
Severity of Peripheral Infection Differentially Affects Brain Functions in Mice via Microglia-Dependent and -Independent Mechanisms

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
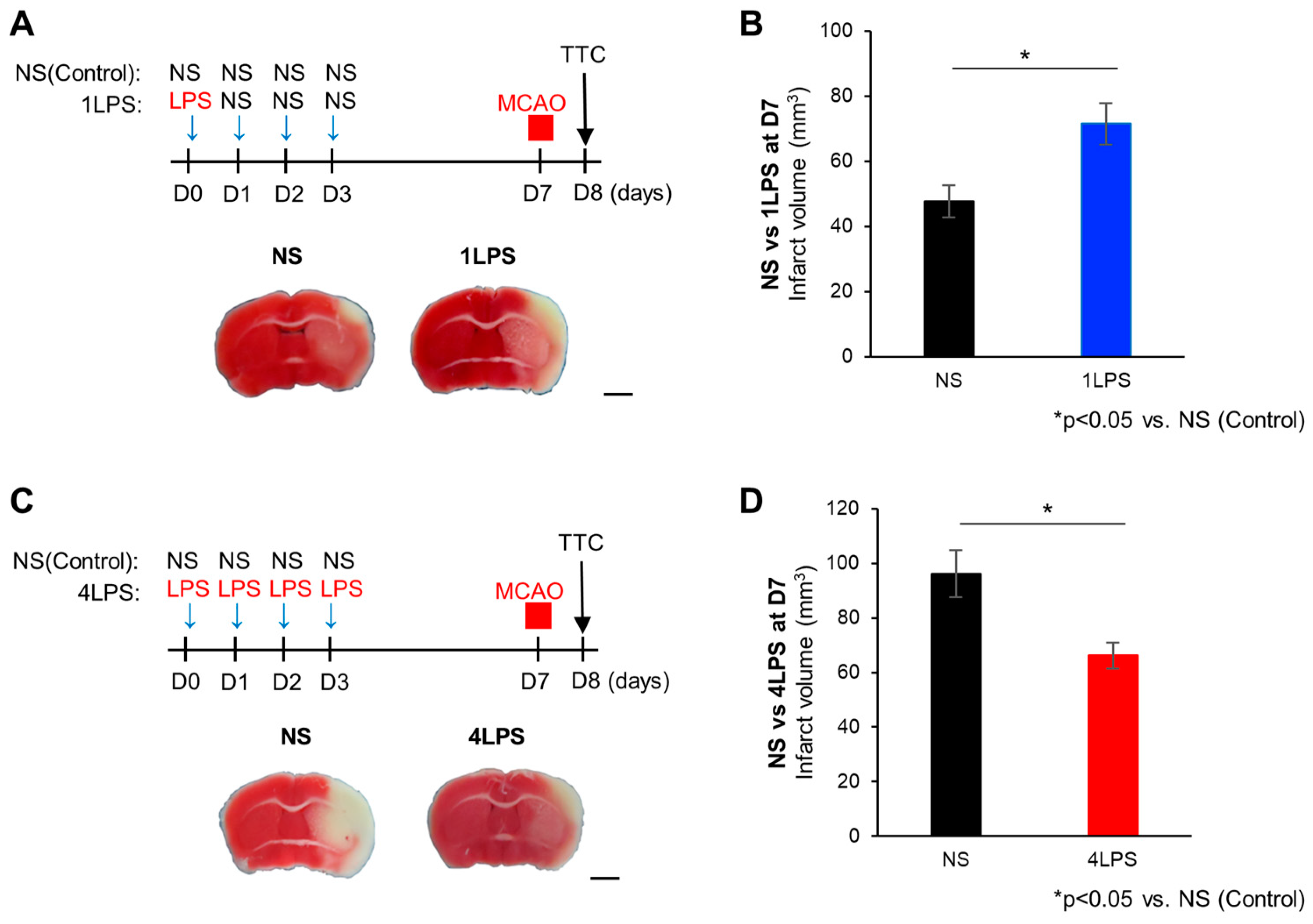
2.1. Opposite Effects of Different Doses of LPS on MCAO-Evoked Brain Damage
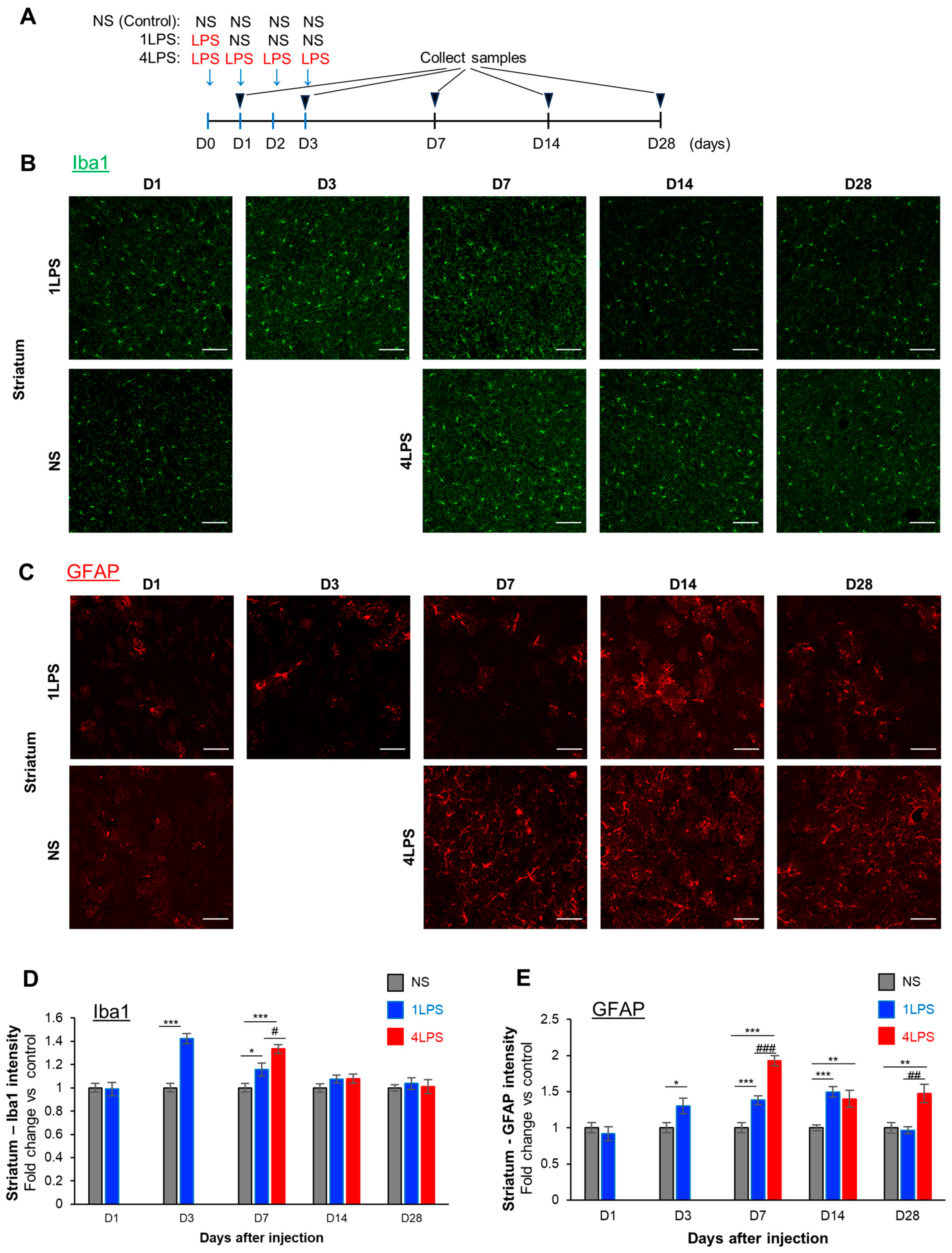
2.2. LPS Administration Induces the Transient Activation of Microglia and Sustained Activation of Astrocytes
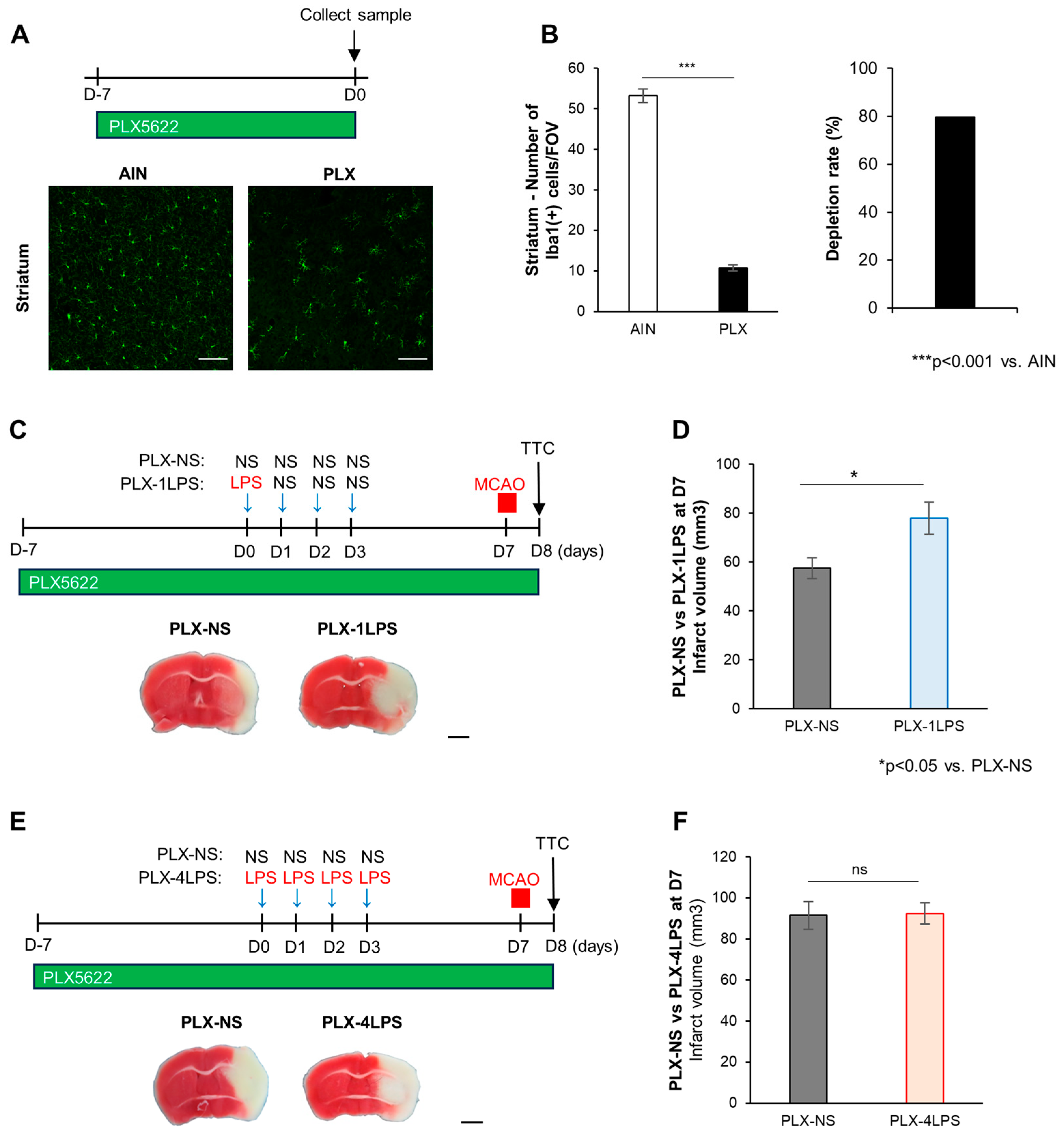
2.3. Severe Inflammation by LPS Protects against MCAO via Microglia
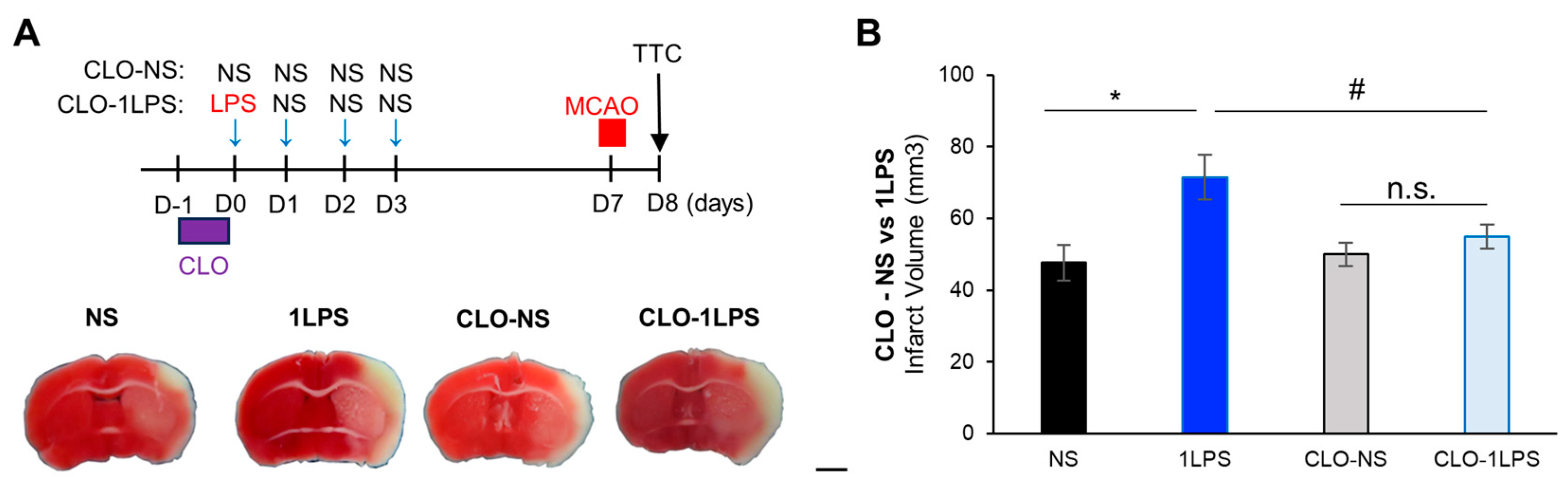
2.4. Peripheral Macrophages Contribute to Mild Inflammation-Related MCAO Exacerbation
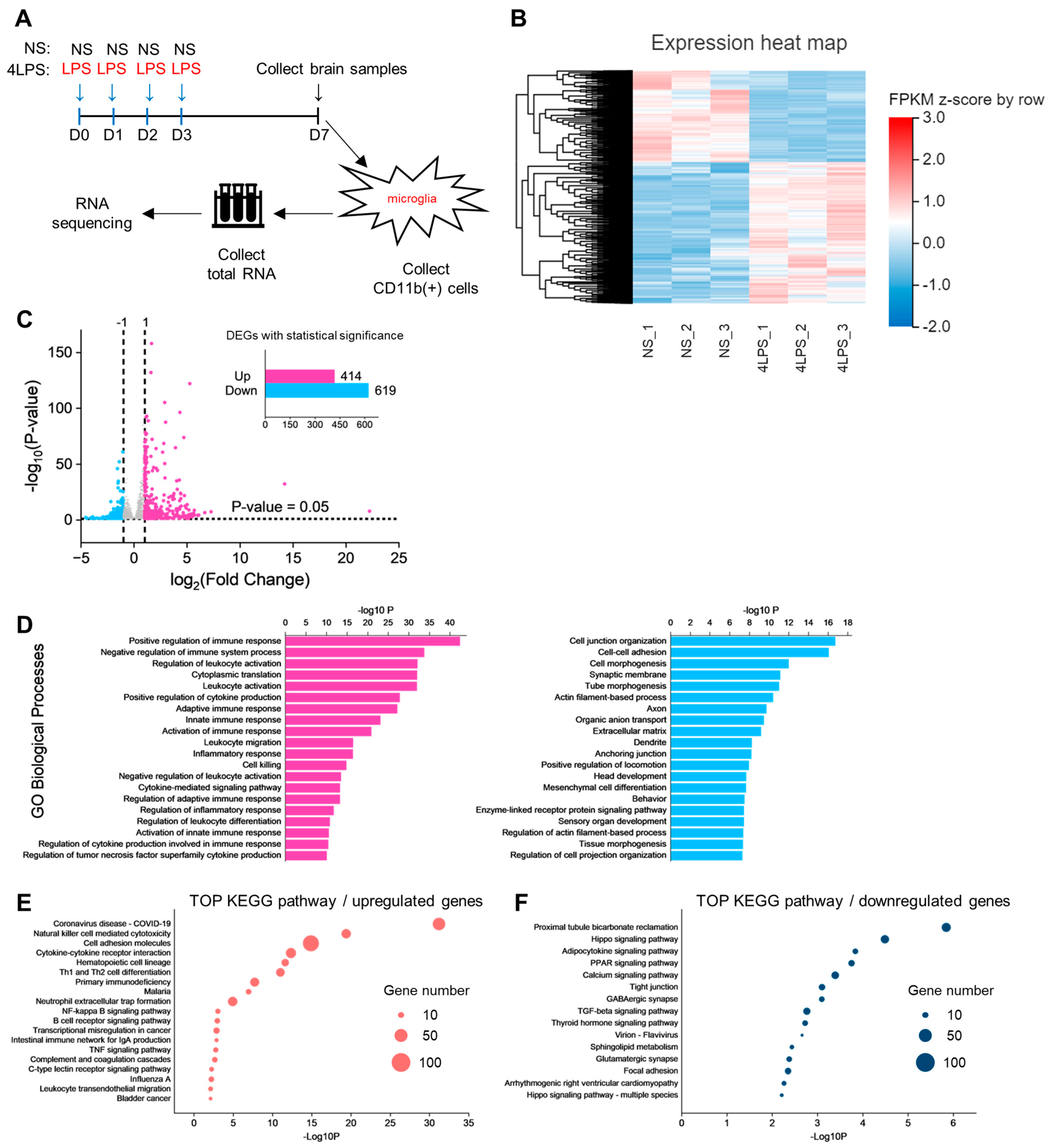
2.5. Transcriptome Analysis of Microglia Identifies Pathways Related to LPS-Induced MCAO Protection
3. Discussion
3.1. Pre-Conditioning of Acute Mild Peripheral Inflammation (1LPS) Causes the Microglia-Independent Exacerbation of MCAO Injury
3.2. Pre-Conditioning of Severe Peripheral Inflammation (4LPS) Causes Microglia-Dependent Inhibition of MCAO Injury
3.3. Molecular Mechanisms Underlying 4LPS-Induced Microglia-Dependent Ischemic Tolerance against MCAO
3.4. Limitations of the Study
3.5. Perspective
4. Materials and Methods
4.1. Mice
4.2. Lipopolysaccharide (LPS) Administration
4.3. Middle Cerebral Artery Occlusion (MCAO)
4.4. Drug Treatment
4.5. Immunohistochemistry
4.6. TTC Staining for Measure of Cerebral Infarct Size
4.7. Magnetic Cell Separation
4.8. RNA Sequencing Analysis
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, I.C.M.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Borst, K.; Dumas, A.A.; Prinz, M. Microglia: Immune and non-immune functions. Immunity 2021, 54, 2194–2208. