
Combined BSA-Seq and RNA-Seq Reveal Genes Associated with the Visual Stay-Green of Maize (Zea mays L.)

Abstract
:1. Introduction
2. Results
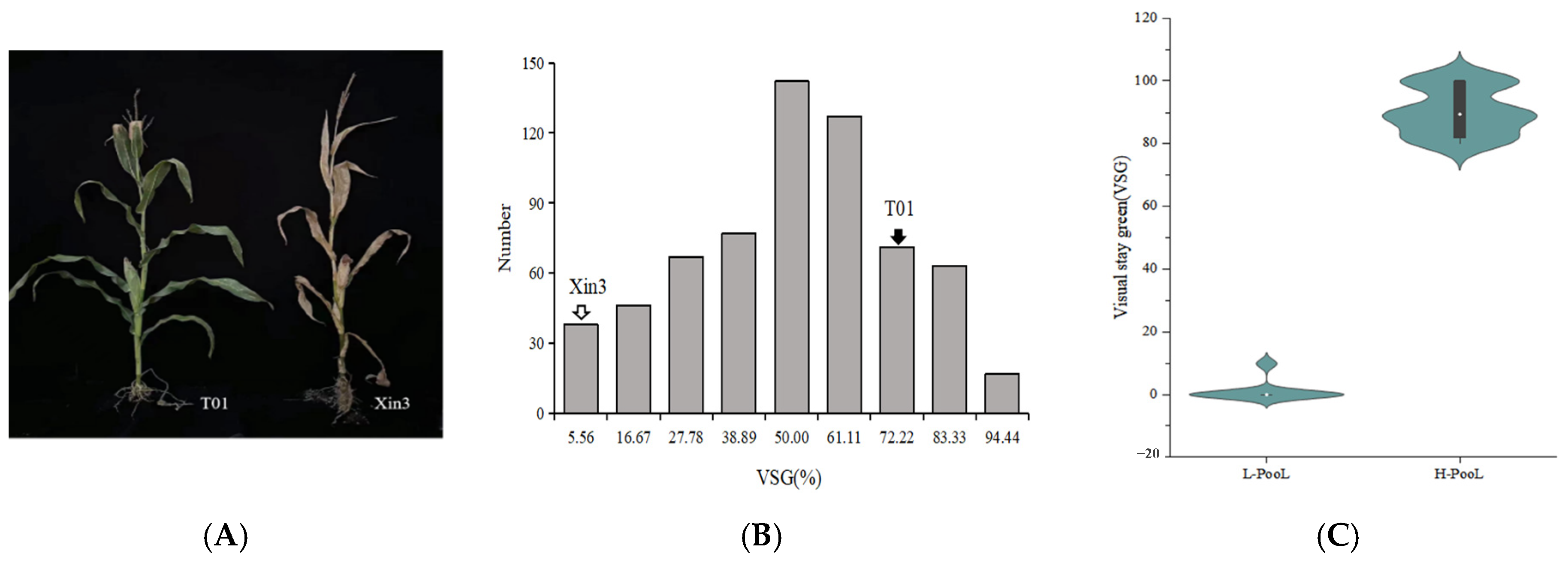
2.1. Analysis of the Visual Stay-Green
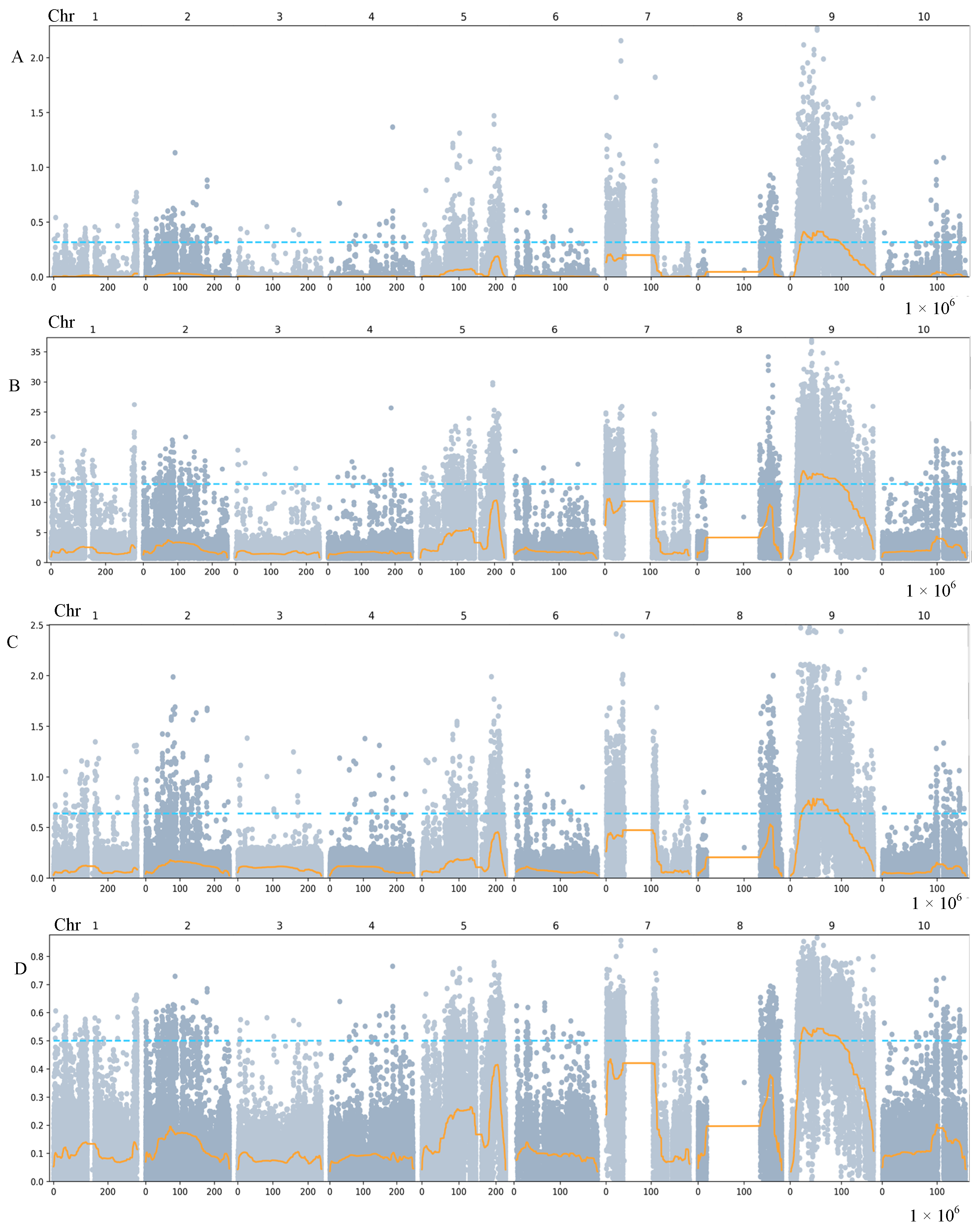
2.2. QTL Mapping Using BSA
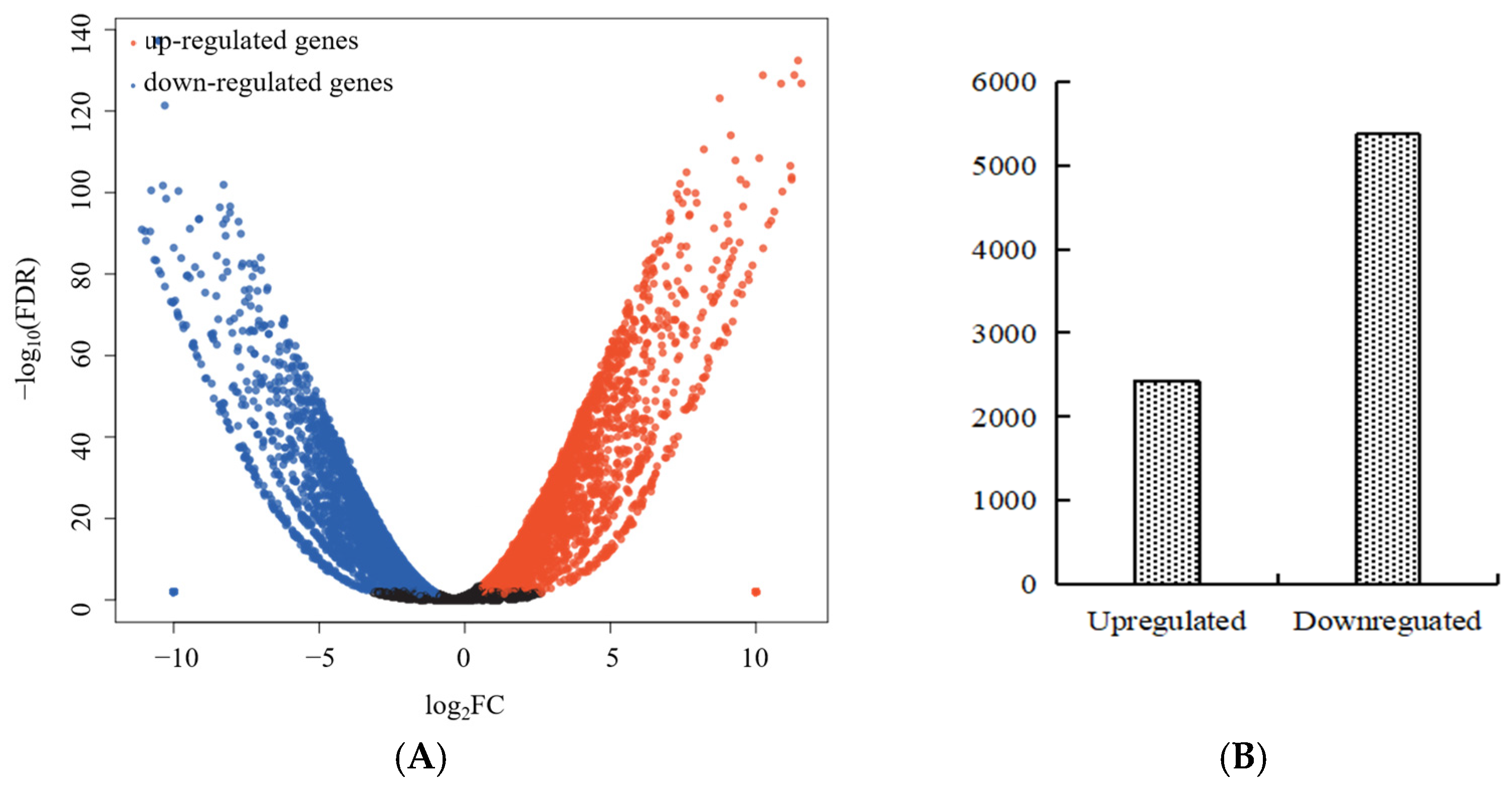
2.3. Transcriptome Sequencing Data Analysis
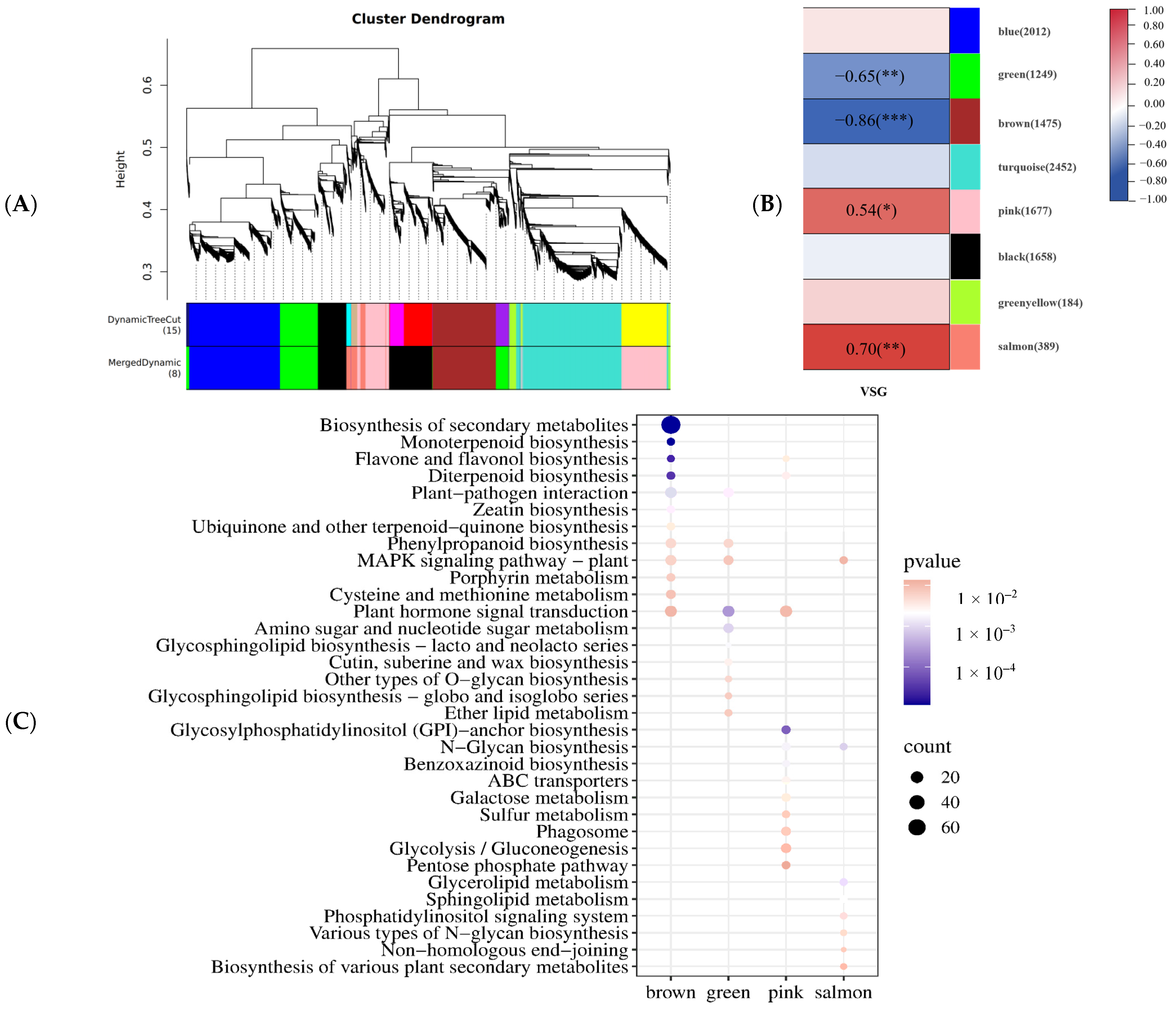
2.4. WGCNA of DEGs in Visual Stay-Green
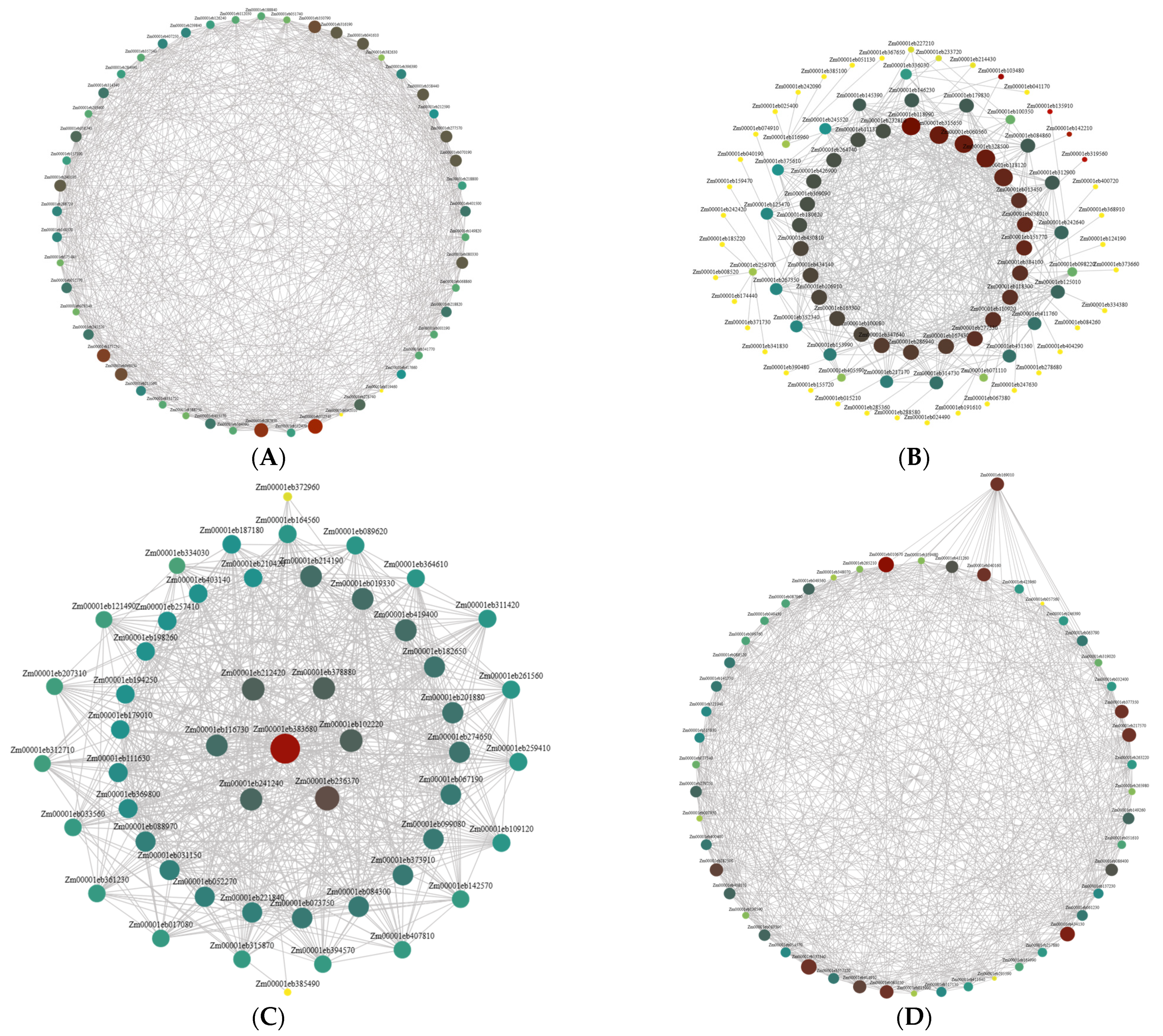
2.5. Candidate Gene Analysis
3. Discussion
3.1. Excavation of Key Genes in the Stay-Green Mutant Contributes to the Promotion of Maize
3.2. Integrating BSA Data and RNA-seq Data for Candidate Gene Prediction
3.3. Chromosome 9 Is Crucial in Controlling Stay-Green-Associated Parameters
3.4. Candidate Gene Analysis
4. Materials and Methods
4.1. Plant Materials
4.2. Statistics of Visual Stay-Green
4.3. DNA Library Construction and BSA-Seq Analysis
4.4. RNA-Seq Analysis
4.5. WGCNA
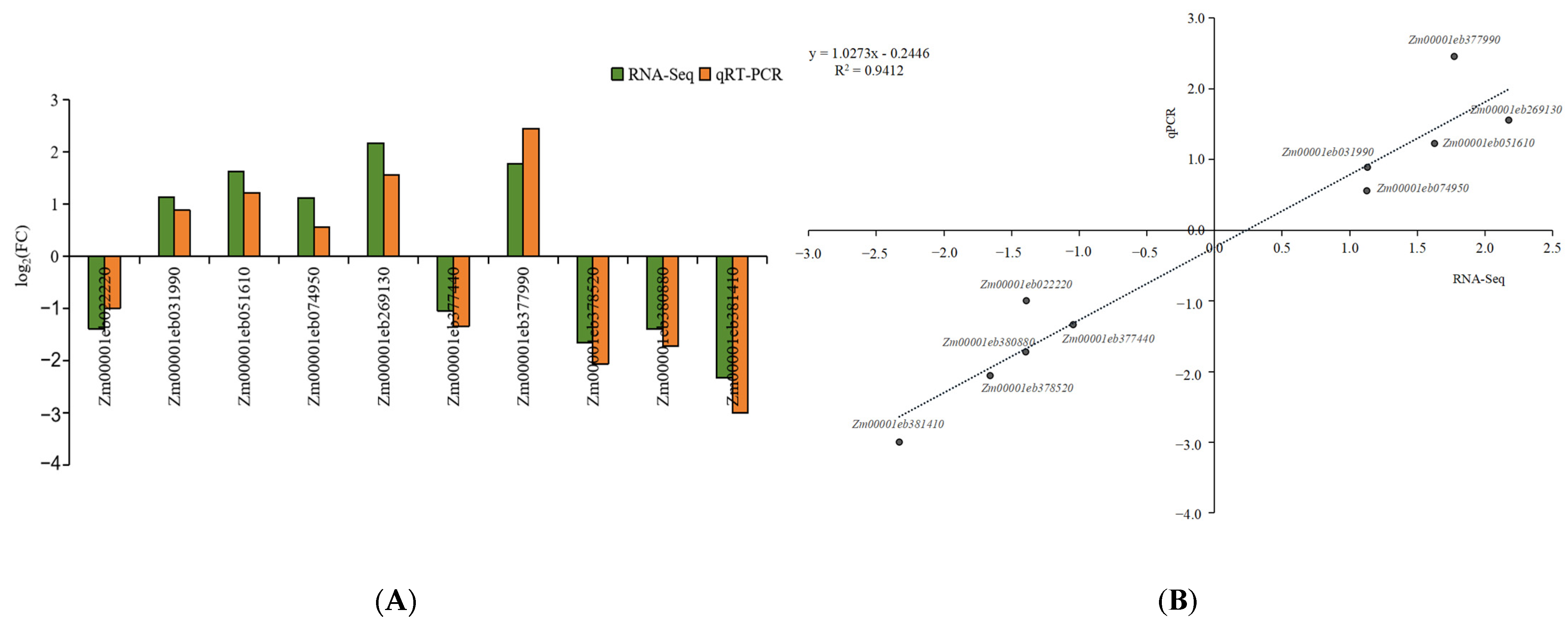
4.6. qRT-PCR Validation of Genes
4.7. Combined Analysis of BSA-Seq and RNA-Seq
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thomas, H.; Howarth, C.J. Five ways to stay green. J. Exp. Bot. 2000, 51, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qiang, C.G.; Wang, X.Q.; Chen, Y.F.; Deng, J.Q.; Jiang, C.H.; Sun, X.M.; Chen, H.Y.; Li, J.; Piao, W.L.; et al. New alleles for chlorophyll content and stay-green traits revealed by a genome wide association study in rice (Oryza sativa). Sci. Rep. 2019, 9, 2541. [Google Scholar] [CrossRef] [PubMed]
- Elsoda, M.; Malosetti, M.; Zwaan, B.J.; Koornneef, M. Genotype × environment interaction QTL mapping in plants: Lessons from Arabidopsis. Trends Plant Sci. 2014, 19, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, Y.; Zhang, H.; Hu, J.; Zhu, W.; Liu, L.; Liu, H.; Fahad, S.; Gao, Q. Comparative physiological and transcriptome analysis of leaf nitrogen fluxes in stay-green maize during the vegetative stage. Plant Physiol. Biochem. 2023, 204, 108147. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.C.; Cho, S.H.; Zhang, H.; Paik, H.C.; Lee, C.H.; Li, J.; Yoo, J.H.; Lee, B.W.; Koh, H.J.; Seo, H.S.; et al. Quantitative trait loci associated with functional stay-green SNU-SG1 in rice. Mol. Cells 2007, 24, 83–94. [Google Scholar]
