
Identification of the Axis β-Catenin–BTK in the Dynamic Adhesion of Chronic Lymphocytic Leukemia Cells to Their Microenvironment
 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
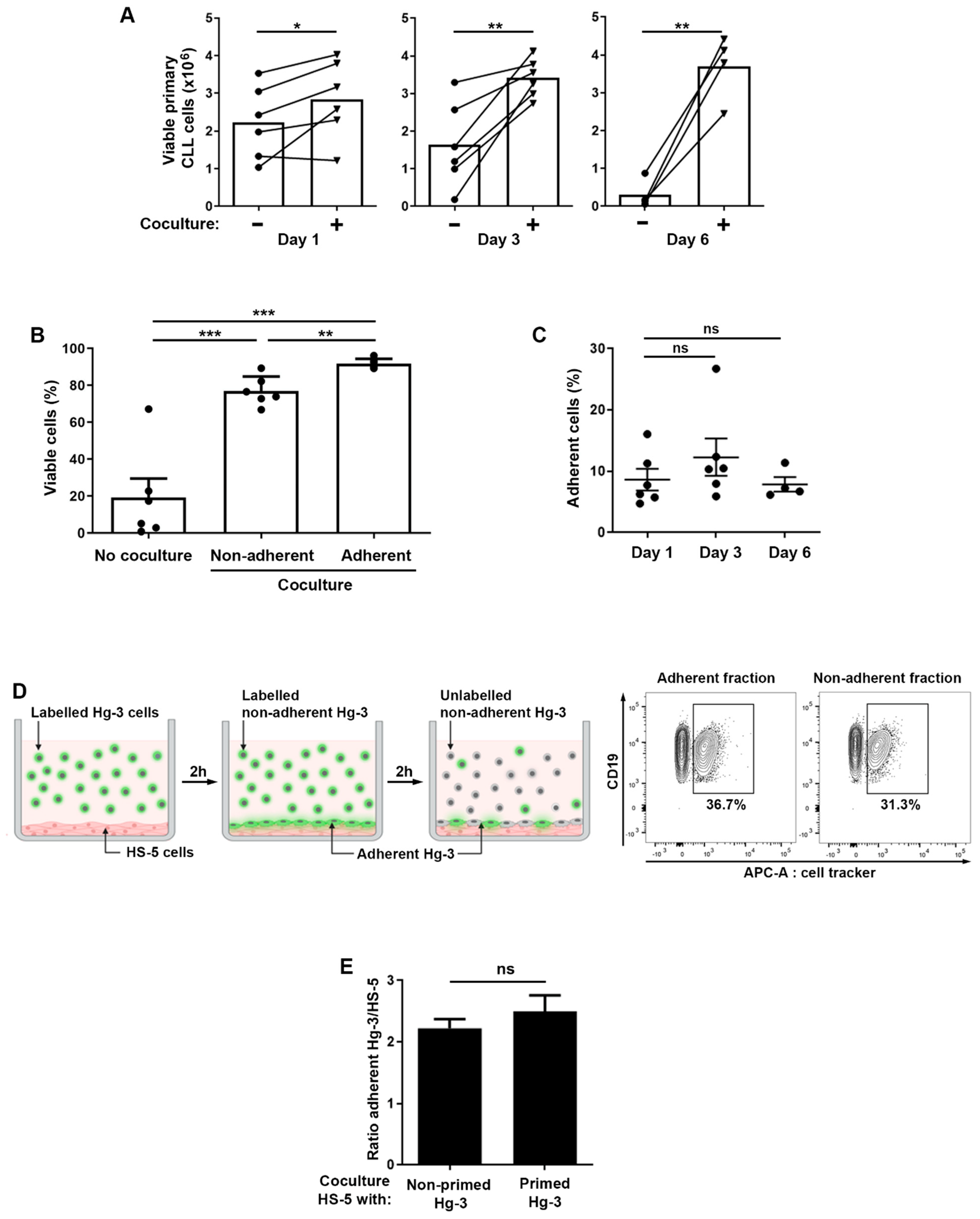
2.1. Relationship between the Adhesion and Survival of CLL B Cells in the Microenvironment
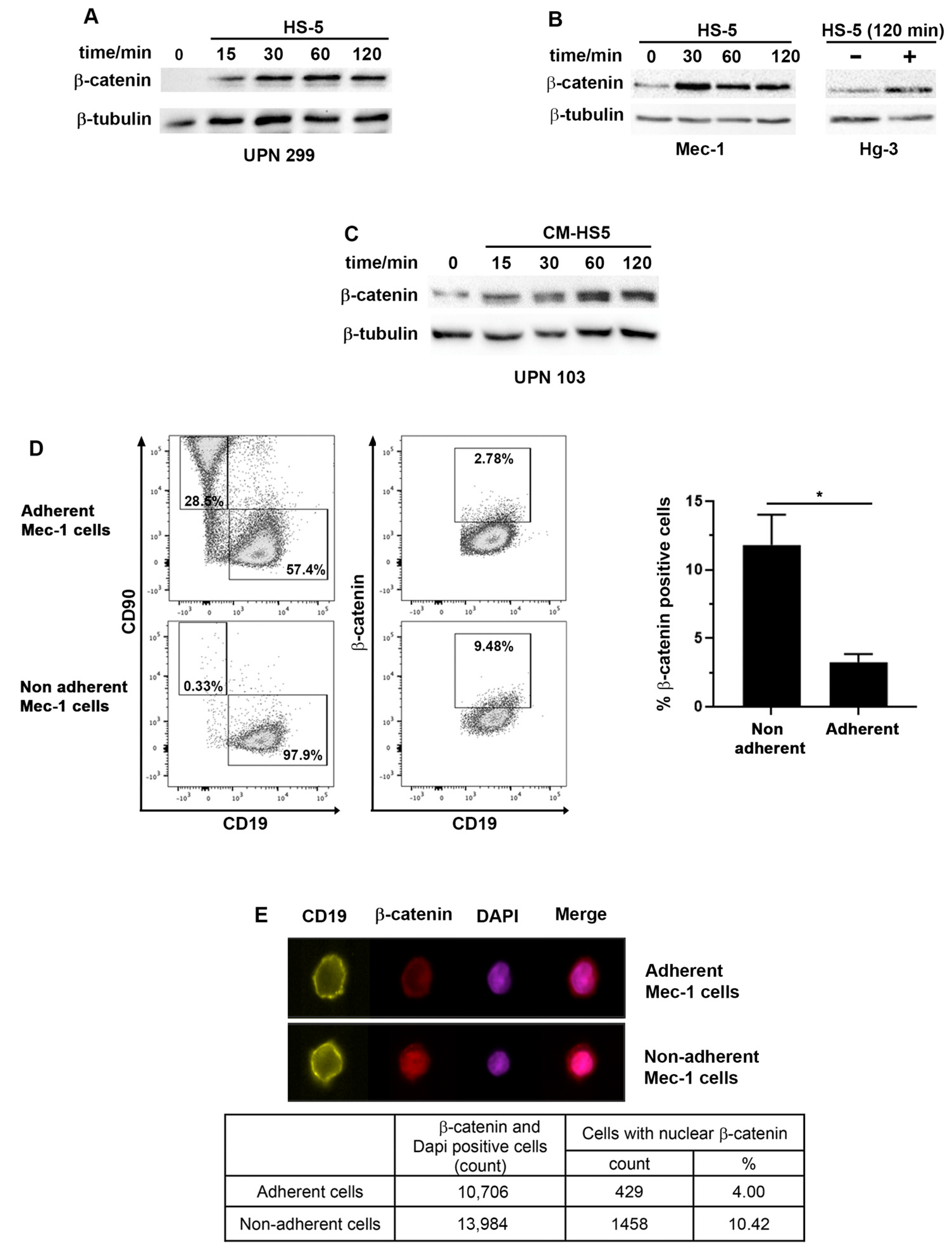
2.2. β-Catenin Levels and Cellular Localization Differ between CLL Cells Adhering or Not to Stromal Cells
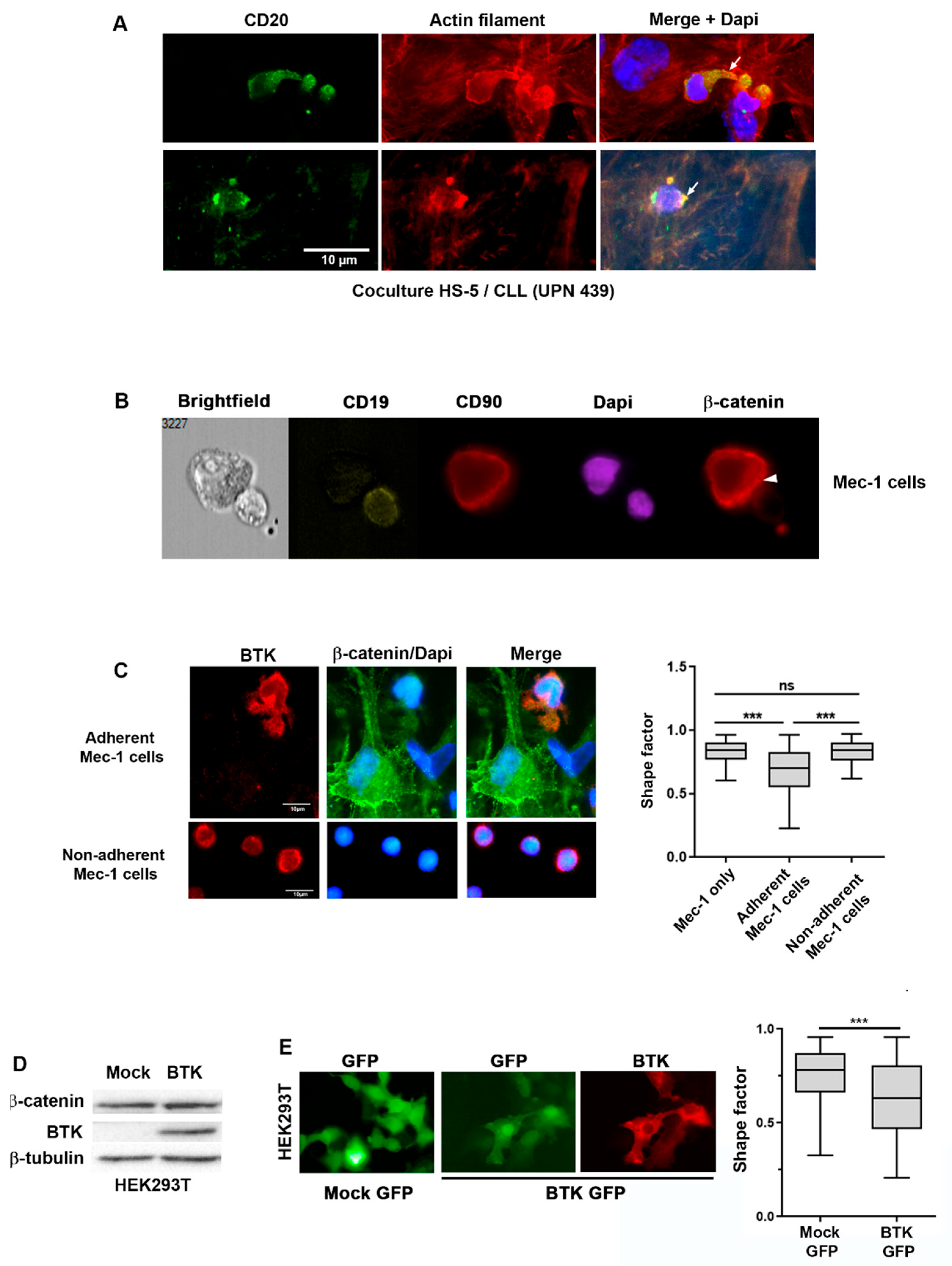
2.3. BTK and β-Catenin Impact Cell Morphology in CLL Cells Co-Cultured with Stromal Cells
2.4. β-Catenin Stabilization Is Dependent on BTK
2.5. β-Catenin Stabilization Induces Tumor Cell Adhesion to Stromal Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Western Blotting, Cell Fractionation and Immunoprecipitation
4.3. Confocal and Fluorescence Microscopy
4.4. Flow Cytometry and ImageStream
4.5. Cell Transfection and Cell Adhesion Assay
4.6. Cell Migration Assay
4.7. Quantification of Cytokine Secretion
4.8. RT-qPCR
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Millán-Salanova, M.; Vicente-Manzanares, M. The interface between biochemical signaling and cell mechanics shapes T lymphocyte migration and activation. Eur. J. Cell Biol. 2022, 101, 151236. [Google Scholar] [CrossRef] [PubMed]
- Mangolini, M.; Ringshausen, I. Bone Marrow Stromal Cells Drive Key Hallmarks of B Cell Malignancies. Int. J. Mol. Sci. 2020, 21, 1466. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.; Danglade, D.; Gardano, L.; Laguillier, C.; Lazarian, G.; Roger, C.; Thieblemont, C.; Marzec, J.; Gribben, J.; Cymbalista, F.; et al. Inhibitors of BCR signalling interrupt the survival signal mediated by the micro-environment in mantle cell lymphoma. Int. J. Cancer 2015, 136, 2761–2774. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primer 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed]
