRNA in Primary Motor Neuron Axons. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 3914–3925. [Google Scholar] [CrossRef]
- Hubers, L.; Valderrama-Carvajal, H.; Laframboise, J.; Timbers, J.; Sanchez, G.; Côté, J. HuD Interacts with Survival Motor Neuron Protein and Can Rescue Spinal Muscular Atrophy-like Neuronal Defects. Hum. Mol. Genet. 2011, 20, 553–579. [Google Scholar] [CrossRef]
- Tadesse, H.; Deschênes-Furry, J.; Boisvenue, S.; Côté, J. KH-Type Splicing Regulatory Protein Interacts with Survival Motor Neuron Protein and Is Misregulated in Spinal Muscular Atrophy. Hum. Mol. Genet. 2008, 17, 506–524. [Google Scholar] [CrossRef]
- Zhang, H.; Xing, L.; Rossoll, W.; Wichterle, H.; Singer, R.H.; Bassell, G.J. Multiprotein Complexes of the Survival of Motor Neuron Protein SMN with Gemins Traffic to Neuronal Processes and Growth Cones of Motor Neurons. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 8622–8632. [Google Scholar] [CrossRef]
- Zhang, H.L.; Pan, F.; Hong, D.; Shenoy, S.M.; Singer, R.H.; Bassell, G.J. Active Transport of the Survival Motor Neuron Protein and the Role of Exon-7 in Cytoplasmic Localization. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 6627–6637. [Google Scholar] [CrossRef]
- Rossoll, W.; Jablonka, S.; Andreassi, C.; Kröning, A.-K.; Karle, K.; Monani, U.R.; Sendtner, M. Smn, the Spinal Muscular Atrophy-Determining Gene Product, Modulates Axon Growth and Localization of Beta-Actin MRNA in Growth Cones of Motoneurons. J. Cell Biol. 2003, 163, 801–812. [Google Scholar] [CrossRef]
- Sharma, A.; Lambrechts, A.; Hao, L.T.; Le, T.T.; Sewry, C.A.; Ampe, C.; Burghes, A.H.M.; Morris, G.E. A Role for Complexes of Survival of Motor Neurons (SMN) Protein with Gemins and Profilin in Neurite-like Cytoplasmic Extensions of Cultured Nerve Cells. Exp. Cell Res. 2005, 309, 185–197. [Google Scholar] [CrossRef]
- von Roretz, C.; Di Marco, S.; Mazroui, R.; Gallouzi, I.-E. Turnover of AU-Rich-Containing MRNAs during Stress: A Matter of Survival. Wiley Interdiscip. Rev. RNA 2011, 2, 336–347. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. R Loops: From Transcription Byproducts to Threats to Genome Stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef]
- Thomas, M.; White, R.L.; Davis, R.W. Hybridization of RNA to Double-Stranded DNA: Formation of R-Loops. Proc. Natl. Acad. Sci. USA 1976, 73, 2294–2298. [Google Scholar] [CrossRef]
- Boguslawski, S.J.; Smith, D.E.; Michalak, M.A.; Mickelson, K.E.; Yehle, C.O.; Patterson, W.L.; Carrico, R.J. Characterization of Monoclonal Antibody to DNA.RNA and Its Application to Immunodetection of Hybrids. J. Immunol. Methods 1986, 89, 123–130. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chédin, F. R-Loop Formation Is a Distinctive Characteristic of Unmethylated Human CpG Island Promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef]
- Hartono, S.R.; Malapert, A.; Legros, P.; Bernard, P.; Chédin, F.; Vanoosthuyse, V. The Affinity of the S9.6 Antibody for Double-Stranded RNAs Impacts the Accurate Mapping of R-Loops in Fission Yeast. J. Mol. Biol. 2017, 430, 272–284. [Google Scholar] [CrossRef]
- Groh, M.; Gromak, N. Out of Balance: R-Loops in Human Disease. PLoS Genet. 2014, 10, e1004630. [Google Scholar] [CrossRef]
- Al-Hadid, Q.; Yang, Y. R-Loop: An Emerging Regulator of Chromatin Dynamics. Acta Biochim. Biophys. Sin. 2016, 48, 623–631. [Google Scholar] [CrossRef]
