
Increased Levels of the Parkinson’s Disease-Associated Gene ITPKB Correlate with Higher Expression Levels of α-Synuclein, Independent of Mutation Status

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results
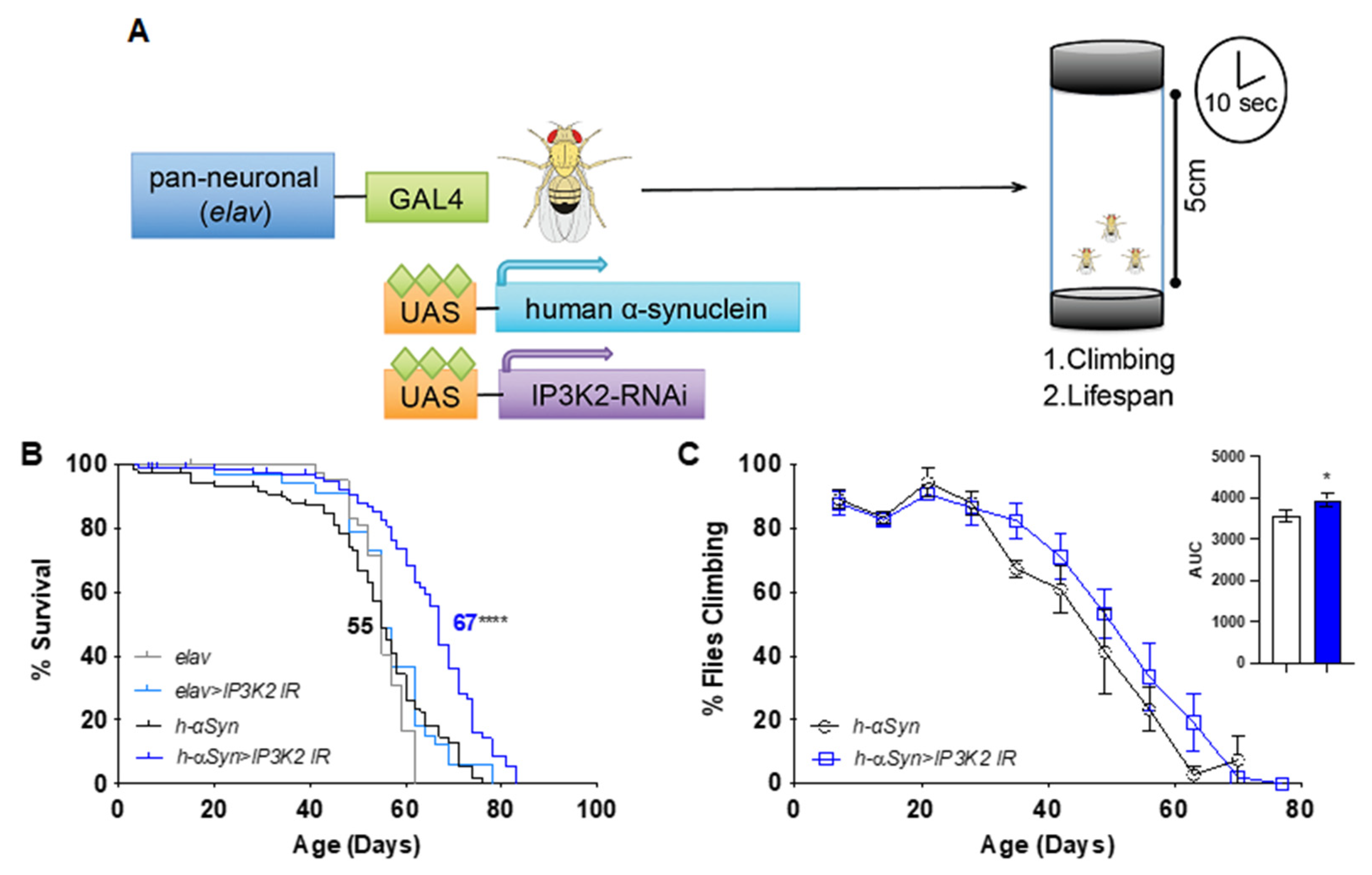
2.1. Neuronal Knockdown of IP3K2 Increases Lifespan and Climbing of Transgenic Flies Expressing Human αSyn in the Brain
2.2. Wild-Type or Mutated α-Synuclein Overexpression in SK-N-SH Cells Increases ITPKB Level
2.3. ITPKB Is Increased in Both Nucleus and Cytoplasm in SK-N-SH Cells Expressing Wild-Type or Mutated α-Synuclein
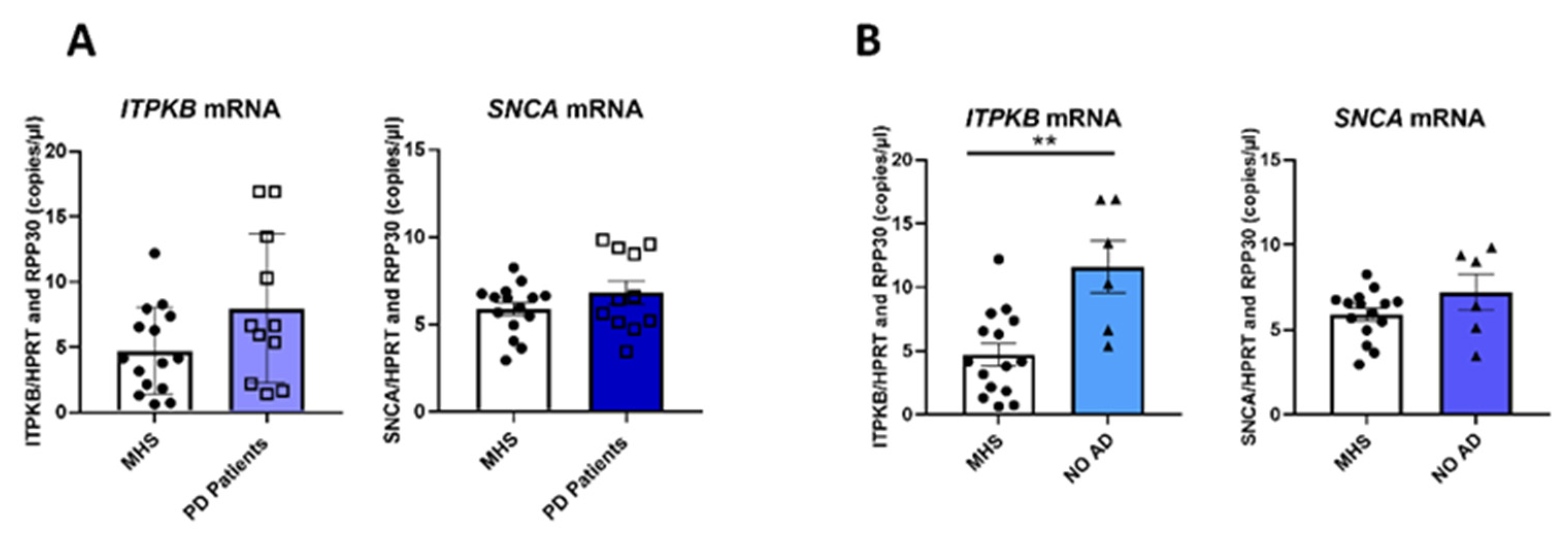
2.4. ITPKB and SNCA Expression Is Also Correlated in the Cortex of Patients with Parkinson’s
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Brain Tissue Samples
4.3. RNA Extraction, cDNA Synthesis and Droplet Digital PCR (ddPCR)
4.4. Protein Extraction and Western Blot
4.5. Immunocytochemistry (ICC) and Images Acquisition
4.6. Fly Husbandry
4.7. Locomotor Function and Lifespan
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s Disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS Clinical Diagnostic Criteria for Parkinson’s Disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-Synuclein Locus Duplication as a Cause of Familial. Lancet 2004, 364, 3. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Kasten, M.; Klein, C. The Many Faces of Alpha-Synuclein Mutations: The Many Faces of Alpha-Synuclein Mutations. Mov. Disord. 2013, 28, 697–701. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Leenders, K.L.; Sprengelmeyer, R.; Muller, T.; Woitalla, D.; Portman, A.T.; Maguire, R.P.; Veenma, L.; Schroder, U.; et al. Familial Parkinsonism with Synuclein Pathology: Clinical and PET Studies of A30P Mutation Carriers. Neurology 2001, 56, 1355–1362. [Google Scholar] [CrossRef]
- Ghosh, D.; Singh, P.K.; Sahay, S.; Jha, N.N.; Jacob, R.S.; Sen, S.; Kumar, A.; Riek, R.; Maji, S.K. Structure Based Aggregation Studies Reveal the Presence of Helix-Rich Intermediate during α-Synuclein Aggregation. Sci. Rep. 2015, 5, 9228. [Google Scholar] [CrossRef] [PubMed]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both Familial Parkinson’s Disease Mutations Accelerate α-Synuclein Aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef]
- Ohgita, T.; Namba, N.; Kono, H.; Shimanouchi, T.; Saito, H. Mechanisms of Enhanced Aggregation and Fibril Formation of Parkinson’s Disease-Related Variants of α-Synuclein. Sci. Rep. 2022, 12, 6770. [Google Scholar] [CrossRef]