- Janovská, P.; Bryja, V. Wnt signalling pathways in chronic lymphocytic leukaemia and B-cell lymphomas. Br. J. Pharmacol. 2017, 174, 4701–4715. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shalek, A.K.; Lawrence, M.; Ding, R.; Gaublomme, J.T.; Pochet, N.; Stojanov, P.; Sougnez, C.; Shukla, S.A.; Stevenson, K.E.; et al. Somatic mutation as a mechanism of Wnt/β-catenin pathway activation in CLL. Blood 2014, 124, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and Other Novel Cancer Genes in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Amin, N. The Wnt signaling pathways and cell adhesion. Front. Biosci. 2012, 17, 784. [Google Scholar] [CrossRef]
- Kaucká, M.; Plevová, K.; Pavlová, S.; Janovská, P.; Mishra, A.; Verner, J.; Procházková, J.; Krejčí, P.; Kotašková, J.; Ovesná, P.; et al. The Planar Cell Polarity Pathway Drives Pathogenesis of Chronic Lymphocytic Leukemia by the Regulation of B-Lymphocyte Migration. Cancer Res. 2013, 73, 1491–1501. [Google Scholar] [CrossRef]
- Yu, J.; Chen, L.; Cui, B.; Widhopf, G.F.; Shen, Z.; Wu, R.; Zhang, L.; Zhang, S.; Briggs, S.P.; Kipps, T.J. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J. Clin. Investig. 2015, 126, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Yu, J.; Chen, L.; Cui, B.; Ii, G.F.W.; Rassenti, L.; Shen, Z.; Briggs, S.P.; Kipps, T.J. Wnt5a induces ROR1 to complex with HS1 to enhance migration of chronic lymphocytic leukemia cells. Leukemia 2017, 31, 2615–2622. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Rassenti, L.; Widhopf, G.F.; Yu, J.; Kipps, T.J. Wnt5a causes ROR1 to complex and activate cortactin to enhance migration of chronic lymphocytic leukemia cells. Leukemia 2019, 33, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. β-catenin is a target for the ubiquitin–proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [PubMed]