- Wahba, L.; Gore, S.K.; Koshland, D. The Homologous Recombination Machinery Modulates the Formation of RNA-DNA Hybrids and Associated Chromosome Instability. eLife 2013, 2, e00505. [Google Scholar] [CrossRef]
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of Topoisomerase I Leads to R-Loop-Mediated Transcriptional Blocks during Ribosomal RNA Synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef]
- Linder, P.; Jankowsky, E. From Unwinding to Clamping—The DEAD Box RNA Helicase Family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human Senataxin Resolves RNA/DNA Hybrids Formed at Transcriptional Pause Sites to Promote Xrn2-Dependent Termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Quinzii, C.M.; Mitsumoto, H.; Hays, A.P.; Roberts, J.K.; Richard, P.; Rowland, L.P. Senataxin Mutations and Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2011, 12, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.-C.; Klur, S.; Watanabe, M.; Németh, A.H.; Le Ber, I.; Moniz, J.-C.; Tranchant, C.; Aubourg, P.; Tazir, M.; Schöls, L.; et al. Senataxin, the Ortholog of a Yeast RNA Helicase, Is Mutant in Ataxia-Ocular Apraxia 2. Nat. Genet. 2004, 36, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.Y.; Gish, G.; Braunschweig, U.; Li, Y.; Ni, Z.; Schmitges, F.W.; Zhong, G.; Liu, K.; Li, W.; Moffat, J.; et al. SMN and Symmetric Arginine Dimethylation of RNA Polymerase II C-Terminal Domain Control Termination. Nature 2016, 529, 48–53. [Google Scholar] [CrossRef]
- Wang, C.H.; Lunn, M.R. Spinal Muscular Atrophy: Advances in Research and Consensus on Care of Patients. Curr. Treat. Options Neurol. 2008, 10, 420–428. [Google Scholar] [CrossRef]
- Crawford, T.O.; Pardo, C.A. The Neurobiology of Childhood Spinal Muscular Atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar] [CrossRef]
- Schmid, A.; DiDonato, C.J. Animal Models of Spinal Muscular Atrophy. J. Child Neurol. 2007, 22, 1004–1012. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Gillingwater, T.H.; Talbot, K. The Contribution of Mouse Models to Understanding the Pathogenesis of Spinal Muscular Atrophy. Dis. Model. Mech. 2011, 4, 457–467. [Google Scholar] [CrossRef]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A Mouse Model for Spinal Muscular Atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar] [CrossRef]
- Monani, U.R.; Sendtner, M.; Coovert, D.D.; Parsons, D.W.; Andreassi, C.; Le, T.T.; Jablonka, S.; Schrank, B.; Rossoll, W.; Rossol, W.; et al. The Human Centromeric Survival Motor Neuron Gene (SMN2) Rescues Embryonic Lethality in Smn(-/-) Mice and Results in a Mouse with Spinal Muscular Atrophy. Hum. Mol. Genet. 2000, 9, 333–339. [Google Scholar] [CrossRef]
- Le, T.T.; Pham, L.T.; Butchbach, M.E.R.; Zhang, H.L.; Monani, U.R.; Coovert, D.D.; Gavrilina, T.O.; Xing, L.; Bassell, G.J.; Burghes, A.H.M. SMNΔ7, the Major Product of the Centromeric Survival Motor Neuron (SMN2) Gene, Extends Survival in Mice with Spinal Muscular Atrophy and Associates with Full-Length SMN. Hum. Mol. Genet. 2005, 14, 845–857. [Google Scholar] [CrossRef]
- Mentis, G.Z.; Blivis, D.; Liu, W.; Drobac, E.; Crowder, M.E.; Kong, L.; Alvarez, F.J.; Sumner, C.J.; O’Donovan, M.J. Early Functional Impairment of Sensory-Motor Connectivity in a Mouse Model of Spinal Muscular Atrophy. Neuron 2011, 69, 453–467. [Google Scholar] [CrossRef]
- Kariya, S.; Park, G.-H.; Maeno-Hikichi, Y.; Leykekhman, O.; Lutz, C.; Arkovitz, M.S.; Landmesser, L.T.; Monani, U.R. Reduced SMN Protein Impairs Maturation of the Neuromuscular Junctions in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2008, 17, 2552–2569. [Google Scholar] [CrossRef]