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Heneka, M.T.; Montana, V.; Oliet, S.H.R.; Schousboe, A.; Haydon, P.G.; Stout, R.F.; Spray, D.C.; Reichenbach, A.; Pannicke, T.; et al. Glial cells in (patho)physiology. J. Neurochem. 2012, 121, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Shigemoto-Mogami, Y.; Koizumi, S.; Mizokoshi, A.; Kohsaka, S.; Salter, M.W.; Inoue, K. P2X(4) receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003, 424, 778–783. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, Y.; Nabekura, J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y(6) receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef]
- Saito, K.; Shigetomi, E.; Shinozaki, Y.; Kobayashi, K.; Parajuli, B.; Kubota, Y.; Sakai, K.; Miyakawa, M.; Horiuchi, H.; Nabekura, J.; et al. Microglia sense astrocyte dysfunction and prevent disease progression in an Alexander disease model. Brain 2023, in press. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.J.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic Analysis of Reactive Astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-Stimulating Factor 1 Receptor Signaling Is Necessary for Microglia Viability, Unmasking a Microglia Progenitor Cell in the Adult Brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Wendeln, A.C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Hasler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A. Heterogeneity of Microglial Activation in the Innate Immune Response in the Brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, M.; Muroi, M.; Tanamoto, K.; Suzuki, T.; Azuma, H.; Ikeda, H. Molecular mechanisms of macrophage activation and deactivation by lipopolysaccharide: Roles of the receptor complex. Pharmacol. Ther. 2003, 100, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.E.; Enos, R.T.; Velazquez, K.T.; Carson, M.S.; Nagarkatti, M.; Nagarkatti, P.S.; Chatzistamou, I.; Davis, J.M.; Carson, J.A.; Robinson, C.M.; et al. Macrophage depletion using clodronate liposomes decreases tumorigenesis and alters gut microbiota in the AOM/DSS mouse model of colon cancer. Am. J. Physiol.