- Harris, K.; Subudhi, P.K.; Borrell, A.; Jordan, D.; Rosenow, D.; Nguyen, H.; Klein, P.; Klein, R.; Mullet, J. Sorghum stay-green QTL individually reduce post-flowering drought-induced leaf senescence. J. Exp. Bot. 2007, 58, 327–338. [Google Scholar] [CrossRef]
- Kiranmayee, K.N.S.U.; Hash, C.T.; Sivasubramani, S.; Ramu, P.; Amindala, B.P.; Rathore, A.; Kishor, P.B.K.; Gupta, R.; Deshpande, S.P. Fine-Mapping of Sorghum Stay-Green QTL on Chromosome10 Revealed Genes Associated with Delayed Senescence. Genes 2020, 11, 1026. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, J.; Ding, D.; Bian, H.; Yin, Y.; Wu, Z.; Zhou, Y.; Zhao, B. A comprehensive meta-analysis of plant morphology, yield, stay-green, and virus disease resistance QTL in maize (Zea mays L.). Planta 2016, 243, 459–471. [Google Scholar] [CrossRef]
- Fu, J.D.; Yan, Y.F.; Kim, M.Y.; Lee, S.H.; Lee, B.W. Population-specific quantitative trait loci mapping for functional stay-green trait in rice (Oryza sativa L.). Genome 2011, 54, 235–243. [Google Scholar] [CrossRef]
- Fang, Y.F.; Li, Y.S.; Bai, J.P.; Mu, P.; Meng, Y.X.; Zhang, J.L.; Wang, H.N.; Shang, X.W. Construction of integration QTL map and identification of candidate genes for stay-green in maize. Acta Pratacult. Sin. 2012, 21, 175–185. [Google Scholar]
- Liu, Z.H.; Xie, H.L.; Wang, C.L.; Tian, G.W.; Wei, X.Y.; Hu, Y.M.; Cui, D.Q. QTL analysis of chlorophyll content of maizeunder N-stress and no N-stress at different development stages. J. Plant Nutr. Fertil. 2008, 5, 845–851. [Google Scholar]
- Wang, A.Y.; Zhang, C.Q. QTL mapping for chlorophyll contentin maize. Hereditas 2008, 8, 1083–1091. [Google Scholar]