- Kong, L.; Wang, X.; Choe, D.W.; Polley, M.; Burnett, B.G.; Bosch-Marcé, M.; Griffin, J.W.; Rich, M.M.; Sumner, C.J. Impaired Synaptic Vesicle Release and Immaturity of Neuromuscular Junctions in Spinal Muscular Atrophy Mice. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 842–851. [Google Scholar] [CrossRef]
- Lee, Y.I.; Mikesh, M.; Smith, I.; Rimer, M.; Thompson, W. Muscles in a Mouse Model of Spinal Muscular Atrophy Show Profound Defects in Neuromuscular Development Even in the Absence of Failure in Neuromuscular Transmission or Loss of Motor Neurons. Dev. Biol. 2011, 356, 432–444. [Google Scholar] [CrossRef]
- Ling, K.K.Y.; Lin, M.-Y.; Zingg, B.; Feng, Z.; Ko, C.-P. Synaptic Defects in the Spinal and Neuromuscular Circuitry in a Mouse Model of Spinal Muscular Atrophy. PloS ONE 2010, 5, e15457. [Google Scholar] [CrossRef]
- Murray, L.M.; Comley, L.H.; Thomson, D.; Parkinson, N.; Talbot, K.; Gillingwater, T.H. Selective Vulnerability of Motor Neurons and Dissociation of Pre- and Post-Synaptic Pathology at the Neuromuscular Junction in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2008, 17, 949–962. [Google Scholar] [CrossRef]
- Ruiz, R.; Casañas, J.J.; Torres-Benito, L.; Cano, R.; Tabares, L. Altered Intracellular Ca2+ Homeostasis in Nerve Terminals of Severe Spinal Muscular Atrophy Mice. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 849–857. [Google Scholar] [CrossRef]
- Fletcher, E.V.; Simon, C.M.; Pagiazitis, J.G.; Chalif, J.I.; Vukojicic, A.; Drobac, E.; Wang, X.; Mentis, G.Z. Reduced Sensory Synaptic Excitation Impairs Motor Neuron Function via Kv2.1 in Spinal Muscular. Atrophy. Nat. Neurosci. 2017, 20, 905–916. [Google Scholar] [CrossRef]
- Bevan, A.K.; Hutchinson, K.R.; Foust, K.D.; Braun, L.; McGovern, V.L.; Schmelzer, L.; Ward, J.G.; Petruska, J.C.; Lucchesi, P.A.; Burghes, A.H.M.; et al. Early Heart Failure in the SMNDelta7 Model of Spinal Muscular Atrophy and Correction by Postnatal ScAAV9-SMN Delivery. Hum. Mol. Genet. 2010, 19, 3895–3905. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, M.; Beauvais, A.; Anderson, C.L.; Kothary, R. Rho-Kinase Inactivation Prolongs Survival of an Intermediate SMA Mouse Model. Hum. Mol. Genet. 2010, 19, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.R.; Satta, R.; Lutz, C.; DiDonato, C.J. Arrhythmia and Cardiac Defects Are a Feature of Spinal Muscular Atrophy Model Mice. Hum. Mol. Genet. 2010, 19, 3906–3918. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN Restoration Is Essential for Long-Term Rescue of a Severe Spinal Muscular Atrophy Mouse Model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Shababi, M.; Habibi, J.; Yang, H.T.; Vale, S.M.; Sewell, W.A.; Lorson, C.L. Cardiac Defects Contribute to the Pathology of Spinal Muscular Atrophy Models. Hum. Mol. Genet. 2010, 19, 4059–4071. [Google Scholar] [CrossRef]
- Hamilton, G.; Gillingwater, T.H. Spinal Muscular Atrophy: Going beyond the Motor Neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Shababi, M.; Lorson, C.L.; Rudnik-Schöneborn, S.S. Spinal Muscular Atrophy: A Motor Neuron Disorder or a Multi-Organ Disease? J. Anat. 2014, 224, 15–28. [Google Scholar] [CrossRef]
- Wan, L.; Battle, D.J.; Yong, J.; Gubitz, A.K.; Kolb, S.J.; Wang, J.; Dreyfuss, G. The Survival of Motor Neurons Protein Determines the Capacity for SnRNP Assembly: Biochemical Deficiency in Spinal Muscular Atrophy. Mol. Cell. Biol. 2005, 25, 5543–5551. [Google Scholar] [CrossRef]
