
Kappa Opioid Receptors Reduce Serotonin Uptake and Escitalopram Efficacy in the Mouse Substantia Nigra Pars Reticulata

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
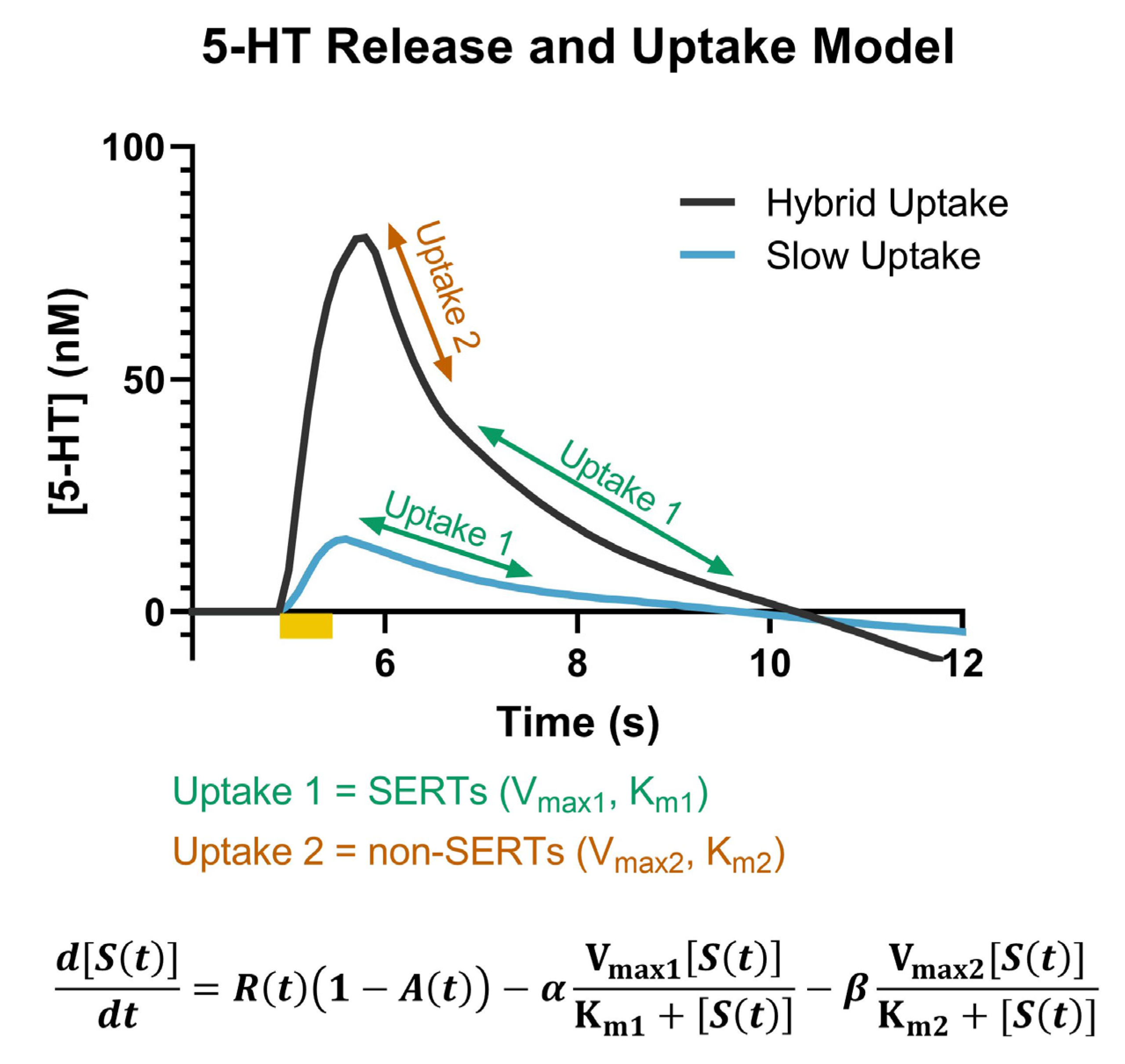
2.1. Ex Vivo Voltammetry Captures Dynamic Response of Serotonin to Escitalopram
2.2. KOR Activation Inhibits Serotonin Release and Uptake via SERTs
2.3. Escitalopram Efficacy Altered by U50 Pretreatment
3. Materials and Methods
3.1. Animals
3.2. Brain Slice Preparation
3.3. Fast-Scan Cyclic Voltammetry
3.4. Data Analysis and Statistics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, S.; Manuck, S.B. Stress, reactivity, and disease. Psychosom. Med. 1995, 57, 423–426. [Google Scholar] [CrossRef]
- Cohen, S.; Gianaros, P.J.; Manuck, S.B. A Stage Model of Stress and Disease. Perspect. Psychol. Sci. 2016, 11, 456–463. [Google Scholar] [CrossRef]
- Turner, A.I.; Smyth, N.; Hall, S.J.; Torres, S.J.; Hussein, M.; Jayasinghe, S.U.; Ball, K.; Clow, A.J. Psychological stress reactivity and future health and disease outcomes: A systematic review of prospective evidence. Psychoneuroendocrinology 2020, 114, 104599. [Google Scholar] [CrossRef]
- Beardsley, P.M.; Howard, J.L.; Shelton, K.L.; Carroll, F.I. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology 2005, 183, 118–126. [Google Scholar] [CrossRef]
- Marchant, N.J.; Li, X.; Shaham, Y. Recent developments in animal models of drug relapse. Curr. Opin. Neurobiol. 2013, 23, 675–683. [Google Scholar] [CrossRef]
- Shaham, Y.; Stewart, J. Stress reinstates heroin-seeking in drug-free animals: An effect mimicking heroin, not withdrawal. Psychopharmacology 1995, 119, 334–341. [Google Scholar] [CrossRef]
- Wee, S.; Koob, G.F. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology 2010, 210, 121–135. [Google Scholar] [CrossRef]
- Kendler, K.S.; Karkowski, L.M.; Prescott, C.A. Causal relationship between stressful life events and the onset of major depression. Am. J. Psychiatry 1999, 156, 837–841. [Google Scholar] [CrossRef]
- Kessler, R.C. The epidemiology of pure and comorbid generalized anxiety disorder: A review and evaluation of recent research. Acta Psychiatr. Scand. Suppl. 2000, 102, 7–13. [Google Scholar] [CrossRef]
- Pine, D.S.; Cohen, P.; Johnson, J.G.; Brook, J.S. Adolescent life events as predictors of adult depression. J. Affect. Disord. 2002, 68, 49–57. [Google Scholar] [CrossRef]
- Hori, H.; Kim, Y. Inflammation and post-traumatic stress disorder. Psychiatry Clin. Neurosci. 2019, 73, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Munhoz, C.D.; Garcia-Bueno, B.; Madrigal, J.L.; Lepsch, L.B.; Scavone, C.; Leza, J.C. Stress-induced neuroinflammation: Mechanisms and new pharmacological targets. Braz. J. Med. Biol. Res. 2008, 41, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bueno, B.; Caso, J.R.; Leza, J.C. Stress as a neuroinflammatory condition in brain: Damaging and protective mechanisms. Neurosci. Biobehav. Rev. 2008, 32, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Z.; Wang, Y.X.; Jiang, C.L. Inflammation: The Common Pathway of Stress-Related Diseases. Front. Hum. Neurosci. 2017, 11, 316. [Google Scholar] [CrossRef]