- Zheng, H.J. Genetic Anylysis and QTL Mapping of Stay-Green Traits in Maize (Zea mays L.). Ph.D. Thesis, Shanghai Jiao Tong University, Shanghai, China, 2008; pp. 1–64. [Google Scholar]
- Yang, Z.; Li, X.; Zhang, N.; Wang, X.L.; Zhang, Y.N.; Ding, Y.L.; Kuai, B.K.; Huang, X.Q. Mapping and validation of the quantitative trait loci for leaf stay-green-associated parameters in maize. Plant Breed. 2017, 136, 188–196. [Google Scholar] [CrossRef]
- Kante, M.; Revilla, P.; De La Fuente, M. Stay-green QTLs in temperate elite maize. Euphytica 2016, 207, 463–473. [Google Scholar] [CrossRef]
- Teng, C.C.; Du, D.Z.; Xiao, L.; Yu, Q.L.; Shang, G.X.; Zhao, Z.G. Mapping and Identifying a Candidate Gene (Bnmfs) for Female-Male Sterility through Whole-Genome Resequencing and RNA-Seq in Rapeseed (Brassica napus L.). Front. Plant Sci. 2017, 8, 2086. [Google Scholar] [CrossRef]
- Xu, J.W.; Liu, J.G.; Zhao, J.; Wang, X.R.; Xiao, S.H. The identification of QTL associated with cotton fruit branch length Suitable for mechanized harvest utilizing BSA-seq. Cotton Sci. 2019, 31, 319–326. [Google Scholar]
- Gu, A.; Meng, C.; Chen, Y.; Wei, L.; Dong, H.; Lu, Y.; Wang, Y.; Chen, X.; Zhao, J.; Shen, S. Coupling Seq-BSA and RNA-Seq Analyses Reveal the Molecular Pathway and Genes Associated with Heading Type in Chinese Cabbage. Front. Genet. 2017, 8, 176. [Google Scholar] [CrossRef]
- Kim, S.; Kim, C.W.; Park, M.; Choi, D. Identification of candidate genes associated with fertility restoration of cytoplasmic male-sterility in onion (Allium cepa L.) using a combination of bulked segregant analysis and RNA-seq. Theor. Appl. Genet. 2015, 128, 2289–2299. [Google Scholar] [CrossRef]
- Ma, Y.; Mackon, E.; Jeazet Dongho Epse Mackon, G.C.; Zhao, Y.; Li, Q.; Dai, X.; Yao, Y.; Xia, X.; Nong, B.; Liu, P. Combined Analysis of BSA-Seq Based Mapping, RNA-Seq, and Metabolomic Unraveled Candidate Genes Associated with Panicle Grain Number in Rice (Oryza sativa L.). Biomolecules 2022, 12, 918. [Google Scholar] [CrossRef]