- Fagotto, F. Looking beyond the Wnt pathway for the deep nature of β-catenin. EMBO Rep. 2013, 14, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Lazarian, G.; Friedrich, C.; Quinquenel, A.; Tran, J.; Ouriemmi, S.; Dondi, E.; Martin, A.; Mihoub, I.; Chiron, D.; Bellanger, C.; et al. Stabilization of β-catenin upon B-cell receptor signaling promotes NF-κB target genes transcription in mantle cell lymphoma. Oncogene 2020, 39, 2934–2947. [Google Scholar] [CrossRef]
- Peifer, M. The vertebrate adhesive junction proteins beta-catenin and plakoglobin and the Drosophila segment polarity gene armadillo form a multigene family with similar properties. J. Cell Biol. 1992, 118, 681–691. [Google Scholar] [CrossRef]
- McCrea, P.D.; Maher, M.T.; Gottardi, C.J. Nuclear Signaling from Cadherin Adhesion Complexes. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 112, pp. 129–196. [Google Scholar]
- Daugherty, R.L.; Gottardi, C.J. Phospho-regulation of β-Catenin Adhesion and Signaling Functions. Physiology 2007, 22, 303–309. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Suriano, G.; Mendes, N.; Gomes, B.; Wen, X.; Carneiro, F.; Seruca, R.; Machado, J.C. E-cadherin impairment increases cell survival through Notch-dependent upregulation of Bcl-2. Hum. Mol. Genet. 2012, 21, 334–343. [Google Scholar] [CrossRef]
- Mangolini, M.; Gotte, F.; Moore, A.; Ammon, T.; Oeslner, M.; Lutzny-Geier, G.; Klein-Hitpass, L.; Williamons, J.C.; Lehner, P.J.; During, J.; et al. Notch2 controls non-autonomous Wnt-signalling in chronic lymphocytic leukaemia. Nat. Commun. 2018, 9, 3839. [Google Scholar] [CrossRef] [PubMed]
- Dadashian, E.L.; McAuley, E.M.; Liu, D.; Shaffer, A.L.; Young, R.M.; Iyer, J.R.; Kruhlak, M.J.; Staudt, L.M.; Wiestner, A.; Herman, S.E.M. TLR Signaling Is Activated in Lymph Node–Resident CLL Cells and Is Only Partially Inhibited by Ibrutinib. Cancer Res. 2019, 79, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Montresor, A.; Toffali, L.; Rigo, A.; Ferrarini, I.; Vinante, F.; Laudanna, C. CXCR4- and BCR-triggered integrin activation in B-cell chronic lymphocytic leukemia cells depends on JAK2-activated Bruton’s tyrosine kinase. Oncotarget 2018, 9, 35123–35140. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.I.; Fluckiger, A.C.; Kato, R.M.; Witte, O.N.; Rawilngs, D.J. Phosphorylation of two regulatory tyrosine residues in the activation of Bruton’s tyrosine kinase via alternative receptors. Proc. Natl. Acad. Sci. USA 1997, 94, 11526–11533. [Google Scholar] [CrossRef] [PubMed]
- Middendorp, S.; Dingjan, G.M.; Maas, A.; Dahlenborg, K.; Hendriks, R.W. Function of Bruton’s Tyrosine Kinase during B Cell Development Is Partially Independent of Its Catalytic Activity. J. Immunol. 2003, 171, 5988–5996. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Delfino, P.; Gatti, A.; Bonato, A.; Kamga, P.T.; Bazzoni, R.; Ugel, S.; Mercuri, A.; Caligola, S.; Krampera, M. HS-5 and HS-27A Stromal Cell Lines to Study Bone Marrow Mesenchymal Stromal Cell-Mediated Support to Cancer Development. Front. Cell Dev. Biol. 2020, 8, 584232. [Google Scholar] [CrossRef] [PubMed]
- Nore, B.F.; Vargas, L.; Mohamed, A.J.; Branden, L.J.; Backesjo, C.-M.; Islam, T.C.; Mattsson, P.T.; Hultenbu, K.; Christensson, B.; Smith, C.I.E. Redistribution of Bruton’s tyrosine kinase by activation of phosphatidylinositol 3-kinase and Rho-family GTPases. Eur. J. Immunol. 2000, 30, 145–154. [Google Scholar] [CrossRef] [PubMed]
- De Gorter, D.J.J.; Beuling, E.A.; Kersseboom, R.; Middendorp, S.; van Gils, J.M.; Hendriks, R.W.; Pals, S.T.; Spaargaren, M. Bruton’s Tyrosine Kinase and Phospholipase Cγ2 Mediate Chemokine-Controlled B Cell Migration and Homing. Immunity 2007, 26, 93–104. [Google Scholar] [CrossRef]
- Corcione, A.; Benvenuto, F.; Ferretti, E.; Giunti, D.; Cappiello, V.; Cazzanti, F.; Risso, M.; Gualandi, F.; Mancardi, G.L.; Pistoia, V.; et al. Human mesenchymal stem cells modulate B-cell functions. Blood 2006, 107, 367–372. [Google Scholar] [CrossRef]