- Carrasco, G.A.; Van de Kar, L.D. Neuroendocrine pharmacology of stress. Eur. J. Pharmacol. 2003, 463, 235–272. [Google Scholar] [CrossRef]
- Tsigos, C.; Chrousos, G.P. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 2002, 53, 865–871. [Google Scholar] [CrossRef]
- Koob, G.F.; Schulkin, J. Addiction and stress: An allostatic view. Neurosci. Biobehav. Rev. 2019, 106, 245–262. [Google Scholar] [CrossRef]
- Land, B.B.; Bruchas, M.R.; Lemos, J.C.; Xu, M.; Melief, E.J.; Chavkin, C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J. Neurosci. 2008, 28, 407–414. [Google Scholar] [CrossRef]
- Bruchas, M.R.; Land, B.B.; Chavkin, C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010, 1314, 44–55. [Google Scholar] [CrossRef]
- Koob, G.F. Brain stress systems in the amygdala and addiction. Brain Res. 2009, 1293, 61–75. [Google Scholar] [CrossRef]
- Liu, S.S.; Pickens, S.; Burma, N.E.; Ibarra-Lecue, I.; Yang, H.; Xue, L.; Cook, C.; Hakimian, J.K.; Severino, A.L.; Lueptow, L.; et al. Kappa Opioid Receptors Drive a Tonic Aversive Component of Chronic Pain. J. Neurosci. 2019, 39, 4162–4178. [Google Scholar] [CrossRef] [PubMed]
- Ragu Varman, D.; Jayanthi, L.D.; Ramamoorthy, S. Kappa Opioid Receptor Mediated Differential Regulation of Serotonin and Dopamine Transporters in Mood and Substance Use Disorder. Handb. Exp. Pharmacol. 2022, 271, 97–112. [Google Scholar] [CrossRef]
- Cahill, C.M.; Taylor, A.M.; Cook, C.; Ong, E.; Moron, J.A.; Evans, C.J. Does the kappa opioid receptor system contribute to pain aversion? Front. Pharmacol. 2014, 5, 253. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Schindler, A.G.; Shankar, H.; Messinger, D.I.; Miyatake, M.; Land, B.B.; Lemos, J.C.; Hagan, C.E.; Neumaier, J.F.; Quintana, A.; et al. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron 2011, 71, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Lalanne, L.; Ayranci, G.; Kieffer, B.L.; Lutz, P.E. The kappa opioid receptor: From addiction to depression, and back. Front. Psychiatry 2014, 5, 170. [Google Scholar] [CrossRef]
- Gutstein, H.B.; Mansour, A.; Watson, S.J.; Akil, H.; Fields, H.L. Mu and kappa opioid receptors in periaqueductal gray and rostral ventromedial medulla. Neuroreport 1998, 9, 1777–1781. [Google Scholar] [CrossRef]
- Winkler, C.W.; Hermes, S.M.; Chavkin, C.I.; Drake, C.T.; Morrison, S.F.; Aicher, S.A. Kappa opioid receptor (KOR) and GAD67 immunoreactivity are found in OFF and NEUTRAL cells in the rostral ventromedial medulla. J. Neurophysiol. 2006, 96, 3465–3473. [Google Scholar] [CrossRef]