-Gastrointest. Liver Physiol. 2018, 314, G22–G31. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, D.; Li, S.Q.; Yu, Y.; Ye, R.D. Suppression of LPS-induced tau hyperphosphorylation by serum amyloid A. J. Neuroinflamm. 2016, 13, 28. [Google Scholar] [CrossRef]
- Boutej, H.; Rahimian, R.; Thammisetty, S.S.; Beland, L.C.; Lalancette-Hebert, M.; Kriz, J. Diverging mRNA and Protein Networks in Activated Microglia Reveal SRSF3 Suppresses Translation of Highly Upregulated Innate Immune Transcripts. Cell Rep. 2017, 21, 3220–3233. [Google Scholar] [CrossRef]
- Nikolakopoulou, A.M.; Dutta, R.; Chen, Z.H.; Miller, R.H.; Trapp, B.D. Activated microglia enhance neurogenesis via trypsinogen secretion. Proc. Natl. Acad. Sci. USA 2013, 110, 8714–8719. [Google Scholar] [CrossRef]
- Hohsfield, L.A.; Najafi, A.R.; Ghorbanian, Y.; Soni, N.; Crapser, J.; Velez, D.X.F.; Jiang, S.; Royer, S.E.; Kim, S.J.; Henningfield, C.M.; et al. Subventricular zone/white matter microglia reconstitute the empty adult microglial niche in a dynamic wave. elife 2021, 10, e66738. [Google Scholar] [CrossRef]
- Karaahmet, B.; Le, L.; Mendes, M.S.; Majewska, A.K.; O’Banion, M.K. Repopulated microglia induce expression of Cxcl13 with differential changes in Tau phosphorylation but do not impact amyloid pathology. J. Neuroinflamm. 2022, 19, 173. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Jiang, Y.; Li, J.Y.; Wang, Y.J.; Tian, Y.; Guo, Q.L.; Cheng, Z.G. DUSP1 Promotes Microglial Polarization toward M2 Phenotype in the Medial Prefrontal Cortex of Neuropathic Pain Rats via Inhibition of MAPK Pathway. Acs Chem. Neurosci. 2021, 12, 966–978. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.M.; Richman, A.R.; Boyd, J.R.; Sabikunnahar, B.; Lahue, K.G.; Montgomery, T.L.; Caldwell, S.; Varnum, S.; Frietze, S.; Krementsov, D.N. p38 MAP Kinase Signaling in Microglia Plays a Sex-Specific Protective Role in CNS Autoimmunity and Regulates Microglial Transcriptional States. Front. Immunol. 2021, 12, 715311. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ma, N.N.; Yu, B.; Zhang, W.; Wan, J. Transcriptomic profiling of microglia and astrocytes throughout aging. J. Neuroinflamm. 2020, 17, 97. [Google Scholar] [CrossRef] [PubMed]
- Shemer, A.; Scheyltjens, I.; Frumer, G.R.; Kim, J.S.; Grozovski, J.; Ayanaw, S.; Dassa, B.; Van Hove, H.; Chappell-Maor, L.; Boura-Halfon, S.; et al. Interleukin-10 Prevents Pathological Microglia Hyperactivation following Peripheral Endotoxin Challenge. Immunity 2020, 53, 1033–1049. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Jalabi, W.; Shpargel, K.B.; Farabaugh, K.T.; Dutta, R.; Yin, X.H.; Kidd, G.J.; Bergmann, C.C.; Stohlman, S.A.; Trapp, B.D. Lipopolysaccharide-Induced Microglial Activation and Neuroprotection against Experimental Brain Injury Is Independent of Hematogenous TLR4. J. Neurosci. 2012, 32, 11706–11715. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef]
- Lei, F.Y.; Cui, N.W.; Zhou, C.X.; Chodosh, J.; Vavvas, D.G.; Paschalis, E.I. CSF1R inhibition by a small-molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proc. Natl. Acad. Sci. USA 2020, 117, 23336–23338. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Gutierrez, E.G. Penetration of Interleukin-6 across the Murine Blood-Brain-Barrier. Neurosci. Lett. 1994, 179, 53–56. [Google Scholar] [CrossRef]