- Petrucci Phenotypic Spectrum of Alpha-Synuclein Mutations: New Insights from Patients and Cellular Models|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S1353802015003545?token=A15EAF7345A76A602CF23B7AED307724791DA338C4C79B0599C9397D84A2CAE3FB7CFE7208602E972B802AA78F650171&originRegion=eu-west-1&originCreation=20220830143837 (accessed on 30 August 2022).
- International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; Van Der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; et al. A Meta-Analysis of Genome-Wide Association Studies Identifies 17 New Parkinson’s Disease Risk Loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Dewaste, V.; Moreau, C.; De SMEDT, F.; Bex, F.; De SMEDT, H.; Wuytack, F.; Missiaen, L.; Erneux, C. The Three Isoenzymes of Human Inositol-1,4,5-Trisphosphate 3-Kinase Show Specific Intracellular Localization but Comparable Ca2+ Responses on Transfection in COS-7 Cells. Biochem. J. 2003, 374, 41–49. [Google Scholar] [CrossRef]
- Decuypere, J.-P.; Monaco, G.; Missiaen, L.; De Smedt, H.; Parys, J.B.; Bultynck, G. IP 3 Receptors, Mitochondria, and Ca2+ Signaling: Implications for Aging. J. Aging Res. 2011, 2011, 920178. [Google Scholar] [CrossRef]
- Pouillon, V.; Hascakova-Bartova, R.; Pajak, B.; Adam, E.; Bex, F.; Dewaste, V.; Van Lint, C.; Leo, O.; Erneux, C.; Schurmans, S. Inositol 1,3,4,5-Tetrakisphosphate Is Essential for T Lymphocyte Development. Nat. Immunol. 2003, 4, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.G.; Pletcher, M.T.; Warashina, M.; Choe, S.H.; Ziaee, N.; Wiltshire, T.; Sauer, K.; Cooke, M.P. Inositol (1,4,5) Trisphosphate 3 Kinase B Controls Positive Selection of T Cells and Modulates Erk Activity. Proc. Natl. Acad. Sci. USA 2004, 101, 5604–5609. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.T.; Sandberg, M.; Huang, Y.H.; Young, M.; Sutton, S.; Sauer, K.; Cooke, M.P. Production of Ins(1,3,4,5)P4 Mediated by the Kinase Itpkb Inhibits Store-Operated Calcium Channels and Regulates B Cell Selection and Activation. Nat. Immunol. 2007, 8, 514–521. [Google Scholar] [CrossRef]
- Emilsson, L.; Saetre, P.; Jazin, E. Alzheimer’s Disease: MRNA Expression Profiles of Multiple Patients Show Alterations of Genes Involved with Calcium Signaling. Neurobiol. Dis. 2006, 21, 618–625. [Google Scholar] [CrossRef]