- Guo, Z.; Cai, L.; Chen, Z.; Wang, R.; Zhang, L.; Guan, S.; Zhang, S.; Ma, W.; Liu, C.; Pan, G. Identification of candidate genes controlling chilling tolerance of rice in the cold region at the booting stage by BSA-Seq and RNA-Seq. R. Soc. Open Sci. 2020, 7, 201081. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Xing, G.; Li, M.; Li, S. Integrating physiology, genetics, and transcriptome to decipher a new thermo-sensitive and light-sensitive virescent leaf gene mutant in cucumber. Front. Plant Sci. 2022, 13, 972620. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Li, W.; Zhou, E.; Xing, Y.; Li, B.; Liu, J.; Zhang, Z.; Ding, D.; Fu, Z.; Xie, H.; et al. Integrating BSA-Seq with RNA-Seq Reveals a Novel Fasciated Ear5 Mutant in Maize. Int. J. Mol. Sci. 2023, 24, 1182. [Google Scholar] [CrossRef]
- Sun, Y.Q.; Chen, S.L.; Chu, J.H.; Li, X.H.; Zhang, C.Y. Mining of QTLs and Candidate Genes for Pod and Seed Traits via Combining BSA-seq and Linkage Mapping in Soybean. J. Agric. Sci. Technol. 2023, 25, 29–42. [Google Scholar]
- Moraga, F.; Alcaíno, M.; Matus, I.; Castillo, D.; Del Pozo, A. Leaf and Canopy Traits Associated with Stay-Green Expression Are Closely Related to Yield Components of Wheat Genotypes with Contrasting Tolerance to Water Stress. Plants 2022, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Lee, S.; Kim, T.H.; Lee, J.H.; Park, J.; Lee, J.; Lee, J.Y.; Cho, L.H.; Choi, J.Y.; Lee, W.; et al. Natural variations at the Stay-Green gene promoter control lifespan and yield in rice cultivars. Nat. Commun. 2020, 4, 2819. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Li, Y.; Shi, Y.; Cui, Z.; Luo, Y.; Zheng, M.; Chen, J.; Li, Y.; Yin, Y.; Wang, Z. Exogenous Cytokinins Increase Grain Yield of Winter Wheat Cultivars by Improving Stay-Green Characteristics under Heat Stress. PLoS ONE 2016, 11, e0155437. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, Y.; Huang, S.; Liu, Z.; Li, C.; Feng, H. Defect in Brnym1, a magnesium-dechelatase protein, causes a stay-green phenotype in an EMS-mutagenized Chinese cabbage (Brassica campestris L. ssp. pekinensis) line. Hortic. Res. 2020, 7, 8. [Google Scholar] [CrossRef]