- Hamada-Kawaguchi, N.; Nore, B.F.; Kuwada, Y.; Smith, C.I.E.; Yamamoto, D. Btk29A Promotes Wnt4 Signaling in the Niche to Terminate Germ Cell Proliferation in Drosophila. Science 2014, 343, 294–297. [Google Scholar] [CrossRef]
- An De Wetering, M.; Barker, N.; Harkes, I.C.; Van Der Heyden, M.; Dijk, N.J.; Hollestelle, A.; Klijn, J.G.; Clevers, H.; Schutte, M. Mutant E-cadherin breast cancer cells do not display constitutive Wnt signaling. Cancer Res. 2001, 61, 278–284. [Google Scholar]
- Lu, D.; Zhao, Y.; Tawatao, R.; Cottam, H.B.; Sen, M.; Leoni, L.M.; Kipps, T.J.; Corr, M.; Carson, D.A. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 3118–3123. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Assoian, R.K. Transcriptional regulation of the cyclin D1 gene at a glance. J. Cell Sci. 2008, 121, 3853–3857. [Google Scholar] [CrossRef] [PubMed]
- Razanadrakoto, L.; Cormier, F.; Laurienté, V.; Dondi, E.; Gardano, L.; Katzav, S.; Guittat, L.; Varin-Blank, N. Mutation of Vav1 adaptor region reveals a new oncogenic activation. Oncotarget 2015, 6, 2524–2538. [Google Scholar] [CrossRef] [PubMed]
- Boudria, R.; Lauriente, V.; Oudar, A.; Rachidi, S.H.; Dondi, E.; Le Roy, C.; Gardano, L.; Varin-Blank, N.; Guittat, L. Regulatory interplay between Vav1, Syk and β-catenin occurs in lung cancer cells. Cell. Signal. 2021, 86, 110079. [Google Scholar] [CrossRef] [PubMed]
- Niemann, C.U.; Herman, S.E.; Maric, I.; Gomez-Rodriguez, J.; Biancotto, A.; Chang, B.Y.; Martyr, S.; Stetler-Stevenson, M.; Yuan, C.M.; Calvo, K.R.; et al. Disruption of in vivo Chronic Lymphocytic Leukemia Tumor–Microenvironment Interactions by Ibrutinib—Findings from an Investigator-Initiated Phase II Study. Clin. Cancer Res. 2016, 22, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Tissino, E.; Benedetti, D.; Herman, S.E.; Hacken, E.T.; Ahn, I.E.; Chaffee, K.G.; Rossi, F.M.; Bo, M.D.; Bulian, P.; Bomben, R.; et al. Functional and clinical relevance of VLA-4 (CD49d/CD29) in ibrutinib-treated chronic lymphocytic leukemia. J. Exp. Med. 2018, 215, 681–697. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihoub, I.; Rharass, T.; Ouriemmi, S.; Oudar, A.; Aubard, L.; Gratio, V.; Lazarian, G.; Ferreira, J.; Dondi, E.; Cymbalista, F.; et al. Identification of the Axis β-Catenin–BTK in the Dynamic Adhesion of Chronic Lymphocytic Leukemia Cells to Their Microenvironment. Int. J. Mol. Sci. 2023, 24, 17623. https://doi.org/10.3390/ijms242417623
Mihoub I, Rharass T, Ouriemmi S, Oudar A, Aubard L, Gratio V, Lazarian G, Ferreira J, Dondi E, Cymbalista F, et al. Identification of the Axis β-Catenin–BTK in the Dynamic Adhesion of Chronic Lymphocytic Leukemia Cells to Their Microenvironment. International Journal of Molecular Sciences. 2023; 24(24):17623. https://doi.org/10.3390/ijms242417623
Chicago/Turabian StyleMihoub, Imane, Tareck Rharass, Souhaïl Ouriemmi, Antonin Oudar, Laure Aubard, Valérie Gratio, Gregory Lazarian, Jordan Ferreira, Elisabetta Dondi, Florence Cymbalista, and et al. 2023. "Identification of the Axis β-Catenin–BTK in the Dynamic Adhesion of Chronic Lymphocytic Leukemia Cells to Their Microenvironment" International Journal of Molecular Sciences 24, no. 24: 17623. https://doi.org/10.3390/ijms242417623
APA StyleMihoub, I., Rharass, T., Ouriemmi, S., Oudar, A., Aubard, L., Gratio, V., Lazarian, G., Ferreira, J., Dondi, E., Cymbalista, F., Levy, V., Baran-Marszak, F., Varin-Blank, N., Ledoux, D., Le Roy, C., & Gardano, L. (2023). Identification of the Axis β-Catenin–BTK in the Dynamic Adhesion of Chronic Lymphocytic Leukemia Cells to Their Microenvironment. International Journal of Molecular Sciences, 24(24), 17623. https://doi.org/10.3390/ijms242417623