
Using a Network-Based Analysis Approach to Investigate the Involvement of S. aureus in the Pathogenesis of Granulomatosis with Polyangiitis

 , , ,
, , ,
Abstract
1. Introduction
2. Results
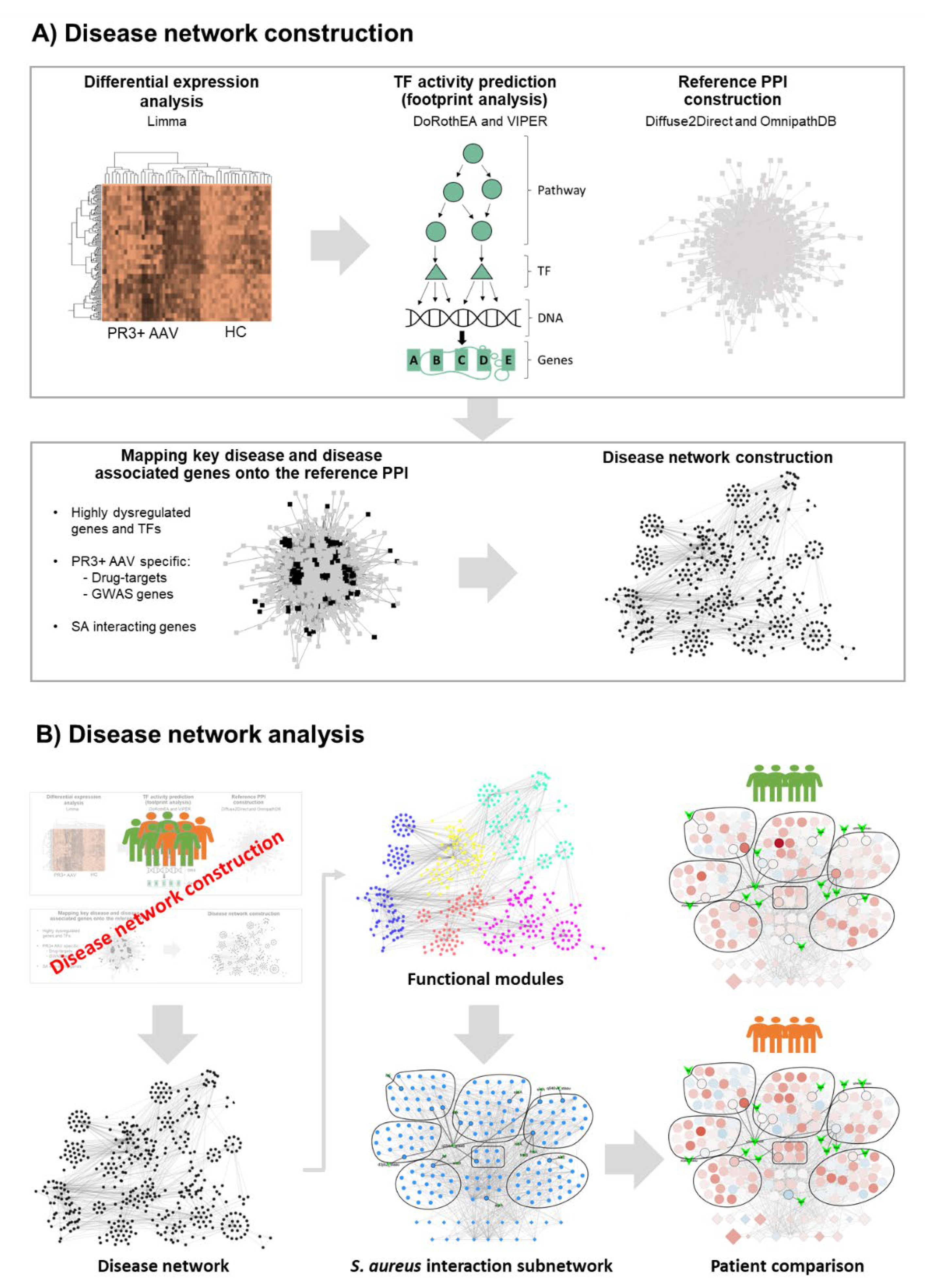
2.1. Generating the PR3+ AAV Disease Network
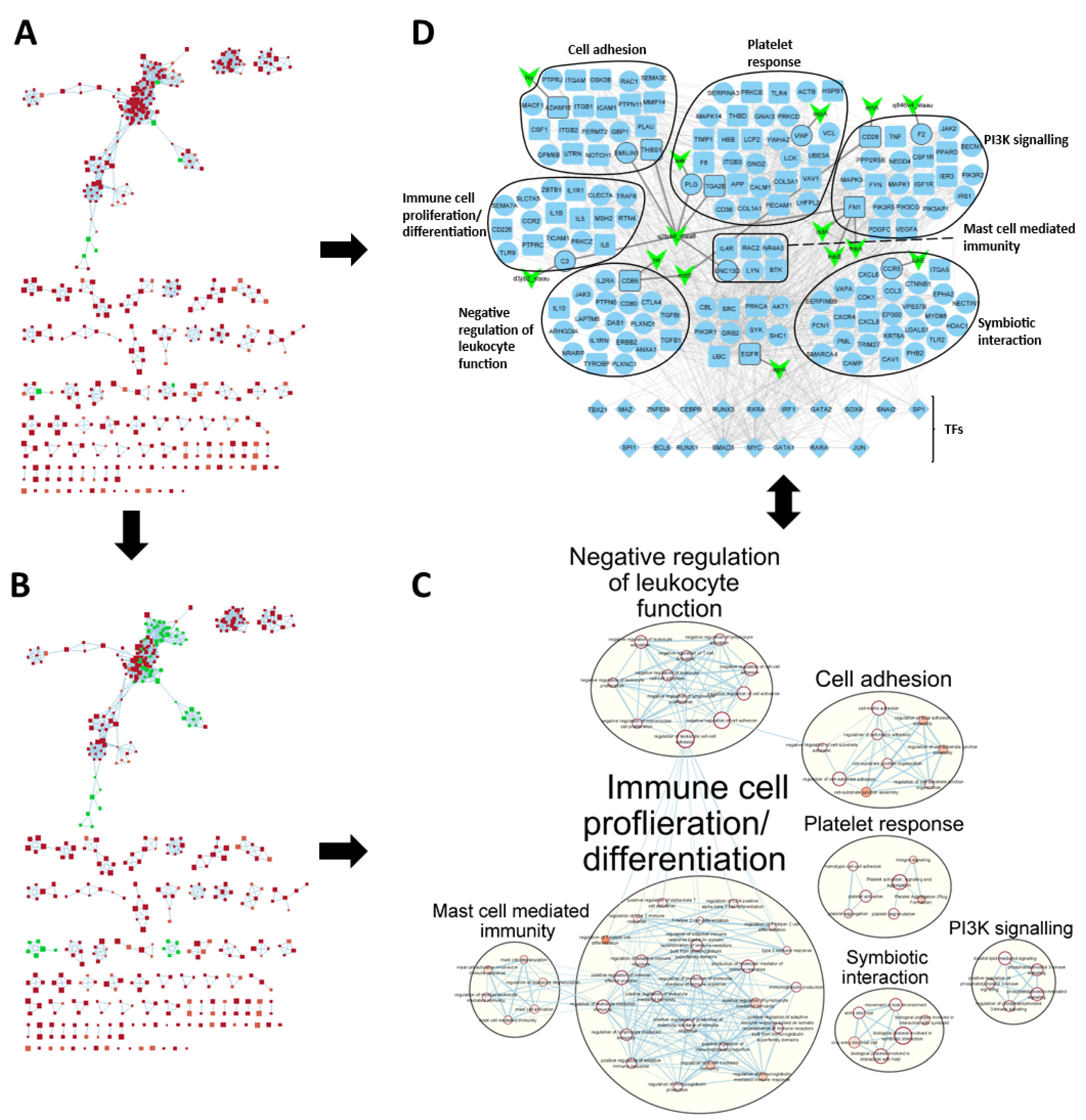
2.2. PR3+ AAV Disease Network Captures Key Disease Genes
2.3. Clustering Reveals Differences between Patient Subgroups
2.4. SA-PR3+ AAV Interaction Network Highlights Potential Impact of SA on Key Functional Modules
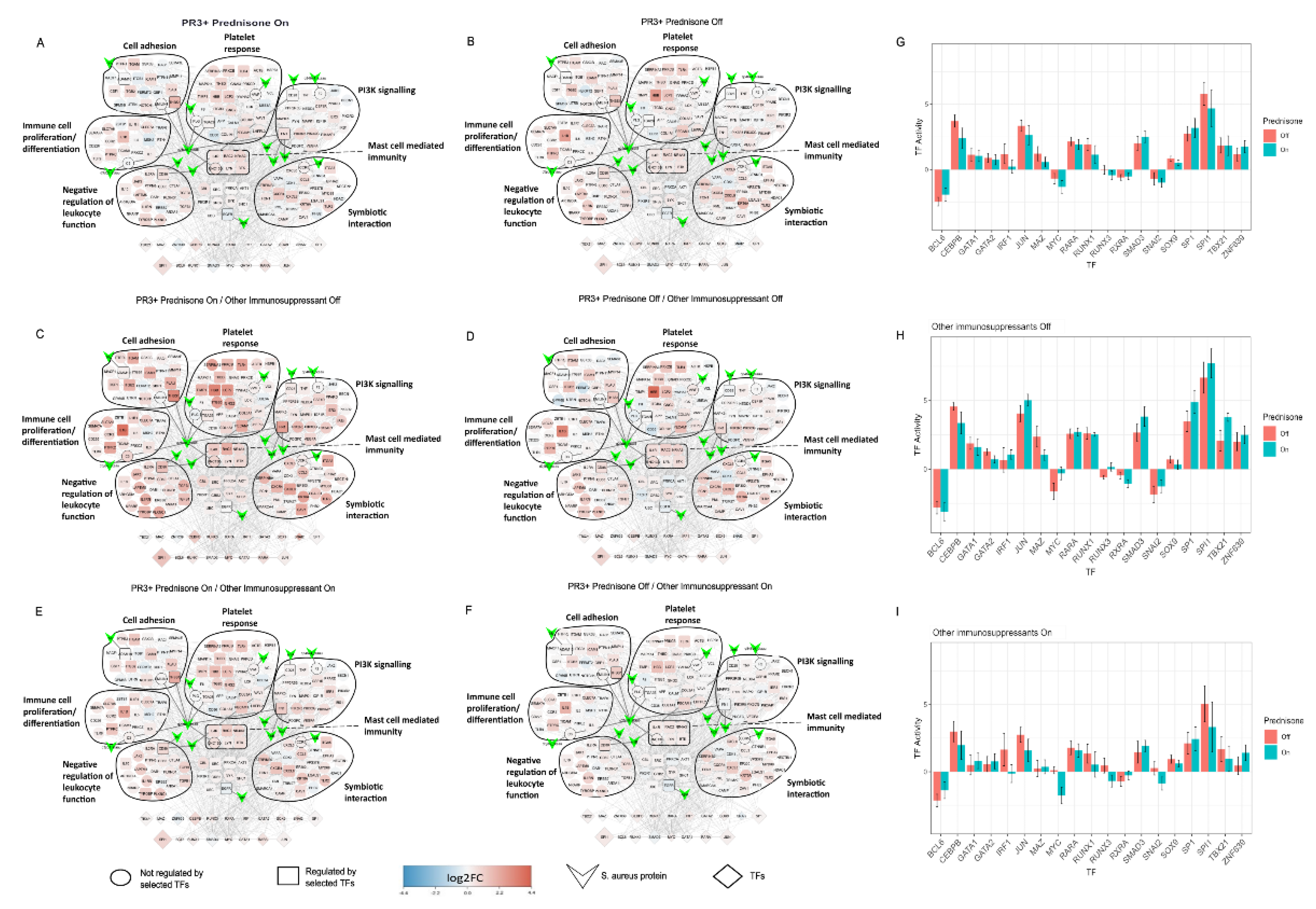
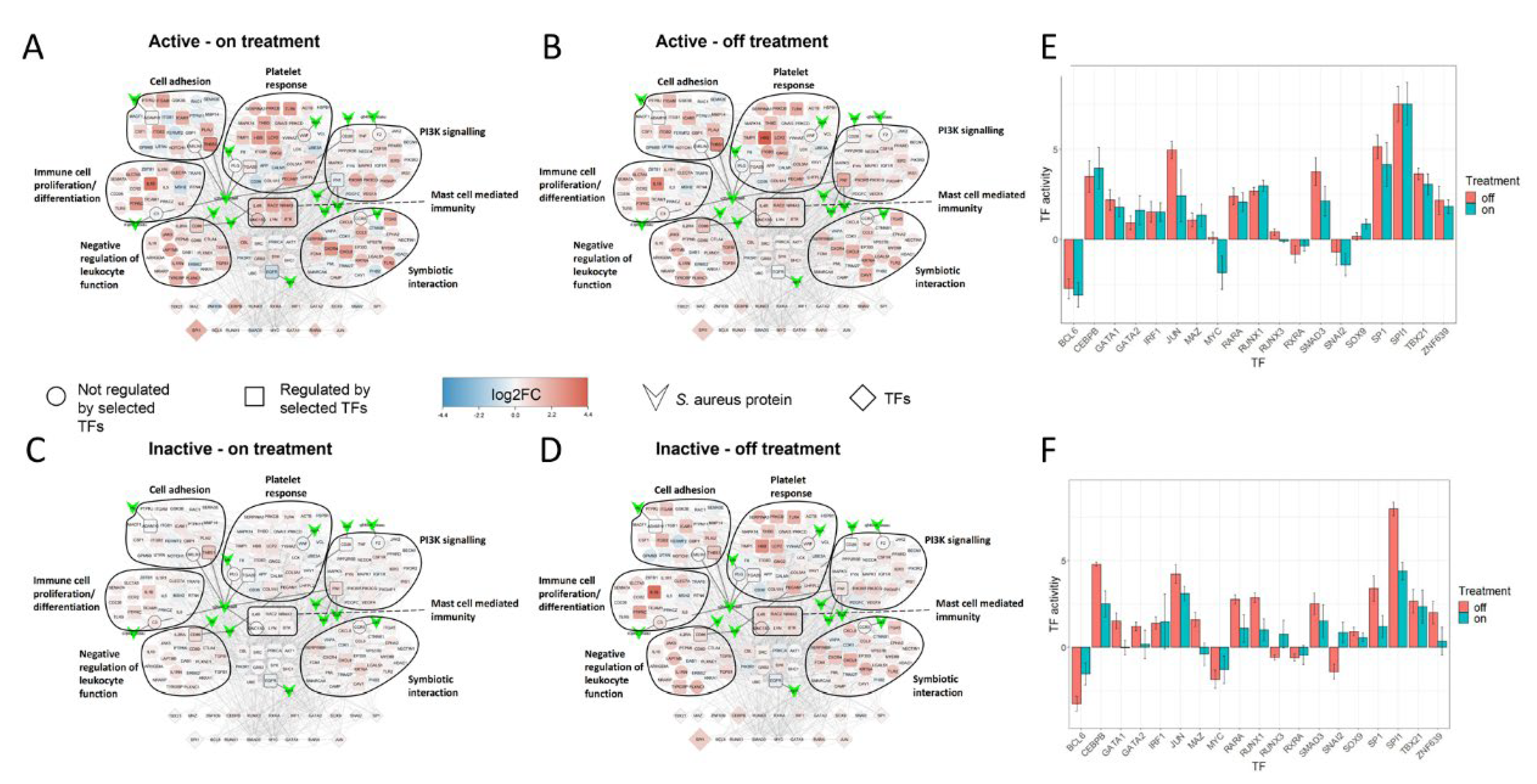
2.5. Treatment Induced Expression Pattern Is Dependent on Nasal Disease Status
3. Discussion
4. Materials and Methods
4.1. Disease Network Construction
Data Acquisition and Pre-Processing
4.2. Microarray Data Analysis
4.3. Predicting TF Activity (Footprint Analysis)
4.4. Finding Genes of Interest
4.5. Constructing a Reference PPI Network
4.6. Selecting Disease Genes
4.7. Augmenting Disease Network
4.8. Disease Network Analysis
Gene Set Enrichment Analysis Using g:Profiler
4.9. GSEA Visualisation, Disease Network Filtering and SA-PR3+ AAV Interaction Network Creation
4.10. Patient Comparisons in SA-PR3+ AAV Interaction Networks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kitching, A.R.; Anders, H.-J.; Basu, N.; Brouwer, E.; Gordon, J.; Jayne, D.R.; Kullman, J.; Lyons, P.A.; Merkel, P.A.; Savage, C.O.S.; et al. ANCA-associated vasculitis. Nat. Rev. Dis. Prim. 2020, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Windpessl, M.; Bettac, E.L.; Gauckler, P.; Shin, J.I.; Geetha, D.; Kronbichler, A. ANCA Status or Clinical Phenotype—What Counts More? Curr. Rheumatol. Rep. 2021, 23, 37. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, F.; Perricone, C.; Olivieri, G.; Cipriano, E.; Spinelli, F.R.; Valesini, G.; Conti, F. Staphylococcus aureus Nasal Carriage and Autoimmune Diseases: From Pathogenic Mechanisms to Disease Susceptibility and Phenotype. Int. J. Mol. Sci. 2019, 20, 5624. [Google Scholar] [CrossRef] [PubMed]