- Stygelbout, V.; Leroy, K.; Pouillon, V.; Ando, K.; D’Amico, E.; Jia, Y.; Luo, H.R.; Duyckaerts, C.; Erneux, C.; Schurmans, S.; et al. Inositol Trisphosphate 3-Kinase B Is Increased in Human Alzheimer Brain and Exacerbates Mouse Alzheimer Pathology. Brain 2014, 137, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; Sierksma, A.; Vanden Eynden, E.; De Strooper, B. MiR-132 Loss De-represses ITPKB and Aggravates Amyloid and TAU Pathology in Alzheimer’s Brain. EMBO Mol. Med. 2016, 8, 1005–1018. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, C.; Nan, Y.; Nan, S. Microglia-Derived Extracellular Vesicles Carrying MiR-711 Alleviate Neurodegeneration in a Murine Alzheimer’s Disease Model by Binding to Itpkb. Front. Cell Dev. Biol. 2020, 8, 566530. [Google Scholar] [CrossRef]
- Apicco, D.J.; Shlevkov, E.; Nezich, C.L.; Tran, D.T.; Guilmette, E.; Nicholatos, J.W.; Bantle, C.M.; Chen, Y.; Glajch, K.E.; Abraham, N.A.; et al. The Parkinson’s Disease-Associated Gene ITPKB Protects against α-Synuclein Aggregation by Regulating ER-to-Mitochondria Calcium Release. Proc. Natl. Acad. Sci. USA 2021, 118, e2006476118. [Google Scholar] [CrossRef] [PubMed]
- Vozdek, R.; Pramstaller, P.P.; Hicks, A.A. Functional Screening of Parkinson’s Disease Susceptibility Genes to Identify Novel Modulators of α-Synuclein Neurotoxicity in Caenorhabditis Elegans. Front. Aging Neurosci. 2022, 14, 806000. [Google Scholar] [CrossRef] [PubMed]
- Volta, M.; Lavdas, A.A.; Obergasteiger, J.; Überbacher, C.; Picard, A.; Pramstaller, P.P.; Hicks, A.A.; Corti, C. Elevated Levels of Alpha-Synuclein Blunt Cellular Signal Transduction Downstream of Gq Protein-Coupled Receptors. Cell. Signal. 2017, 30, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Obergasteiger, J.; Frapporti, G.; Lamonaca, G.; Pizzi, S.; Picard, A.; Lavdas, A.A.; Pischedda, F.; Piccoli, G.; Hilfiker, S.; Lobbestael, E.; et al. Kinase Inhibition of G2019S-LRRK2 Enhances Autolysosome Formation and Function to Reduce Endogenous Alpha-Synuclein Intracellular Inclusions. Cell Death Discov. 2020, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, L.; Mingozzi, F.; Cigni, C.; Marzi, R.; Di Gioia, M.; Garrè, M.; Parazzoli, D.; Sironi, L.; Collini, M.; Sakaguchi, R.; et al. Inositol 1,4,5-Trisphosphate 3-Kinase B Promotes Ca2+ Mobilization and the Inflammatory Activity of Dendritic Cells. Sci. Signal. 2021, 14, eaaz2120. [Google Scholar] [CrossRef] [PubMed]