- Stefanucci, A.; Iobbi, V.; Della Valle, A.; Scioli, G.; Pieretti, S.; Minosi, P.; Mirzaie, S.; Novellino, E.; Mollica, A. In Silico Identification of Tripeptides as Lead Compounds for the Design of KOR Ligands. Molecules 2021, 26, 4767. [Google Scholar] [CrossRef]
- Stefanucci, A.; Della Valle, A.; Scioli, G.; Marinaccio, L.; Pieretti, S.; Minosi, P.; Szucs, E.; Benyhe, S.; Masci, D.; Tanguturi, P.; et al. Discovery of kappa Opioid Receptor (KOR)-Selective d-Tetrapeptides with Improved In Vivo Antinociceptive Effect after Peripheral Administration. ACS Med. Chem. Lett. 2022, 13, 1707–1714. [Google Scholar] [CrossRef]
- Brust, T.F.; Morgenweck, J.; Kim, S.A.; Rose, J.H.; Locke, J.L.; Schmid, C.L.; Zhou, L.; Stahl, E.L.; Cameron, M.D.; Scarry, S.M.; et al. Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci. Signal. 2016, 9, ra117. [Google Scholar] [CrossRef]
- Berger, B.; Rothmaier, A.K.; Wedekind, F.; Zentner, J.; Feuerstein, T.J.; Jackisch, R. Presynaptic opioid receptors on noradrenergic and serotonergic neurons in the human as compared to the rat neocortex. Br. J. Pharmacol. 2006, 148, 795–806. [Google Scholar] [CrossRef]
- Kalyuzhny, A.E.; Wessendorf, M.W. Serotonergic and GABAergic neurons in the medial rostral ventral medulla express kappa-opioid receptor immunoreactivity. Neuroscience 1999, 90, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.D.; Fontaine, H.M.; Song, A.J.; Andrews, M.M.; Baird, M.A.; Kieffer, B.L.; Land, B.B.; Chavkin, C. kappa-Opioid Receptor Activation in Dopamine Neurons Disrupts Behavioral Inhibition. Neuropsychopharmacol 2018, 43, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, J.M.; Messinger, D.I.; Knakal, C.R.; Kuhar, J.R.; Schattauer, S.S.; Bruchas, M.R.; Zweifel, L.S.; Kieffer, B.L.; Phillips, P.E.; Chavkin, C. Kappa Opioid Receptor-Induced Aversion Requires p38 MAPK Activation in VTA Dopamine Neurons. J. Neurosci. 2015, 35, 12917–12931. [Google Scholar] [CrossRef] [PubMed]
- Knoll, A.T.; Carlezon, W.A., Jr. Dynorphin, stress, and depression. Brain Res. 2010, 1314, 56–73. [Google Scholar] [CrossRef]
- Pourhamzeh, M.; Moravej, F.G.; Arabi, M.; Shahriari, E.; Mehrabi, S.; Ward, R.; Ahadi, R.; Joghataei, M.T. The Roles of Serotonin in Neuropsychiatric Disorders. Cell. Mol. Neurobiol. 2022, 42, 1671–1692. [Google Scholar] [CrossRef] [PubMed]
- Lemos, J.C.; Roth, C.A.; Messinger, D.I.; Gill, H.K.; Phillips, P.E.; Chavkin, C. Repeated stress dysregulates kappa-opioid receptor signaling in the dorsal raphe through a p38alpha MAPK-dependent mechanism. J. Neurosci. 2012, 32, 12325–12336. [Google Scholar] [CrossRef]