- Laudien, M.; Gadola, S.D.; Podschun, R.; Hedderich, J.; Paulsen, J.; Reinhold-Keller, E.; Csernok, E.; Ambrosch, P.; Hellmich, B.; Moosig, F.; et al. Nasal carriage of Staphylococcus aureus and endonasal activity in Wegener s granulomatosis as compared to rheumatoid arthritis and chronic Rhinosinusitis with nasal polyps. Clin. Exp. Rheumatol. 2010, 28, 51–55. [Google Scholar] [PubMed]
- Salmela, A.; Rasmussen, N.; Tervaert, J.W.C.; Jayne, D.R.W.; Ekstrand, A. Chronic nasal Staphylococcus aureus carriage identifies a subset of newly diagnosed granulomatosis with polyangiitis patients with high relapse rate. Rheumatology 2017, 56, 965–972. [Google Scholar] [CrossRef]
- Popa, E.R.; Stegeman, C.A.; Abdulahad, W.H.; van der Meer, B.; Arends, J.; Manson, W.M.; Bos, N.A.; Kallenberg, C.G.M.; Cohen Tervaert, J.-W. Staphylococcal toxic-shock-syndrome-toxin-1 as a risk factor for disease relapse in Wegener’s granulomatosis. Rheumatology 2007, 46, 1029–1033. [Google Scholar] [CrossRef]
- Stegeman, C.A.; Cohen Tervaert, J.W.; de Jong, P.E.; Kallenberg, C.G.M. Trimethoprim–Sulfamethoxazole (Co-Trimoxazole) for the Prevention of Relapses of Wegener’s Granulomatosis. N. Engl. J. Med. 1996, 335, 16–20. [Google Scholar] [CrossRef]
- Zycinska, K.; Wardyn, K.A.; Zielonka, T.M.; Krupa, R.; Lukas, W. Co-trimoxazole and prevention of relapses of PR3-ANCA positive vasculitis with pulmonary involvement. Eur. J. Med. Res. 2009, 14, 265–267. [Google Scholar] [CrossRef]
- Chelliah, V.; van der Graaf, P. Model-informed target identification and validation through combining quantitative systems pharmacology with network-based analysis. CPT Pharmacomet. Syst. Pharm. 2022, 11, 399–402. [Google Scholar]
- Grayson, P.C.; Steiling, K.; Platt, M.; Berman, J.S.; Zhang, X.; Xiao, J.; Alekseyev, Y.O.; Liu, G.; Monach, P.A.; Kaplan, M.J.; et al. Defining the nasal transcriptome in granulomatosis with polyangiitis (Wegener’s). Arthritis Rheumatol. 2015, 67, 2233–2239. [Google Scholar] [CrossRef]