- Nalaskowski, M.M.; Fliegert, R.; Ernst, O.; Brehm, M.A.; Fanick, W.; Windhorst, S.; Lin, H.; Giehler, S.; Hein, J.; Lin, Y.-N.; et al. Human Inositol 1,4,5-Trisphosphate 3-Kinase Isoform B (IP3KB) Is a Nucleocytoplasmic Shuttling Protein Specifically Enriched at Cortical Actin Filaments and at Invaginations of the Nuclear Envelope. J. Biol. Chem. 2011, 286, 4500–4510. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kim, H.; Jo, A.; Khang, R.; Park, C.-H.; Park, S.-J.; Kwag, E.; Shin, J.-H. α-Synuclein A53T Binds to Transcriptional Adapter 2-Alpha and Blocks Histone H3 Acetylation. Int. J. Mol. Sci. 2021, 22, 5392. [Google Scholar] [CrossRef]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. α-Synuclein Acts in the Nucleus to Inhibit Histone Acetylation and Promote Neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Genome Wide Association Study. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Ding, J.; Lian, Y.; Meng, Y.; He, Y.; Fan, H.; Li, C.; Qiu, P. The Effect of α-Synuclein and Tau in Methamphetamine Induced Neurotoxicity in Vivo and in Vitro. Toxicol. Lett. 2020, 319, 213–224. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1 α, A Potential Therapeutic Target for Early Intervention in Parkinson’s Disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.C. Update on Major Neurocognitive Disorders. Focus J. Life Long Learn. Psychiatry 2021, 19, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The Genetic Architecture of Parkinson’s Disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Gialluisi, A.; Reccia, M.G.; Modugno, N.; Nutile, T.; Lombardi, A.; Di Giovannantonio, L.G.; Pietracupa, S.; Ruggiero, D.; Scala, S.; Gambardella, S.; et al. Identification of Sixteen Novel Candidate Genes for Late Onset Parkinson’s Disease. Mol. Neurodegener. 2021, 16, 35. [Google Scholar] [CrossRef] [PubMed]
- Saetre, P.; Jazin, E.; Emilsson, L. Age-Related Changes in Gene Expression Are Accelerated in Alzheimer’s Disease. Synapse 2011, 65, 971–974. [Google Scholar] [CrossRef]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the Open-Access Database of Transcription Factor Binding Profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Jia, C.; Cheng, C.; Li, T.; Chen, X.; Yang, Y.; Liu, X.; Li, S.; Le, W. α-Synuclein Up-Regulates Monoamine Oxidase A Expression and Activity via Trans-Acting Transcription Factor 1. Front. Aging Neurosci. 2021, 13, 653379. [Google Scholar] [CrossRef]
- Visanji, N.P.; Lang, A.E.; Kovacs, G.G. Beyond the Synucleinopathies: Alpha Synuclein as a Driving Force in Neurodegenerative Comorbidities. Transl. Neurodegener. 2019, 8, 28. [Google Scholar] [CrossRef]