- Banks, W.A.; Erickson, M.A. The blood-brain barrier and immune function and dysfunction. Neurobiol. Dis. 2010, 37, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Larke, L.E.C.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Culemann, S.; Knab, K.; Euler, M.; Wegner, A.; Garibagaoglu, H.; Ackermann, J.; Fischer, K.; Kienhöefer, D.; Crainiciuc, G.; Hahn, J.; et al. Stunning of neutrophils accounts for the anti-inflammatory effects of clodronate liposomes. J. Exp. Med. 2023, 220, e20220525. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with Ischemia-A Delay of Lethal Cell Injury in Ischemic Myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Matsumoto, M.; Tagaya, M.; Hata, R.; Ueda, H.; Niinobe, M.; Handa, N.; Fukunaga, R.; Kimura, K.; Mikoshiba, K.; et al. Ischemic Tolerance Phenomenon Found in the Brain. Brain Res. 1990, 528, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Matsumoto, M.; Kuwabara, K.; Tagaya, M.; Ohtsuki, T.; Hata, R.; Ueda, H.; Handa, N.; Kimura, K.; Kamada, T. Ischemic Tolerance Phenomenon Detected in Various Brain-Regions. Brain Res. 1991, 561, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Kirino, T. Ischemic tolerance. J. Cereb. Blood Flow Metab. 2002, 22, 1283–1296. [Google Scholar] [CrossRef] [PubMed]
- Prass, K.; Scharff, A.; Ruscher, K.; Löwl, D.; Muselmann, C.; Victorov, I.; Kapinya, K.; Dirnagl, U.; Meisel, A. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 2003, 34, 1981–1986. [Google Scholar] [CrossRef]
- Bernaudin, M.; Nedelec, A.S.; Divoux, D.; MacKenzie, E.T.; Petit, E.; Schumann-Bard, P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J. Cereb. Blood Flow Metab. 2002, 22, 393–403. [Google Scholar] [CrossRef]
- Liu, Y.; Kato, H.; Nakata, N.; Kogure, K. Temporal Profile of Heat-Shock Protein-70 Synthesis in Ischemic Tolerance Induced by Preconditioning Ischemia in Rat Hippocampus. Neuroscience 1993, 56, 921–927. [Google Scholar] [CrossRef]