- Kronbichler, A.; Lee, K.H.; Denicolò, S.; Choi, D.; Lee, H.; Ahn, D.; Kim, K.H.; Lee, J.H.; Kim, H.; Hwang, M.; et al. Immunopathogenesis of ANCA-Associated Vasculitis. Int. J. Mol. Sci. 2020, 21, 7319. [Google Scholar] [CrossRef]
- Ammari, M.G.; Gresham, C.R.; McCarthy, F.M.; Nanduri, B. HPIDB 2.0: A curated database for host–pathogen interactions. Database 2016, 2016, baw103. [Google Scholar] [CrossRef]
- Dugourd, A.; Saez-Rodriguez, J. Footprint-based functional analysis of multiomic data. Curr. Opin. Syst. Biol. 2019, 15, 82–90. [Google Scholar] [CrossRef]
- Schreiber, A.; Kettritz, R. The neutrophil in antineutrophil cytoplasmic autoantibody-associated vasculitis. J. Leukoc. Biol. 2013, 94, 623–631. [Google Scholar] [CrossRef]
- Adamik, J.; Wang, K.Z.Q.; Unlu, S.; Su, A.-J.A.; Tannahill, G.M.; Galson, D.L.; O’Neill, L.A.; Auron, P.E. Distinct Mechanisms for Induction and Tolerance Regulate the Immediate Early Genes Encoding Interleukin 1β and Tumor Necrosis Factor α. PLoS ONE 2013, 8, e70622. [Google Scholar] [CrossRef]
- Kominato, Y.; Galson, D.; Waterman, W.R.; Webb, A.C.; Auron, P.E. Monocyte expression of the human prointerleukin 1 beta gene (IL1B) is dependent on promoter sequences which bind the hematopoietic transcription factor Spi-1/PU.1. Mol. Cell. Biol. 1995, 15, 59–68. [Google Scholar] [CrossRef]
- Pulugulla, S.H.; Workman, R.; Rutter, N.W.; Yang, Z.; Adamik, J.; Lupish, B.; Macar, D.A.; el Abdouni, S.; Esposito, E.X.; Galson, D.L.; et al. A combined computational and experimental approach reveals the structure of a C/EBPβ–Spi1 interaction required for IL1B gene transcription. J. Biol. Chem. 2018, 293, 19942–19956. [Google Scholar] [CrossRef]
- Abdgawad, M.; Pettersson, Å.; Gunnarsson, L.; Bengtsson, A.A.; Geborek, P.; Nilsson, L.; Segelmark, M.; Hellmark, T. Decreased Neutrophil Apoptosis in Quiescent ANCA-Associated Systemic Vasculitis. PLoS ONE 2012, 7, e32439. [Google Scholar] [CrossRef]
