

Myeloproliferative Neoplasms: Contemporary Review and Molecular Landscape

Abstract
:1. Introduction and Brief Epidemiology
2. Molecular and Mutational Landscape
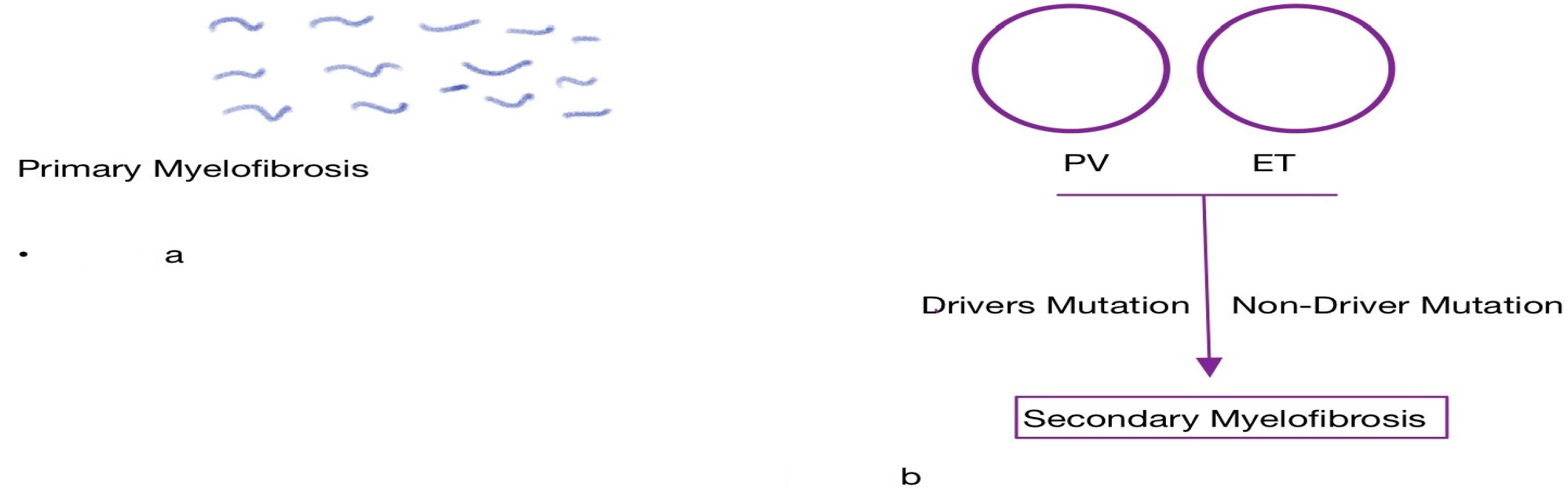
2.1. Role of Driver Mutations
2.2. Role of Non-Driver Mutations
2.3. Role of Megakaryocytes
2.4. Role of Endothelial Cells
2.5. Role of Innate and Acquired Immunity
3. Clinical Features of Myelofibrosis
4. MDS/MPN
5. Molecular and Mutational Landscape of MDS/MPNs
Differences in the Mutational Landscape of MDS/MPN and MF/MPN
6. Therapeutic Overview and Treatment Paradigm
6.1. Treatment of Chronic Phase MPN
6.2. Role of Cytoreductive Agents
6.3. Role of Interferons
6.4. Role of JAK Kinase Inhibitors
6.5. Accelerated MPN, Blast Phase, and Leukemic Transformation
6.6. Treatment of Accelerated MPN and MPN Blast Phase
6.7. Future Therapeutic Landscape
7. Conclusions
Funding
Conflicts of Interest
References
- Dameshek, W. Some speculations on the myeloproliferative syndromes [editorial]. Blood. 1951;6(4):372-375. Blood 2016, 127, 663. [Google Scholar] [CrossRef]
- Gangat, N.; Tefferi, A. Myelofibrosis biology and contemporary management. Br. J. Haematol. 2020, 191, 152–170. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Yu, J.; Kish, J.K.; Paranagama, D.; Kaufman, J.; Myerscough, C.; Grunwald, M.R.; Colucci, P.; Mesa, R. Real-world risk assessment and treatment initiation among patients with myelofibrosis at community oncology practices in the United States. Ann. Hematol. 2020, 99, 2555–2564. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Saeed, L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N. Application of current prognostic models for primary myelofibrosis in the setting of post-polycythemia vera or post-essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2851–2852. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Gleitz, H.F.; Chifotides, H.T.; Harrison, C.N.; Verstovsek, S.; Vannucchi, A.M.; Rampal, R.K.; Kiladjian, J.J.; Vainchenker, W.; Hoffman, R.; et al. Biological Drivers of Clinical Phenotype in Myelofibrosis. Leukemia 2023, 37, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.-L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef]
- Mehta, J.; Wang, H.; Iqbal, S.U.; Mesa, R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk. Lymphoma. 2014, 55, 595–600. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef]
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef]
- Rotunno, G.; Pacilli, A.; Artusi, V.; Rumi, E.; Maffioli, M.; Delaini, F.; Brogi, G.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Epidemiology and clinical relevance of mutations in postpolycythemia vera and postessential thrombocythemia myelofibrosis: A study on 359 patients of the AGIMM group. Am. J. Hematol. 2016, 91, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Barosi, G.; Specchia, G.; Rambaldi, A.; Coco, F.L.; Antonioli, E.; Pieri, L.; Pancrazzi, A.; Ponziani, V.; Delaini, F.; et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood 2009, 114, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Rumi, E. Clinical relevance of JAK2 (V617F) mutant allele burden. Haematologica 2009, 94, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martínez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghamdi, Y.A.; Lake, J.; Bagg, A.; Thakral, B.; Wang, S.A.; Bueso-Ramos, C.; Masarova, L.; Verstovsek, S.; Rogers, H.J.; Hsi, E.D.; et al. Triple-Negative Primary Myelofibrosis: A Bone Marrow Pathology Group Study. Mod. Pathol. 2023, 36, 100016. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Pacilli, A.; Rotunno, G.; Rumi, E.; Rosti, V.; Delaini, F.; Maffioli, M.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood 2017, 129, 3227–3236. [Google Scholar] [CrossRef]
- Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Szuber, N.; Begna, K.H.; Patnaik, M.M.; Gangat, N.; Pardanani, A.; et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018, 2, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Easwar, A.; Siddon, A.J. Genetic landscape of myeloproliferative neoplasms with an emphasis on molecular diagnostic laboratory testing. Life 2021, 11, 1158. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.; Gangat, N.; A Hanson, C.; Ketterling, R.P.; Pardanani, A. Prognostic significance of ASXL1 mutation types and allele burden in myelofibrosis. Leukemia 2018, 32, 837–839. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; A Belachew, A.; Pancrazzi, A.; Wassie, E.A.; Ketterling, R.P.; et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. U2AF1 mutation types in primary myelofibrosis: Phenotypic and prognostic distinctions. Leukemia 2018, 32, 2274–2278. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Reiter, A.; et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef]
- Luque Paz, D.; Riou, J.; Verger, E.; Cassinat, B.; Chauveau, A.; Ianotto, J.-C.; Dupriez, B.; Boyer, F.; Renard, M.; Mansier, O.; et al. Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: A FIM study. Blood Adv. 2021, 5, 1442–1451. [Google Scholar] [CrossRef]
- Lasho, T.L.; Jimma, T.; Finke, C.M.; Patnaik, M.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SRSF2 mutations in primary myelofibrosis: Significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012, 120, 4168–4171. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.P.; Daltro De Oliveira, R.; Marcault, C.; Soret, J.; Gauthier, N.; Verger, E.; Maslah, N.; Roux, B.; Parquet, N.; Dosquet, C.; et al. SF3B1 mutations in the Driver Clone Increase the Risk of Evolution to Myelofibrosis in Patients with Myeloproliferative Neoplasms (MPN). Blood 2020, 136 (Suppl. S1), 1. [Google Scholar] [CrossRef]
- Tamari, R.; Rapaport, F.; Zhang, N.; McNamara, C.; Kuykendall, A.; Sallman, D.A.; Komrokji, R.; Arruda, A.; Najfeld, V.; Sandy, L.; et al. Impact of High-Molecular-Risk Mutations on Transplantation Outcomes in Patients with Myelofibrosis. Biol. Blood Marrow Transpl. 2019, 25, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Melo-Cardenas, J.; Migliaccio, A.R.; Crispino, J.D. The Role of Megakaryocytes in Myelofibrosis. Hematol. Oncol. Clin. 2021, 35, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Perry, J.M.; Marshall, H.; Venkatraman, A.; Qian, P.; He, X.C.; Ahamed, J.; Li, L. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med. 2014, 20, 1321–1326. [Google Scholar] [CrossRef]
- Gilles, L.; Arslan, A.D.; Marinaccio, C.; Wen, Q.J.; Arya, P.; McNulty, M.; Yang, Q.; Zhao, J.C.; Konstantinoff, K.; Lasho, T.; et al. Downregulation of GATA1 drives impaired hematopoiesis in primary myelofibrosis. J. Clin. Investig. 2017, 127, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat. Commun. 2020, 11, 73. [Google Scholar] [CrossRef]