- Terasaki, Y.; Sasaki, T.; Yagita, Y.; Okazaki, S.; Sugiyama, Y.; Oyama, N.; Omura-Matsuoka, E.; Sakoda, S.; Kitagawa, K. Activation of NR2A receptors induces ischemic tolerance through CREB signaling. J. Cereb. Blood Flow Metab. 2010, 30, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, T.; Kis, B.; Rajapakse, N.; Shimizu, K.; Busija, D.W. Opening of mitochondrial ATP-sensitive potassium channels is a trigger of 3-nitropropionic acid-induced tolerance to transient focal cerebral ischemia in rats. Stroke 2003, 34, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.P.; Dave, K.R.; Pérez-Pinzón, M.A. Resveratrol mimics ischemic preconditioning in the brain. J. Cereb. Blood Flow Metab. 2006, 26, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Jalabi, W.; Jin, Y.; Dadabayev, A.; Penn, M.S.; Trapp, B.D. Preconditioning paradigms and pathways in the brain. Clevel. Clin. J. Med. 2008, 75, S77–S82. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.A.; Furuya, K.; Gotoh, J.; Nakao, Y.; Hallenbeck, J.M. Cerebrovascular hemodynamics and ischemic tolerance: Lipopolysaccharide-induced resistance to focal cerebral ischemia is not due to changes in severity of the initial ischemic insult, but is associated with preservation of microvascular perfusion. J. Cereb. Blood Flow Metab. 1999, 19, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, H.L.; Lessov, N.S.; Henshall, D.C.; Minami, M.; Simon, R.P.; Stenzel-Poore, M.P. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 2004, 35, 2576–2581. [Google Scholar] [CrossRef] [PubMed]
- Furuya, K.; Zhu, L.D.; Kawahara, N.; Abe, O.; Kirino, T. Differences in infarct evolution between lipopolysaccharide-induced tolerant and nontolerant conditions to focal cerebral ischemia. J. Neurosurg. 2005, 103, 715–723. [Google Scholar] [CrossRef]
- Tasaki, K.; Ruetzler, C.A.; Ohtsuki, T.; Martin, D.; Nawashiro, H.; Hallenbeck, J.M. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997, 748, 267–270. [Google Scholar] [CrossRef]