- Falvo, J.V.; Uglialoro, A.M.; Brinkman, B.M.; Merika, M.; Parekh, B.S.; Tsai, E.Y.; King, H.C.; Morielli, A.D.; Peralta, E.G.; Maniatis, T.; et al. Stimulus-specific assembly of enhancer complexes on the tumor necrosis factor alpha gene promoter. Mol. Cell. Biol. 2000, 20, 2239–2247. [Google Scholar] [CrossRef]
- Larsson, L.; Rymo, L.; Berglundh, T. Sp1 binds to the G allele of the−1087 polymorphism in the IL-10 promoter and promotes IL-10 mRNA transcription and protein production. Genes Immun. 2010, 11, 181–187. [Google Scholar] [CrossRef]
- Popa, E.R.; Franssen, C.F.; Limburg, P.C.; Huitema, M.G.; Kallenberg, C.G.; Tervaert, J.W. In vitro cytokine production and proliferation of T cells from patients with anti-proteinase 3- and antimyeloperoxidase-associated vasculitis, in response to proteinase 3 and myeloperoxidase. Arthritis Rheum. 2002, 46, 1894–1904. [Google Scholar] [CrossRef]
- Kanada, S.; Nishiyama, C.; Nakano, N.; Suzuki, R.; Maeda, K.; Hara, M.; Kitamura, N.; Ogawa, H.; Okumura, K. Critical role of transcription factor PU.1 in the expression of CD80 and CD86 on dendritic cells. Blood 2011, 117, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Boehme, L.; Nel, L.; Casian, A.; Sangle, S.; Nova-Lamperti, E.; Seitan, V.; Spencer, J.; Lavender, P.; D’Cruz, D.P.; et al. Defective STAT5 Activation and Aberrant Expression of BCL6 in Naive CD4 T Cells Enhances Follicular Th Cell-like Differentiation in Patients with Granulomatosis with Polyangiitis. J. Immunol. 2022, 208, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell Adhesion by Integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H. Integrin signalling and function in immune cells. Immunology 2012, 135, 268–275. [Google Scholar] [CrossRef]
- Wikman, A.; Lundahl, J.; Jacobson, S.H. Sustained Monocyte Activation in Clinical Remission of Systemic Vasculitis. Inflammation 2008, 31, 384–390. [Google Scholar] [CrossRef]