- Sundaramurthy, S.; Annamalai, B.; Samuvel, D.J.; Shippenberg, T.S.; Jayanthi, L.D.; Ramamoorthy, S. Modulation of serotonin transporter function by kappa-opioid receptor ligands. Neuropharmacology 2017, 113, 281–292. [Google Scholar] [CrossRef]
- Di Giovanni, G.; Di Matteo, V.; Pierucci, M.; Benigno, A.; Esposito, E. Serotonin involvement in the basal ganglia pathophysiology: Could the 5-HT2C receptor be a new target for therapeutic strategies? Curr. Med. Chem. 2006, 13, 3069–3081. [Google Scholar] [CrossRef]
- Chen, C.; Willhouse, A.H.; Huang, P.; Ko, N.; Wang, Y.; Xu, B.; Huang, L.H.M.; Kieffer, B.; Barbe, M.F.; Liu-Chen, L.Y. Characterization of a Knock-In Mouse Line Expressing a Fusion Protein of kappa Opioid Receptor Conjugated with tdTomato: 3-Dimensional Brain Imaging via CLARITY. eNeuro 2020, 7, ENEURO.0028-20.2020. [Google Scholar] [CrossRef]
- Hormigo, S.; Vega-Flores, G.; Castro-Alamancos, M.A. Basal Ganglia Output Controls Active Avoidance Behavior. J. Neurosci. 2016, 36, 10274–10284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Meng, S.; Chen, W.; Chen, Y.; Huang, E.; Zhang, G.; Liang, Y.; Ding, Z.; Xue, Y.; Chen, Y.; et al. High-Frequency Deep Brain Stimulation of the Substantia Nigra Pars Reticulata Facilitates Extinction and Prevents Reinstatement of Methamphetamine-Induced Conditioned Place Preference. Front. Pharmacol. 2021, 12, 705813. [Google Scholar] [CrossRef] [PubMed]
- Yamane, F.; Okazawa, H.; Blier, P.; Diksic, M. Reduction in serotonin synthesis following acute and chronic treatments with paroxetine, a selective serotonin reuptake inhibitor, in rat brain: An autoradiographic study with alpha-[14C]methyl-L-tryptophan(2). Biochem. Pharmacol. 2001, 62, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Chaouloff, F.; Berton, O.; Mormede, P. Serotonin and stress. Neuropsychopharmacol 1999, 21, 28S–32S. [Google Scholar] [CrossRef]
- Muck-Seler, D.; Jevric-Causevic, A.; Diksic, M. Influence of fluoxetine on regional serotonin synthesis in the rat brain. J. Neurochem. 1996, 67, 2434–2442. [Google Scholar] [CrossRef]
- Jackson, B.P.; Dietz, S.M.; Wightman, R.M. Fast-Scan Cyclic Voltammetry of 5-Hydroxytryptamine. Anal. Chem. 1995, 67, 1115–1120. [Google Scholar] [CrossRef]
- Hashemi, P.; Dankoski, E.C.; Petrovic, J.; Keithley, R.B.; Wightman, R.M. Voltammetric detection of 5-hydroxytryptamine release in the rat brain. Anal. Chem. 2009, 81, 9462–9471. [Google Scholar] [CrossRef]
- Wood, K.M.; Zeqja, A.; Nijhout, H.F.; Reed, M.C.; Best, J.; Hashemi, P. Voltammetric and mathematical evidence for dual transport mediation of serotonin clearance in vivo. J. Neurochem. 2014, 130, 351–359. [Google Scholar] [CrossRef]
- Daws, L.C.; Montanez, S.; Owens, W.A.; Gould, G.G.; Frazer, A.; Toney, G.M.; Gerhardt, G.A. Transport mechanisms governing serotonin clearance in vivo revealed by high-speed chronoamperometry. J. Neurosci. Methods 2005, 143, 49–62. [Google Scholar] [CrossRef]