- Nam, A.S.; Kim, K.T.; Chaligne, R.; Izzo, F.; Ang, C.; Taylor, J.; Myers, R.M.; Abu-Zeinah, G.; Brand, R.; Omans, N.D.; et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 2019, 571, 355–360. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Pancrazzi, A.; Guglielmelli, P.; Di Lollo, S.; Bogani, C.; Baroni, G.; Bianchi, L.; Migliaccio, A.R.; Bosi, A.; Paoletti, F. Abnormalities of GATA-1 in Megakaryocytes from Patients with Idiopathic Myelofibrosis. Am. J. Pathol. 2005, 167, 849–858. [Google Scholar] [CrossRef]
- Farina, M.; Bernardi, S.; Polverelli, N.; D’adda, M.; Malagola, M.; Bosio, K.; Re, F.; Almici, C.; Dunbar, A.; Levine, R.L.; et al. Comparative Mutational Profiling of Hematopoietic Progenitor Cells and Circulating Endothelial Cells (CECs) in Patients with Primary Myelofibrosis. Cells 2021, 10, 2764. [Google Scholar] [CrossRef]
- Strickland, M.; Quek, L.; Psaila, B. The immune landscape in BCR-ABL negative myeloproliferative neoplasms: Inflammation, infections and opportunities for immunotherapy. Br. J. Haematol. 2022, 196, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Skov, V.; Riley, C.H.; Thomassen, M.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Whole blood transcriptional profiling reveals significant down-regulation of human leukocyte antigen class I and II genes in essential thrombocythemia, polycythemia vera and myelofibrosis. Leuk. Lymphoma 2013, 54, 2269–2273. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Sollazzo, D.; Trabanelli, S.; Barone, M.; Polverelli, N.; Perricone, M.; Forte, D.; Luatti, S.; Cavo, M.; Vianelli, N.; et al. Mutations in JAK2 and Calreticulin genes are associated with specific alterations of the immune system in myelofibrosis. Oncoimmunology 2017, 6, e1345402. [Google Scholar] [CrossRef] [PubMed]
- Landtblom, A.R.; Andersson, T.M.L.; Dickman, P.W.; Smedby, K.E.; Eloranta, S.; Batyrbekova, N.; Samuelsson, J.; Björkholm, M.; Hultcrantz, M. Risk of infections in patients with myeloproliferative neoplasms-a population-based cohort study of 8363 patients. Leukemia 2021, 35, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Alimam, S.; Ann Timms, J.; Harrison, C.N.; Dillon, R.; Mare, T.; DeLavallade, H.; Radia, D.; Woodley, C.; Francis, Y.; Sanchez, K.; et al. Altered immune response to the annual influenza A vaccine in patients with myeloproliferative neoplasms. Br. J. Haematol. 2021, 193, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef]
- Tefferi, A. Myelofibrosis with Myeloid Metaplasia. N. Engl. J. Med. 2000, 342, 1255–1265. [Google Scholar] [CrossRef]
- Visani, G.; Finelli, C.; Castelli, U.; Petti, M.C.; Ricci, P.; Vianelli, N.; Gianni, L.; Zuffa, E.; Spiriti, M.A.A.; Latagliata, R.; et al. Myelofibrosis with myeloid metaplasia: Clinical and haematological parameters predicting survival in a series of 133 patients. Br. J. Haematol. 1990, 75, 4–9. [Google Scholar] [CrossRef]
- Tefferi, A. Primary Myelofibrosis: 2023 update on diagnosis, riskstratification, and management. Am. J. Hematol. 2023, 98, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Carobbio, A.; Cervantes, F.; Vannucchi, A.M.; Guglielmelli, P.; Antonioli, E.; Alvarez-Larrán, A.; Rambaldi, A.; Finazzi, G.; Barosi, G. Thrombosis in primary myelofibrosis: Incidence and risk factors. Blood. 2010, 115, 778–782. [Google Scholar] [CrossRef]
- Dores, G.M.; Curtis, R.E.; Linet, M.S.; Morton, L.M. Cause-specific mortality following polycythemia vera, essential thrombocythemia, and primary myelofibrosis in the US population, 2001–2017. Am. J. Hematol. 2021, 96, E451. [Google Scholar] [CrossRef]
- Ott, G.; Hsi, E.D.; Delabie, J.; Rodig, S. Principles of the pathology and biology of malignant lymphomas. Rare Lymphomas 2014, 3–16. [Google Scholar] [CrossRef]