- Wikman, A.; Fagergren, A.; Gunnar, O.; Johansson, S.; Lundahl, J.; Jacobson, S.H. Monocyte activation and relationship to anti-proteinase 3 in acute vasculitis. Nephrol. Dial. Transplant. 2003, 18, 1792–1799. [Google Scholar] [CrossRef]
- Kobold, A.C.M.; Kallenberg, C.G.M.; Tervaert, J.W.C. Monocyte activation in patients with Wegener’s granulomatosis. Ann. Rheum. Dis. 1999, 58, 237–245. [Google Scholar] [CrossRef]
- Nowack, R.; Schwalbe, K.; Flores-Suarez, L.-F.; Yard, B.; Van Der Woude, F.J. Upregulation of CD14 and CD18 on Monocytes In Vitro by Antineutrophil Cytoplasmic Autoantibodies. J. Am. Soc. Nephrol. 2000, 11, 1639–1646. [Google Scholar] [CrossRef]
- Matsumoto, K.; Kurasawa, T.; Yoshimoto, K.; Suzuki, K.; Takeuchi, T. Identification of neutrophil β2-integrin LFA-1 as a potential mechanistic biomarker in ANCA-associated vasculitis via microarray and validation analyses. Arthritis Res. Ther. 2021, 23, 136. [Google Scholar] [CrossRef]
- Jerke, U.; Rolle, S.; Dittmar, G.; Bayat, B.; Santoso, S.; Sporbert, A.; Luft, F.; Kettritz, R. Complement receptor Mac-1 is an adaptor for NB1 (CD177)-mediated PR3-ANCA neutrophil activation. J. Biol. Chem. 2011, 286, 7070–7081. [Google Scholar] [CrossRef]
- Beekhuizen, H.; Corsèl-Van Tilburg, A.J.; Blokland, I.; Van Furth, R. Characterization of the adherence of human monocytes to cytokine-stimulated human macrovascular endothelial cells. Immunology 1991, 74, 661–669. [Google Scholar]
- Pahl, H.L.; Scheibe, R.J.; Zhang, D.E.; Chen, H.M.; Galson, D.L.; Maki, R.A.; Tenen, D.G. The proto-oncogene PU.1 regulates expression of the myeloid-specific CD11b promoter. J. Biol. Chem. 1993, 268, 5014–5020. [Google Scholar] [CrossRef]
- Rosmarin, A.G.; Caprio, D.; Levy, R.; Simkevich, C. CD18 (beta 2 leukocyte integrin) promoter requires PU.1 transcription factor for myeloid activity. Proc. Natl. Acad. Sci. USA 1995, 92, 801–805. [Google Scholar] [CrossRef]