- Winkler, C.; Eggert, C.; Gradl, D.; Meister, G.; Giegerich, M.; Wedlich, D.; Laggerbauer, B.; Fischer, U. Reduced U SnRNP Assembly Causes Motor Axon Degeneration in an Animal Model for Spinal Muscular Atrophy. Genes Dev. 2005, 19, 2320–2330. [Google Scholar] [CrossRef]
- Workman, E.; Saieva, L.; Carrel, T.L.; Crawford, T.O.; Liu, D.; Lutz, C.; Beattie, C.E.; Pellizzoni, L.; Burghes, A.H.M. A SMN Missense Mutation Complements SMN2 Restoring SnRNPs and Rescuing SMA Mice. Hum. Mol. Genet. 2009, 18, 2215–2229. [Google Scholar] [CrossRef]
- Borg, R.M.; Fenech Salerno, B.; Vassallo, N.; Bordonne, R.; Cauchi, R.J. Disruption of SnRNP Biogenesis Factors Tgs1 and PICln Induces Phenotypes That Mirror Aspects of SMN-Gemins Complex Perturbation in Drosophila, Providing New Insights into Spinal Muscular Atrophy. Neurobiol. Dis. 2016, 94, 245–258. [Google Scholar] [CrossRef]
- Gabanella, F.; Butchbach, M.E.R.; Saieva, L.; Carissimi, C.; Burghes, A.H.M.; Pellizzoni, L. Ribonucleoprotein Assembly Defects Correlate with Spinal Muscular Atrophy Severity and Preferentially Affect a Subset of Spliceosomal SnRNPs. PloS ONE 2007, 2, e921. [Google Scholar] [CrossRef]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN Deficiency Causes Tissue-Specific Perturbations in the Repertoire of SnRNAs and Widespread Defects in Splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef]
- Boulisfane, N.; Choleza, M.; Rage, F.; Neel, H.; Soret, J.; Bordonné, R. Impaired Minor Tri-SnRNP Assembly Generates Differential Splicing Defects of U12-Type Introns in Lymphoblasts Derived from a Type I SMA Patient. Hum. Mol. Genet. 2011, 20, 641–648. [Google Scholar] [CrossRef]
- Doktor, T.K.; Hua, Y.; Andersen, H.S.; Brøner, S.; Liu, Y.H.; Wieckowska, A.; Dembic, M.; Bruun, G.H.; Krainer, A.R.; Andresen, B.S. RNA-Sequencing of a Mouse-Model of Spinal Muscular Atrophy Reveals Tissue-Wide Changes in Splicing of U12-Dependent Introns. Nucleic Acids Res. 2017, 45, 395–416. [Google Scholar] [CrossRef]
- Jangi, M.; Fleet, C.; Cullen, P.; Gupta, S.V.; Mekhoubad, S.; Chiao, E.; Allaire, N.; Bennett, C.F.; Rigo, F.; Krainer, A.R.; et al. SMN Deficiency in Severe Models of Spinal Muscular Atrophy Causes Widespread Intron Retention and DNA Damage. Proc. Natl. Acad. Sci. USA 2017, 114, E2347–E2356. [Google Scholar] [CrossRef]
- Lotti, F.; Imlach, W.L.; Saieva, L.; Beck, E.S.; Hao, L.T.; Li, D.K.; Jiao, W.; Mentis, G.Z.; Beattie, C.E.; McCabe, B.D.; et al. A SMN-Dependent U12 Splicing Event Essential for Motor Circuit Function. Cell 2012, 151, 440–454. [Google Scholar] [CrossRef]
- Ruggiu, M.; McGovern, V.L.; Lotti, F.; Saieva, L.; Li, D.K.; Kariya, S.; Monani, U.R.; Burghes, A.H.M.; Pellizzoni, L. A Role for SMN Exon 7 Splicing in the Selective Vulnerability of Motor Neurons in Spinal Muscular Atrophy. Mol. Cell. Biol. 2012, 32, 126–138. [Google Scholar] [CrossRef]
- Jodelka, F.M.; Ebert, A.D.; Duelli, D.M.; Hastings, M.L. A Feedback Loop Regulates Splicing of the Spinal Muscular Atrophy-Modifying Gene, SMN2. Hum. Mol. Genet. 2010, 19, 4906–4917. [Google Scholar] [CrossRef]
- Li, L.; Ding, Z.; Pang, T.-L.; Zhang, B.; Li, C.-H.; Liang, A.-M.; Wang, Y.-R.; Zhou, Y.; Fan, Y.-J.; Xu, Y.-Z. Defective Minor Spliceosomes Induce SMA-Associated Phenotypes through Sensitive Intron-Containing Neural Genes in Drosophila. Nat. Commun. 2020, 11, 5608. [Google Scholar] [CrossRef]