- Fu, W.; Wang, Y.; Ye, Y.; Zhen, S.; Zhou, B.; Wang, Y.; Hu, Y.; Zhao, Y.; Huang, Y. Grain Yields and Nitrogen Use Efficiencies in Different Types of Stay-Green Maize in Response to Nitrogen Fertilizer. Plants 2020, 9, 474. [Google Scholar] [CrossRef]
- Zhu, Y.; Ren, Y.; Liu, J.; Liang, W.; Zhang, Y.; Shen, F.; Ling, J.; Zhang, C. New Genes Identified as Modulating Salt Tolerance in Maize Seedlings Using the Combination of Transcriptome Analysis and BSA. Plants 2023, 12, 1331. [Google Scholar] [CrossRef]
- Wang, A.Y.; Li, Y.; Zhang, C.Q. QTL mapping for staygreen in maize (Zea mays). Plant Sci. 2012, 92, 249–256. [Google Scholar]
- Beavis, W.D.; Smith, O.S.; Grant, D.; Fincher, R. Identification of quantitative trait loci using a small sample of topcrossed and F4 progeny from maize. Crop Sci. 1994, 34, 882–896. [Google Scholar] [CrossRef]
- Zheng, H.J.; Wu, A.Z.; Zheng, C.C.; Wang, Y.F.; Cai, R.; Shen, X.F.; Xu, R.R.; Liu, P.; Kong, L.J.; Dong, S.T. QTL mapping of maize (Zea mays) stay-green traits and their relationship to yield. Plant Breed 2009, 128, 54–62. [Google Scholar] [CrossRef]
- Li, H.; Wang, G.; Liu, S.; An, Q.; Zheng, Q.; Li, B.; Li, Z. Comparative changes in the antioxidant system in the flag leaf of early and normally senescing near-isogenic lines of wheat (Triticum aestivum L.). Plant Cell Rep. 2014, 33, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Bowler, C.; Slooten, L.; Vandenbranden, S.; De Rycke, R.; Botterman, J.; Sybesma, C.; Van Montagu, M.; Inzé, D. Manganese superoxide dismutase can reduce cellular damage mediated by oxygen radicals in transgenic plants. EMBO J. 1991, 10, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Mckerise, B.D.; Chen, Y.; De, B.; Bowley, S.R.; Bowler, C.; Inzé, D.; Dhalluin, K.; Botterman, J. Superoxide dismutase enhances to lerance of freez ing stress in transgenica-l falfa (Medicago sativa L.). Plant Physiol. 1993, 103, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Breusegem, F.; Slooten, L.; Stassart, J.; Botterman, J.; Moens, T.; Montagu, M.; Inzé, D. Effects of overproduction of tobacco MnSOD in maize chloroplasts on foliar tolerance to cold and oxidative stress. J. Exp. Bot. 1993, 50, 71–78. [Google Scholar] [CrossRef]