- Li, C.; Li, J.; Ni, H. Crosstalk Between Platelets and Microbial Pathogens. Front. Immunol. 2020, 11, 1962. [Google Scholar] [CrossRef]
- Laudien, M.; Häsler, R.; Wohlers, J.; Böck, J.; Lipinski, S.; Bremer, L.; Podschun, R.; Ambrosch, P.; Lamprecht, P.; Rosenstiel, P.; et al. Molecular signatures of a disturbed nasal barrier function in the primary tissue of Wegener’s granulomatosis. Mucosal Immunol. 2011, 4, 564–573. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Glasner, C.; de Goffau, M.C.; van Timmeren, M.M.; Schulze, M.L.; Jansen, B.; Tavakol, M.; van Wamel, W.J.B.; Stegeman, C.A.; Kallenberg, C.G.M.; Arends, J.P.; et al. Genetic loci of Staphylococcus aureus associated with anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitides. Sci. Rep. 2017, 7, 12211. [Google Scholar] [CrossRef]
- Fijołek, J.; Wiatr, E.; Petroniec, V.; Augustynowicz-Kopec, E.; Bednarek, M.; Gawryluk, D.; Martusewicz-Boros, M.M.; Modrzewska, K.; Radzikowska, E.; Roszkowski-Sliz, K. The presence of staphylococcal superantigens in nasal swabs and correlation with activity of granulomatosis with polyangiitis in own material. Clin. Exp. Rheumatol. 2018, 36, 40–45. [Google Scholar]
- Rhee, R.L.; Sreih, A.G.; Najem, C.E.; Grayson, P.C.; Zhao, C.; Bittinger, K.; Collman, R.G.; Merkel, P.A. Characterisation of the nasal microbiota in granulomatosis with polyangiitis. Ann. Rheum. Dis. 2018, 77, 1448–1453. [Google Scholar] [CrossRef]
- Rowland, G.; van der Graaf, P.; Chelliah, V. Model-informed drug target selection and validation through combining quantitative systems pharmacology with network-based analysis. Br. J. Pharmacol. 2021, 178, 4987–4988. [Google Scholar]
- Jansen, M.; Rowland, G.; van der Graaf, P.; Chelliah, V. Network-based analysis for model-informed drug target selection and validation: Application to Covid-19 induced inflammatory response. Br. J. Pharmacol. 2021, 178, 4988–4990. [Google Scholar]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alonso, L.; Holland, C.H.; Ibrahim, M.M.; Turei, D.; Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019, 29, 1363–1375. [Google Scholar] [CrossRef]