- Hasserjian, R.P.; Buckstein, R.; Patnaik, M.M. Navigating Myelodysplastic and Myelodysplastic/Myeloproliferative Overlap Syndromes. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Pati, H.; Kundil Veetil, K. Myelodysplastic Syndrome/Myeloproliferative Neoplasm (MDS/MPN) Overlap Syndromes: Molecular Pathogenetic Mechanisms and Their Implications. Indian J. Hematol. Blood Transfus. 2019, 35, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Roman, E.; Smith, A.; Appleton, S.; Crouch, S.; Kelly, R.; Kinsey, S.; Cargo, C.; Patmore, R. Myeloid malignancies in the real-world: Occurrence, progression and survival in the UK’s population-based Haematological Malignancy Research Network 2004–15. Cancer Epidemiol. 2016, 42, 186–198. [Google Scholar] [CrossRef]
- Hunter, A.M.; Padron, E. Molecular genetics of MDS/MPN overlap syndromes. Best. Pract. Res. Clin. Haematol. 2020, 33, 101195. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Itzykson, R.; Lasho, T.L.; Kosmider, O.; Finke, C.M.; Hanson, C.A.; Knudson, R.A.; Ketterling, R.P.; Tefferi, A.; Solary, E. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: A two-center study of 466 patients. Leukemia 2014, 28, 2206–2212. [Google Scholar] [CrossRef] [PubMed]
- Faisal, M.; Stark, H.; Büsche, G.; Schlue, J.; Teiken, K.; Kreipe, H.H.; Lehmann, U.; Bartels, S. Comprehensive mutation profiling and mRNA expression analysis in atypical chronic myeloid leukemia in comparison with chronic myelomonocytic leukemia. Cancer Med. 2019, 8, 742–750. [Google Scholar] [CrossRef]
- Itzykson, R.; Kosmider, O.; Renneville, A.; Morabito, M.; Preudhomme, C.; Berthon, C.; Adès, L.; Fenaux, P.; Platzbecker, U.; Gagey, O.; et al. Clonal architecture of chronic myelomonocytic leukemias. Blood 2013, 121, 2186–2198. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.; Fermo, E.; Corti, S.; Molteni, M.; Faricciotti, A.; Cortelezzi, A.; Deliliers, G.L.; Beran, M.; Onida, F. RAS Mutations Contribute to Evolution of Chronic Myelomonocytic Leukemia to the Proliferative Variant. Clin. Cancer Res. 2010, 16, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Yu, C.; Xia, S.; Liang, T.; Gu, X.; Liu, Z. A rare atypical chronic myeloid leukemia BCR-ABL1 negative with concomitant JAK2 V617F and SETBP1 mutations: A case report and literature review. Ther. Adv. Hematol. 2020, 11, 204062072092710. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Barraco, D.; Lasho, T.L.; Finke, C.M.; Reichard, K.; Hoversten, K.P.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Targeted next generation sequencing and identification of risk factors in World Health Organization defined atypical chronic myeloid leukemia. Am. J. Hematol. 2017, 92, 542–548. [Google Scholar] [CrossRef]
- Mughal, T.I.; Cross, N.C.P.; Padron, E.; Tiu, R.V.; Savona, M.; Malcovati, L.; Tibes, R.; Komrokji, R.S.; Kiladjian, J.-J.; Garcia-Manero, G.; et al. An International MDS/MPN Working Group’s perspective and recommendations on molecular pathogenesis, diagnosis and clinical characterization of myelodysplastic/myeloproliferative neoplasms. Haematologica 2015, 100, 1117–1130. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Lasho, T.L.; Finke, C.M.; Hanson, C.A.; King, R.L.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Predictors of survival in refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) and the role of next-generation sequencing. Am. J. Hematol. 2016, 91, 492–498. [Google Scholar] [CrossRef]