- Keo, A.; Mahfouz, A.; Ingrassia, A.M.T.; Meneboo, J.-P.; Villenet, C.; Mutez, E.; Comptdaer, T.; Lelieveldt, B.P.F.; Figeac, M.; Chartier-Harlin, M.-C.; et al. Transcriptomic Signatures of Brain Regional Vulnerability to Parkinson’s Disease. Commun. Biol. 2020, 3, 101. [Google Scholar] [CrossRef]
- Hawrylycz, M.; Miller, J.A.; Menon, V.; Feng, D.; Dolbeare, T.; Guillozet-Bongaarts, A.L.; Jegga, A.G.; Aronow, B.J.; Lee, C.-K.; Bernard, A.; et al. Canonical Genetic Signatures of the Adult Human Brain. Nat. Neurosci. 2015, 18, 1832–1844. [Google Scholar] [CrossRef]
- Pallos, J.; Jeng, S.; McWeeney, S.; Martin, I. Dopamine Neuron-Specific LRRK2 G2019S Effects on Gene Expression Revealed by Translatome Profiling. Neurobiol. Dis. 2021, 155, 105390. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.T.; Dahlberg, C.; Sandberg, M.L.; Wen, B.G.; Beisner, D.R.; Hoerter, J.A.H.; Parker, A.; Schmedt, C.; Stinson, M.; Avis, J.; et al. Inhibition of the Inositol Kinase Itpkb Augments Calcium Signaling in Lymphocytes and Reveals a Novel Strategy to Treat Autoimmune Disease. PLoS ONE 2015, 10, e0131071. [Google Scholar] [CrossRef] [PubMed]
- Thangavelu, G.; Du, J.; Paz, K.G.; Loschi, M.; Zaiken, M.C.; Flynn, R.; Taylor, P.A.; Kirchmeier, A.K.; Panoskaltsis-Mortari, A.; Luznik, L.; et al. Inhibition of Inositol Kinase B Controls Acute and Chronic Graft-versus-Host Disease. Blood 2020, 135, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Dietzl, G.; Chen, D.; Schnorrer, F.; Su, K.-C.; Barinova, Y.; Fellner, M.; Gasser, B.; Kinsey, K.; Oppel, S.; Scheiblauer, S.; et al. A Genome-Wide Transgenic RNAi Library for Conditional Gene Inactivation in Drosophila. Nature 2007, 448, 151–156. [Google Scholar] [CrossRef]
- Le Bourg, E.; Lints, F.A. Hypergravity and Aging in Drosophila Melanogaster. 4. Climbing Activity. Gerontology 1992, 38, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Ganetzky, B.; Flanagan, J.R. On the Relationship between Senescence and Age-Related Changes in Two Wild-Type Strains of Drosophila Melanogaster. Exp. Gerontol. 1978, 13, 189–196. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age at Death (Years) | Sex | AD | LB | PMI (h) |
|---|---|---|---|---|---|
| P1 | 73 | F | No | Diffuse | 22.3 |
| P2 | 81 | F | No | No | 78.3 |
| P3 | 86 | F | Early Alzheimer’s | Diffuse | NA |
| P4 | 84 | M | Alzheimer’s | No | NA |