- Lee, K.S.; Kronbichler, A.; Pereira Vasconcelos, D.F.; Pereira da Silva, F.R.; Ko, Y.; Oh, Y.S.; Eisenhut, M.; Merkel, P.A.; Jayne, D.; Amos, C.I.; et al. Genetic Variants in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis: A Bayesian Approach and Systematic Review. J. Clin. Med. 2019, 8, 266. [Google Scholar] [CrossRef]
- Ochoa, D.; Hercules, A.; Carmona, M.; Suveges, D.; Gonzalez-Uriarte, A.; Malangone, C.; Miranda, A.; Fumis, L.; Carvalho-Silva, D.; Spitzer, M.; et al. Open Targets Platform: Supporting systematic drug–target identification and prioritisation. Nucleic Acids Res. 2020, 49, D1302–D1310. [Google Scholar] [CrossRef]
- Silverbush, D.; Sharan, R. A systematic approach to orient the human protein-protein interaction network. Nat. Commun. 2019, 10, 3015. [Google Scholar] [CrossRef]
- Türei, D.; Korcsmáros, T.; Saez-Rodriguez, J. OmniPath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 2016, 13, 966–967. [Google Scholar] [CrossRef]
- Han, H.; Lee, S.; Lee, I. NGSEA: Network-Based Gene Set Enrichment Analysis for Interpreting Gene Expression Phenotypes with Functional Gene Sets. Mol. Cells 2019, 42, 579–588. [Google Scholar] [CrossRef]
- Ghiassian, S.D.; Menche, J.; Barabási, A.-L. A DIseAse MOdule Detection (DIAMOnD) Algorithm Derived from a Systematic Analysis of Connectivity Patterns of Disease Proteins in the Human Interactome. PLOS Comput. Biol. 2015, 11, e1004120. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Nasal Disease (8 Patients) | Inactive Nasal Disease (11 Patients) | No Nasal Involvement (7 Patients) | |
|---|---|---|---|
| Mean age (years) | 41.5 | 48 | 60.3 |
| Disease duration (years) | 2.3 | 5.6 | 7 |
| BVAS (WG; mean) | 5.3 | 0 | 0 |
| VDI | 1.3 | 1.5 | 1 |