- Hirayama, Y.; Ikeda-Matsuo, Y.; Notomi, S.; Enaida, H.; Kinouchi, H.; Koizumi, S. Astrocyte-Mediated Ischemic Tolerance. J. Neurosci. 2015, 35, 3794–3805. [Google Scholar] [CrossRef]
- Hirayama, Y.; Koizumi, S. Hypoxia-Independent Mechanisms of HIF-1α Expression in Astrocytes after Ischemic Preconditioning. Glia 2017, 65, 523–530. [Google Scholar] [CrossRef]
- Hirayama, Y.; Anzai, N.; Koizumi, S. Mechanisms underlying sensitization of P2X7 receptors in astrocytes for induction of ischemic tolerance. Glia 2021, 69, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Lecordier, S.; Pons, V.; Rivest, S.; ElAli, A. Multifocal Cerebral Microinfarcts Modulate Early Alzheimer’s Disease Pathology in a Sex-Dependent Manner. Front. Immunol. 2022, 12, 813536. [Google Scholar] [CrossRef] [PubMed]
- Gliem, M.; Mausberg, A.K.; Lee, J.I.; Simiantonakis, I.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol. 2012, 71, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Okazaki, R.; Morioka, K.; Nakamura, K.; Tanaka, S.; Ogata, T. Lipopolysaccharide Preconditioning Facilitates M2 Activation of Resident Microglia After Spinal Cord Injury. J. Neurosci. Res. 2014, 92, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Spera, P.A.; Ellison, J.A.; Feuerstein, G.Z.; Barone, F.C. IL-10 reduces rat brain injury following focal stroke. Neurosci. Lett. 1998, 251, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Kohno, K.; Shirasaka, R.; Yoshihara, K.; Mikuriya, S.; Tanaka, K.; Takanami, K.; Inoue, K.; Sakamoto, H.; Ohkawa, Y.; Masuda, T.; et al. A spinal microglia population involved in remitting and relapsing neuropathic pain. Science 2022, 376, 86–90. [Google Scholar] [CrossRef]