- Cao, S.Q.; Xu, Q.T.; Zhou, H.J.; Cao, Y.J.; Zhu, Y.; Yu, F.; Kuai, B.K. Screening for lifespan-extension mutants with paraquat in Arabidopsis. Shi Yan Sheng Wu Xue Bao 2003, 36, 233–237. [Google Scholar]
- Punyasu, N.; Kalapanulak, S.; Saithong, T. CO2 recycling by phosphoenolpyruvate carboxylase enables cassava leaf metabolism to tolerate low water availability. Front. Plant Sci. 2023, 14, 1159247. [Google Scholar] [CrossRef]
- Shojaee, S.; Ravash, R.; Shiran, B.; Ebrahimie, E. Meta-analysis highlights the key drought responsive genes in genes: PEPC and TaSAG7 are hubs response networks. J. Genet. Eng. Biotechnol. 2022, 20, 127. [Google Scholar] [CrossRef]
- Behera, D.; Swain, A.; Karmakar, S.; Dash, M.; Swain, P.; Baig, M.J.; Molla, K.A. Overexpression of Setaria italica phosphoenolpyruvate carboxylase gene in rice positively impacts photosynthesis and agronomic traits. Plant Physiol. Biochem. 2023, 194, 169–181. [Google Scholar] [CrossRef]
- De la Osa, C.; Pérez-López, J.; Feria, A.B.; Baena, G.; Marino, D.; Coleto, I.; Pérez-Montaño, F.; Gandullo, J.; Echevarría, C.; García-Mauriño, S.; et al. Knock-down of phosphoenolpyruvate carboxylase 3 negatively impacts growth, productivity, and responses to salt stress in sorghum (Sorghum bicolor L.). Plant J. 2022, 111, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Chapple, C. Molecular-genetic analysis of plant cytochrome p450-dependent monooxygenases. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 311–343. [Google Scholar] [CrossRef] [PubMed]