| Disease characteristic (limited/severe) (n; %) | 5 limited (62.5) 3 severe (37.5) | 4 limited (36.4) 7 severe (63.6) | 1 limited (14.3) 6 severe (85.7) |
| Relapsing disease (n; %) | 3; 37.5 (range 1–6) | 6; 54.5 (range 1–7) | 3; 42.9 (range 1–2) |
| Female sex (%) | 62.5 | 18.2 | 57.1 |
| Smoking status | Never: 6 Former: 2 | Never: 8 Former: 2 Current: 1 | Never: 1 Former: 5 Current: 1 |
| On prednisone (n; %) | 7; 87.5 | 3; 27.3 | 3; 42.9 |
| Mean prednisone dose, mg | 24.9 | 1.3 | 2.9 |
| Number taking other immunosuppressants (n; %) | 5; 62.5 | 5; 45.5 | 6; 85.7 |
| Rituximab use (n; %) | 1; 12.5 | 0; 0 | 2; 28.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rowland, G.; Kronbichler, A.; Smith, R.; Jayne, D.; van der Graaf, P.H.; Chelliah, V. Using a Network-Based Analysis Approach to Investigate the Involvement of S. aureus in the Pathogenesis of Granulomatosis with Polyangiitis. Int. J. Mol. Sci. 2023, 24, 1822. https://doi.org/10.3390/ijms24031822
Rowland G, Kronbichler A, Smith R, Jayne D, van der Graaf PH, Chelliah V. Using a Network-Based Analysis Approach to Investigate the Involvement of S. aureus in the Pathogenesis of Granulomatosis with Polyangiitis. International Journal of Molecular Sciences. 2023; 24(3):1822. https://doi.org/10.3390/ijms24031822
Chicago/Turabian StyleRowland, Gregory, Andreas Kronbichler, Rona Smith, David Jayne, Piet H. van der Graaf, and Vijayalakshmi Chelliah. 2023. "Using a Network-Based Analysis Approach to Investigate the Involvement of S. aureus in the Pathogenesis of Granulomatosis with Polyangiitis" International Journal of Molecular Sciences 24, no. 3: 1822. https://doi.org/10.3390/ijms24031822
APA StyleRowland, G., Kronbichler, A., Smith, R., Jayne, D., van der Graaf, P. H., & Chelliah, V. (2023). Using a Network-Based Analysis Approach to Investigate the Involvement of S. aureus in the Pathogenesis of Granulomatosis with Polyangiitis. International Journal of Molecular Sciences, 24(3), 1822. https://doi.org/10.3390/ijms24031822