| P5 | 77 | M | Early Alzheimer’s | Yes | 7 |
| P6 | 71 | M | Alzheimer’s | No | NA |
| P7 | 70 | F | No | Diffuse | 8 |
| P8 | 79 | F | Alzheimer’s type pathology | No | NA |
| P9 | 86 | M | No | Diffuse | NA |
| P10 | 78 | M | No | No | 23 |
| P11 | 85 | F | No | Yes | 13.3 |
| C1 | 76 | M | - | - | 23.3 |
| C2 | 88 | M | - | - | NA |
| C3 | 73 | M | - | - | 13 |
| C4 | 69 | W | - | - | 54 |
| C5 | 69 | M | - | - | 55 |
| C6 | 85 | F | - | - | 24 |
| C7 | 61 | M | - | - | 28.3 |
| C8 | 61 | F | - | - | 71 |
| C9 | 78 | M | - | - | 77 |
| C10 | 76 | F | - | - | 6.5 |
| C11 | 72 | M | - | - | 38.3 |
| C12 | 76 | F | - | - | 24 |
| C13 | 76 | M | - | - | 41.3 |
| C14 | 72 | F | - | - | 64.3 |
| C15 | 76 | M | - | - | 32.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Leva, F.; Filosi, M.; Oyston, L.; Silvestri, E.; Picard, A.; Lavdas, A.A.; Lobbestael, E.; Baekelandt, V.; Neely, G.G.; Pramstaller, P.P.; et al. Increased Levels of the Parkinson’s Disease-Associated Gene ITPKB Correlate with Higher Expression Levels of α-Synuclein, Independent of Mutation Status. Int. J. Mol. Sci. 2023, 24, 1984. https://doi.org/10.3390/ijms24031984
Di Leva F, Filosi M, Oyston L, Silvestri E, Picard A, Lavdas AA, Lobbestael E, Baekelandt V, Neely GG, Pramstaller PP, et al. Increased Levels of the Parkinson’s Disease-Associated Gene ITPKB Correlate with Higher Expression Levels of α-Synuclein, Independent of Mutation Status. International Journal of Molecular Sciences. 2023; 24(3):1984. https://doi.org/10.3390/ijms24031984
Chicago/Turabian StyleDi Leva, Francesca, Michele Filosi, Lisa Oyston, Erica Silvestri, Anne Picard, Alexandros A. Lavdas, Evy Lobbestael, Veerle Baekelandt, G. Gregory Neely, Peter P. Pramstaller, and et al. 2023. "Increased Levels of the Parkinson’s Disease-Associated Gene ITPKB Correlate with Higher Expression Levels of α-Synuclein, Independent of Mutation Status" International Journal of Molecular Sciences 24, no. 3: 1984. https://doi.org/10.3390/ijms24031984
APA StyleDi Leva, F., Filosi, M., Oyston, L., Silvestri, E., Picard, A., Lavdas, A. A., Lobbestael, E., Baekelandt, V., Neely, G. G., Pramstaller, P. P., Hicks, A. A., & Corti, C. (2023). Increased Levels of the Parkinson’s Disease-Associated Gene ITPKB Correlate with Higher Expression Levels of α-Synuclein, Independent of Mutation Status. International Journal of Molecular Sciences, 24(3), 1984. https://doi.org/10.3390/ijms24031984