- Paquette, S.M.; Bak, S.; Feyereisen, R. Intron-exon organization and phylogeny in a large superfamily, the paralogous cytochrome P450 genes of Arabidopsis thaliana. DNA Cell Biol. 2000, 19, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Christ, B.; Süssenbacher, I.; Moser, S.; Bichsel, N.; Egert, A.; Müller, T.; Kräutler, B.; Hörtensteiner, S. Cytochrome P450 CYP89A9 is involved in the formation of major chlorophyll catabolites during leaf senescence in Arabidopsis. Plant Cell 2013, 25, 1868–1880. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Number of Samples | Min (%) | Max (%) | Mean (%) | CV (%) | Kurt | Skew |
|---|---|---|---|---|---|---|---|
| F2 | 786 | 0.00 | 100.00 | 51.57 | 48.92 | −0.454 | −0.374 |
| H-pool | 50 | 80.00 | 100.00 | 90.23 | 8.05 | 0.200 | −1.324 |
| L-pool | 50 | 0.00 | 10.00 | 1.18 | - | 2.418 | 4.017 |
| Associated Analysis Method | Chromosome | Start of Associated Regions (bp) | End of Associated Regions (bp) | Gene Number in the Associated Regions |
|---|---|---|---|---|
| Results of ED correlation | Chr9 | 21,439,311 | 97,423,834 | 841 |
| Results of Ridit correlation | Chr9 | 20,699,310 | 98,235,998 | 895 |
| Results of SmoothG correlation | Chr9 | 22,466,899 | 94,938,048 | 778 |
| Results of SNP correlation | Chr9 | 20,964,571 | 98,003,843 | 878 |
| The intersection of the four association results | Chr9 | 22,466,899 | 94,938,048 | 778 |
| Gene | Annotation |
|---|---|
| Zm00001eb378880 * | superoxide dismutase |
| Zm00001eb380180 | autophagy 2 |
| Zm00001eb380210 | ribulose 1,5-bisphosphate carboxylase/oxygenase large subunit |
| Zm00001eb380300 | senescence-specific cysteine protease SAG12 |
| Zm00001eb382070 | cryptochrome-2 |
| Zm00001eb383680 * | phosphoenolpyruvate carboxylase |
| Zm00001eb384100 * | cytochrome P450 monooxygenase |
| Module | Gene | Annotation |
|---|---|---|
| green | Zm00001eb240100 | putative GATA transcription factor 22 isoform X1 |
| brown | Zm00001eb384100 * | cytochrome P450 monooxygenase |
| Zm00001eb405590 | NAC transcription factor 29 | |
| Zm00001eb116960 | early light inducible protein1 | |
| Zm00001eb118120 | agamous-like MADS-box protein AGL8 | |
| pink | Zm00001eb383680 * | phosphoenolpyruvate carboxylase |
| Zm00001eb378880 * | superoxide dismutase | |