- Mena, S.; Dietsch, S.; Berger, S.N.; Witt, C.E.; Hashemi, P. Novel, User-Friendly Experimental and Analysis Strategies for Fast Voltammetry: 1. The Analysis Kid for FSCV. ACS Meas. Sci. Au 2021, 1, 11–19. [Google Scholar] [CrossRef]
- Wood, K.M.; Hashemi, P. Fast-scan cyclic voltammetry analysis of dynamic serotonin reponses to acute escitalopram. ACS Chem. Neurosci. 2013, 4, 715–720. [Google Scholar] [CrossRef] [PubMed]
- John, C.E.; Jones, S.R. Voltammetric characterization of the effect of monoamine uptake inhibitors and releasers on dopamine and serotonin uptake in mouse caudate-putamen and substantia nigra slices. Neuropharmacology 2007, 52, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Venton, B.J.; Seipel, A.T.; Phillips, P.E.; Wetsel, W.C.; Gitler, D.; Greengard, P.; Augustine, G.J.; Wightman, R.M. Cocaine increases dopamine release by mobilization of a synapsin-dependent reserve pool. J Neurosci. 2006, 26, 3206–3209. [Google Scholar] [CrossRef]
- Kile, B.M.; Guillot, T.S.; Venton, B.J.; Wetsel, W.C.; Augustine, G.J.; Wightman, R.M. Synapsins differentially control dopamine and serotonin release. J. Neurosci. 2010, 30, 9762–9770. [Google Scholar] [CrossRef] [PubMed]
- Tassan Mazzocco, M.; Guarnieri, F.C.; Monzani, E.; Benfenati, F.; Valtorta, F.; Comai, S. Dysfunction of the serotonergic system in the brain of synapsin triple knockout mice is associated with behavioral abnormalities resembling synapsin-related human pathologies. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 105, 110135. [Google Scholar] [CrossRef] [PubMed]
- Holmes, J.; Lau, T.; Saylor, R.; Fernandez-Novel, N.; Hersey, M.; Keen, D.; Hampel, L.; Horschitz, S.; Ladewig, J.; Parke, B.; et al. Voltammetric Approach for Characterizing the Biophysical and Chemical Functionality of Human Induced Pluripotent Stem Cell-Derived Serotonin Neurons. Anal. Chem. 2022, 94, 8847–8856. [Google Scholar] [CrossRef]
- Zhong, H.; Haddjeri, N.; Sanchez, C. Escitalopram, an antidepressant with an allosteric effect at the serotonin transporter—A review of current understanding of its mechanism of action. Psychopharmacology 2012, 219, 1–13. [Google Scholar] [CrossRef]
- Lau, T.; Proissl, V.; Ziegler, J.; Schloss, P. Visualization of neurotransmitter uptake and release in serotonergic neurons. J. Neurosci. Methods 2015, 241, 10–17. [Google Scholar] [CrossRef]
- Lau, T.; Horschitz, S.; Berger, S.; Bartsch, D.; Schloss, P. Antidepressant-induced internalization of the serotonin transporter in serotonergic neurons. FASEB J. 2008, 22, 1702–1714. [Google Scholar] [CrossRef]