- Murray, L.M.; Beauvais, A.; Gibeault, S.; Courtney, N.L.; Kothary, R. Transcriptional Profiling of Differentially Vulnerable Motor Neurons at Pre-Symptomatic Stage in the Smn (2b/-) Mouse Model of Spinal Muscular Atrophy. Acta Neuropathol. Commun. 2015, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.M.; Dai, Y.; Van Alstyne, M.; Koutsioumpa, C.; Pagiazitis, J.G.; Chalif, J.I.; Wang, X.; Rabinowitz, J.E.; Henderson, C.E.; Pellizzoni, L.; et al. Converging Mechanisms of P53 Activation Drive Motor Neuron Degeneration in Spinal Muscular Atrophy. Cell Rep. 2017, 21, 3767–3780. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pinto, A.M.; Wan, L.; Wang, W.; Berg, M.G.; Oliva, I.; Singh, L.N.; Dengler, C.; Wei, Z.; Dreyfuss, G. Dysregulation of Synaptogenesis Genes Antecedes Motor Neuron Pathology in Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 19348–19353. [Google Scholar] [CrossRef] [PubMed]
- Osman, E.Y.; Van Alstyne, M.; Yen, P.-F.; Lotti, F.; Feng, Z.; Ling, K.K.; Ko, C.-P.; Pellizzoni, L.; Lorson, C.L. Minor SnRNA Gene Delivery Improves the Loss of Proprioceptive Synapses on SMA Motor Neurons. JCI Insight 2020, 5, e130574, 130574. [Google Scholar] [CrossRef]
- See, K.; Yadav, P.; Giegerich, M.; Cheong, P.S.; Graf, M.; Vyas, H.; Lee, S.G.P.; Mathavan, S.; Fischer, U.; Sendtner, M.; et al. SMN Deficiency Alters Nrxn2 Expression and Splicing in Zebrafish and Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2014, 23, 1754–1770. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Barreiro-Iglesias, A.; Oliver, P.L.; Biba, A.; Becker, T.; Davies, K.E.; Becker, C.G.; Talbot, K. Chondrolectin Affects Cell Survival and Neuronal Outgrowth in in Vitro and in Vivo Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2014, 23, 855–869. [Google Scholar] [CrossRef]
- Wang, W.; Dimatteo, D.; Funanage, V.L.; Scavina, M. Increased Susceptibility of Spinal Muscular Atrophy Fibroblasts to Camptothecin-Induced Cell Death. Mol. Genet. Metab. 2005, 85, 38–45. [Google Scholar] [CrossRef]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and Cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-Capetillo, O.; Chen, H.T.; Sedelnikova, O.A.; Reina-San-Martin, B.; Coppola, V.; Meffre, E.; Difilippantonio, M.J.; et al. Genomic Instability in Mice Lacking Histone H2AX. Science 2002, 296, 922–927. [Google Scholar] [CrossRef]
- Tisdale, S.; Van Alstyne, M.; Simon, C.M.; Mentis, G.Z.; Pellizzoni, L. SMN Controls Neuromuscular Junction Integrity through U7 SnRNP. Cell Rep. 2022, 40, 111393. [Google Scholar] [CrossRef]
- Kim, J.-K.; Caine, C.; Awano, T.; Herbst, R.; Monani, U.R. Motor Neuronal Repletion of the NMJ Organizer, Agrin, Modulates the Severity of the Spinal Muscular Atrophy Disease Phenotype in Model Mice. Hum. Mol. Genet. 2017, 26, 2377–2385. [Google Scholar] [CrossRef]
- Rossoll, W.; Kröning, A.-K.; Ohndorf, U.-M.; Steegborn, C.; Jablonka, S.; Sendtner, M. Specific Interaction of Smn, the Spinal Muscular Atrophy Determining Gene Product, with HnRNP-R and Gry-Rbp/HnRNP-Q: A Role for Smn in RNA Processing in Motor Axons? Hum. Mol. Genet. 2002, 11, 93–105. [Google Scholar] [CrossRef]
- Donlin-Asp, P.G.; Fallini, C.; Campos, J.; Chou, C.-C.; Merritt, M.E.; Phan, H.C.; Bassell, G.J.; Rossoll, W. The Survival of Motor Neuron Protein Acts as a Molecular Chaperone for MRNP Assembly. Cell Rep. 2017, 18, 1660–1673. [Google Scholar] [CrossRef]