| salmon | Zm00001eb040160 | MYB transcription factor |
| Zm00001eb411260 | diacylglycerol kinase 2 | |
| Zm00001eb414910 | ZIM-transcription factor 40 | |
| Zm00001eb332340 | MYB-RELATED transcription factor |
| ID | Sense Primer | Anti-Sense Primer |
|---|---|---|
| Zm00001eb022220 | GGCCTCAATTCCGTCACTCA | CTTTTTAGCACTGCCGCGTT |
| Zm00001eb031990 | AACGGTGTTCTACCCGATGC | TCCATTGGGCTCTTCCTTGG |
| Zm00001eb051610 | AGGGATAGTGATGGGCGTCT | TGAGATTTCCTGACATAGCTTTAGA |
| Zm00001eb074950 | CGCAGGGGTCTTGAGAGAAG | CTAGCTAGAGCTATCGCGGC |
| Zm00001eb269130 | CCGAGAGGGACAGTCCAGTA | GTCGACAAGCTCACGCTTCA |
| Zm00001eb377440 | GCGCGCGGGAAATTATAGTG | TGGCCTTCCTCACCTAGCTT |
| Zm00001eb377990 | AACCTCGAGAAAGTCTGCGG | CTCAAAGCGATCCCAGGTGT |
| Zm00001eb378520 | CGTCAACTGCAACGGGTG | AGCGACGACATGTTCAGAGC |
| Zm00001eb380880 | GGCTATGGAAGATGACGCGA | CGACGTCATAAAGTACATAGCGA |
| Zm00001eb381410 | TGGTGATGGAGGCTATGGGA | ACAAGGGAGAAGGGTCAACG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, R.; Deng, M.; Lv, D.; Tong, B.; Liu, Y.; Luo, H. Combined BSA-Seq and RNA-Seq Reveal Genes Associated with the Visual Stay-Green of Maize (Zea mays L.). Int. J. Mol. Sci. 2023, 24, 17617. https://doi.org/10.3390/ijms242417617
Zheng R, Deng M, Lv D, Tong B, Liu Y, Luo H. Combined BSA-Seq and RNA-Seq Reveal Genes Associated with the Visual Stay-Green of Maize (Zea mays L.). International Journal of Molecular Sciences. 2023; 24(24):17617. https://doi.org/10.3390/ijms242417617
Chicago/Turabian StyleZheng, Ran, Min Deng, Dan Lv, Bo Tong, Yuqing Liu, and Hongbing Luo. 2023. "Combined BSA-Seq and RNA-Seq Reveal Genes Associated with the Visual Stay-Green of Maize (Zea mays L.)" International Journal of Molecular Sciences 24, no. 24: 17617. https://doi.org/10.3390/ijms242417617
APA StyleZheng, R., Deng, M., Lv, D., Tong, B., Liu, Y., & Luo, H. (2023). Combined BSA-Seq and RNA-Seq Reveal Genes Associated with the Visual Stay-Green of Maize (Zea mays L.). International Journal of Molecular Sciences, 24(24), 17617. https://doi.org/10.3390/ijms242417617