- Dhawan, B.N.; Cesselin, F.; Raghubir, R.; Reisine, T.; Bradley, P.B.; Portoghese, P.S.; Hamon, M. International Union of Pharmacology. XII. Classification of opioid receptors. Pharmacol. Rev. 1996, 48, 567–592. [Google Scholar]
- Tao, R.; Auerbach, S.B. Opioid receptor subtypes differentially modulate serotonin efflux in the rat central nervous system. J. Pharmacol. Exp. Ther. 2002, 303, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Schindler, A.G.; Messinger, D.I.; Smith, J.S.; Shankar, H.; Gustin, R.M.; Schattauer, S.S.; Lemos, J.C.; Chavkin, N.W.; Hagan, C.E.; Neumaier, J.F.; et al. Stress produces aversion and potentiates cocaine reward by releasing endogenous dynorphins in the ventral striatum to locally stimulate serotonin reuptake. J. Neurosci. 2012, 32, 17582–17596. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, T.N.; Christensen, P.M.; Gether, U. Serotonin-induced down-regulation of cell surface serotonin transporter. Neurochem. Int. 2014, 73, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Mauterer, M.I.; Estave, P.M.; Holleran, K.M.; Jones, S.R. Measurement of Dopamine Using Fast Scan Cyclic Voltammetry in Rodent Brain Slices. Bio Protoc. 2018, 8, e2473. [Google Scholar] [CrossRef]
- Yorgason, J.T.; Espana, R.A.; Jones, S.R. Demon voltammetry and analysis software: Analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J. Neurosci. Methods 2011, 202, 158–164. [Google Scholar] [CrossRef]
- Abdalla, A.; West, A.; Jin, Y.; Saylor, R.A.; Qiang, B.; Pena, E.; Linden, D.J.; Nijhout, H.F.; Reed, M.C.; Best, J.; et al. Fast serotonin voltammetry as a versatile tool for mapping dynamic tissue architecture: I. Responses at carbon fibers describe local tissue physiology. J. Neurochem. 2020, 153, 33–50. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
West, A.M.; Holleran, K.M.; Jones, S.R. Kappa Opioid Receptors Reduce Serotonin Uptake and Escitalopram Efficacy in the Mouse Substantia Nigra Pars Reticulata. Int. J. Mol. Sci. 2023, 24, 2080. https://doi.org/10.3390/ijms24032080
West AM, Holleran KM, Jones SR. Kappa Opioid Receptors Reduce Serotonin Uptake and Escitalopram Efficacy in the Mouse Substantia Nigra Pars Reticulata. International Journal of Molecular Sciences. 2023; 24(3):2080. https://doi.org/10.3390/ijms24032080
Chicago/Turabian StyleWest, Alyssa M., Katherine M. Holleran, and Sara R. Jones. 2023. "Kappa Opioid Receptors Reduce Serotonin Uptake and Escitalopram Efficacy in the Mouse Substantia Nigra Pars Reticulata" International Journal of Molecular Sciences 24, no. 3: 2080. https://doi.org/10.3390/ijms24032080
APA StyleWest, A. M., Holleran, K. M., & Jones, S. R. (2023). Kappa Opioid Receptors Reduce Serotonin Uptake and Escitalopram Efficacy in the Mouse Substantia Nigra Pars Reticulata. International Journal of Molecular Sciences, 24(3), 2080. https://doi.org/10.3390/ijms24032080