- Fallini, C.; Donlin-Asp, P.G.; Rouanet, J.P.; Bassell, G.J.; Rossoll, W. Deficiency of the Survival of Motor Neuron Protein Impairs MRNA Localization and Local Translation in the Growth Cone of Motor Neurons. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 3811–3820. [Google Scholar] [CrossRef]
- Hao le, T.; Duy, P.Q.; An, M.; Talbot, J.; Iyer, C.C.; Wolman, M.; Beattie, C.E. HuD and the Survival Motor Neuron Protein Interact in Motoneurons and Are Essential for Motoneuron Development, Function, and MRNA Regulation. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 11559–11571. [Google Scholar] [CrossRef]
- Shukla, S.; Parker, R. Hypo- and Hyper-Assembly Diseases of RNA-Protein Complexes. Trends Mol. Med. 2016, 22, 615–628. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA Helicase Gene Mutations in a Form of Juvenile Amyotrophic Lateral Sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef]
- Gandhi, G.; Abdullah, S.; Foead, A.I.; Yeo, W.W.Y. The Potential Role of MiRNA Therapies in Spinal Muscle Atrophy. J. Neurol. Sci. 2021, 427, 117485. [Google Scholar] [CrossRef]
- Wang, L.-T.; Chiou, S.-S.; Liao, Y.-M.; Jong, Y.-J.; Hsu, S.-H. Survival of Motor Neuron Protein Downregulates MiR-9 Expression in Patients with Spinal Muscular Atrophy. Kaohsiung J. Med. Sci. 2014, 30, 229–234. [Google Scholar] [CrossRef]
- Sison, S.L.; Patitucci, T.N.; Seminary, E.R.; Villalon, E.; Lorson, C.L.; Ebert, A.D. Astrocyte-Produced MiR-146a as a Mediator of Motor Neuron Loss in Spinal Muscular Atrophy. Hum. Mol. Genet. 2017, 26, 3409–3420. [Google Scholar] [CrossRef]
- Kye, M.J.; Niederst, E.D.; Wertz, M.H.; Gonçalves, I.d.C.G.; Akten, B.; Dover, K.Z.; Peters, M.; Riessland, M.; Neveu, P.; Wirth, B.; et al. SMN Regulates Axonal Local Translation via MiR-183/MTOR Pathway. Hum. Mol. Genet. 2014, 23, 6318–6331. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, V.; Boido, M.; De Amicis, E.; Piras, A.; Vercelli, A. Expression of Muscle-Specific MiRNA 206 in the Progression of Disease in a Murine SMA Model. PloS ONE 2015, 10, e0128560. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W.; Luo, D.; Seo, J.; Singh, N.N.; Singh, R.N. Human Survival Motor Neuron Genes Generate a Vast Repertoire of Circular RNAs. Nucleic Acids Res. 2019, 47, 2884–2905. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W.; Singh, R.N. Characteristics of Circular RNAs Generated by Human Survival Motor Neuron Genes. Cell. Signal. 2020, 73, 109696. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faravelli, I.; Riboldi, G.M.; Rinchetti, P.; Lotti, F. The SMN Complex at the Crossroad between RNA Metabolism and Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 2247. https://doi.org/10.3390/ijms24032247
Faravelli I, Riboldi GM, Rinchetti P, Lotti F. The SMN Complex at the Crossroad between RNA Metabolism and Neurodegeneration. International Journal of Molecular Sciences. 2023; 24(3):2247. https://doi.org/10.3390/ijms24032247
Chicago/Turabian StyleFaravelli, Irene, Giulietta M. Riboldi, Paola Rinchetti, and Francesco Lotti. 2023. "The SMN Complex at the Crossroad between RNA Metabolism and Neurodegeneration" International Journal of Molecular Sciences 24, no. 3: 2247. https://doi.org/10.3390/ijms24032247
APA StyleFaravelli, I., Riboldi, G. M., Rinchetti, P., & Lotti, F. (2023). The SMN Complex at the Crossroad between RNA Metabolism and Neurodegeneration. International Journal of Molecular Sciences, 24(3), 2247. https://doi.org/10.3390/ijms24032247