- Misharin, A.V.; Cuda, C.M.; Saber, R.; Turner, J.D.; Gierut, A.K.; Haines, G.K.; Berdnikovs, S.; Filer, A.; Clark, A.R.; Buckley, C.D.; et al. Nonclassical Ly6C− Monocytes Drive the Development of Inflammatory Arthritis in Mice. Cell Rep. 2014, 9, 591–604. [Google Scholar] [CrossRef]
- Sharma, K.; Schmitt, S.; Bergner, C.G.; Tyanova, S.; Kannaiyan, N.; Manrique-Hoyos, N.; Kongi, K.; Cantuti, L.; Hanisch, U.K.; Philips, M.A.; et al. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 2015, 18, 1819–1831. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, Y.-P.; Saito, K.; Parajuli, B.; Sakai, K.; Kubota, Y.; Miyakawa, M.; Shinozaki, Y.; Shigetomi, E.; Koizumi, S. Severity of Peripheral Infection Differentially Affects Brain Functions in Mice via Microglia-Dependent and -Independent Mechanisms. Int. J. Mol. Sci. 2023, 24, 17597. https://doi.org/10.3390/ijms242417597
Le Y-P, Saito K, Parajuli B, Sakai K, Kubota Y, Miyakawa M, Shinozaki Y, Shigetomi E, Koizumi S. Severity of Peripheral Infection Differentially Affects Brain Functions in Mice via Microglia-Dependent and -Independent Mechanisms. International Journal of Molecular Sciences. 2023; 24(24):17597. https://doi.org/10.3390/ijms242417597
Chicago/Turabian StyleLe, Yen-Phung, Kozo Saito, Bijay Parajuli, Kent Sakai, Yuto Kubota, Miho Miyakawa, Youichi Shinozaki, Eiji Shigetomi, and Schuichi Koizumi. 2023. "Severity of Peripheral Infection Differentially Affects Brain Functions in Mice via Microglia-Dependent and -Independent Mechanisms" International Journal of Molecular Sciences 24, no. 24: 17597. https://doi.org/10.3390/ijms242417597
APA StyleLe, Y.-P., Saito, K., Parajuli, B., Sakai, K., Kubota, Y., Miyakawa, M., Shinozaki, Y., Shigetomi, E., & Koizumi, S. (2023). Severity of Peripheral Infection Differentially Affects Brain Functions in Mice via Microglia-Dependent and -Independent Mechanisms. International Journal of Molecular Sciences, 24(24), 17597. https://doi.org/10.3390/ijms242417597