- Padron, E.; Painter, J.S.; Kunigal, S.; Mailloux, A.W.; McGraw, K.; McDaniel, J.M.; Kim, E.; Bebbington, C.; Baer, M.; Yarranton, G.; et al. GM-CSF–dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood 2013, 121, 5068–5077. [Google Scholar] [CrossRef]
- Emanuel, P.; Bates, L.; Castleberry, R.; Gualtieri, R.; Zuckerman, K. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood 1991, 77, 925–929. [Google Scholar] [CrossRef]
- Sakashita, K.; Matsuda, K.; Koike, K. Diagnosis and treatment of juvenile myelomonocytic leukemia. Pediatr. Int. 2016, 58, 681–690. [Google Scholar] [CrossRef]
- Loh, M.L. Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br. J. Haematol. 2011, 152, 677–687. [Google Scholar] [CrossRef]
- Mangaonkar, A.A.; Swoboda, D.M.; Coltro, G.; Lasho, T.L.; Novotny, P.J.; Pophali, P.; Carr, R.M.; Binder, M.; Finke, C.M.; Gangat, N.; et al. Clinicopathologic characteristics, prognostication and treatment outcomes for myelodysplastic/myeloproliferative neoplasm, unclassifiable (MDS/MPN-U): Mayo Clinic-Moffitt Cancer Center study of 135 consecutive patients. Leukemia 2020, 34, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Meggendorfer, M.; Jeromin, S.; Haferlach, C.; Kern, W.; Haferlach, T. The mutational landscape of 18 investigated genes clearly separates four subtypes of myelodysplastic/myeloproliferative neoplasms. Haematologica 2018, 103, e192–e195. [Google Scholar] [CrossRef]
- Bose, P.; Nazha, A.; Komrokji, R.S.; Patel, K.P.; Pierce, S.A.; Al-Ali, N.; Sochacki, A.; Shaver, A.; Ma, W.; Su, X.; et al. Mutational landscape of myelodysplastic/myeloproliferative neoplasm–unclassifiable. Blood 2018, 132, 2100–2103. [Google Scholar] [CrossRef] [PubMed]
- Such, E.; Cervera, J.; Costa, D.; Solé, F.; Vallespí, T.; Luño, E.; Collado, R.; Calasanz, M.J.; Hernández-Rivas, J.M.; Cigudosa, J.C.; et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica 2011, 96, 375–383. [Google Scholar] [CrossRef]
- Wang, S.A.; Hasserjian, R.P.; Fox, P.S.; Rogers, H.J.; Geyer, J.T.; Chabot-Richards, D.; Weinzierl, E.; Hatem, J.; Jaso, J.; Kanagal-Shamanna, R.; et al. Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms. Blood 2014, 123, 2645–2651. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Han, B. Comparison and Implications of Mutational Profiles of Myelodysplastic Syndromes, Myeloproliferative Neoplasms, and Myelodysplastic/Myeloproliferative Neoplasms: A Meta-Analysis. Front. Oncol. 2020, 10, 579221. [Google Scholar] [CrossRef]
- Coltro, G.; Mangaonkar, A.A.; Lasho, T.L.; Finke, C.M.; Pophali, P.; Carr, R.; Gangat, N.; Binder, M.; Pardanani, A.; Fernandez-Zapico, M.; et al. Clinical, molecular, and prognostic correlates of number, type, and functional localization of TET2 mutations in chronic myelomonocytic leukemia (CMML)—A study of 1084 patients. Leukemia 2020, 34, 1407–1421. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, W.; Wang, D.; Yang, E.; Li, Y.; Li, Y.; Sun, Y.; Wang, M.; Lv, Y.; Hu, X. TET2 Mutation May Be More Valuable in Predicting Thrombosis in ET Patients Compared to PV Patients: A Preliminary Report. J. Clin. Med. 2022, 11, 6615. [Google Scholar] [CrossRef]
- Tefferi, A.; Pardanani, A.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Gangat, N.; Finke, C.M.; Schwager, S.; Mullally, A.; et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009, 23, 905–911. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Kanagal-Shamanna, R.; Orazi, A.; Hasserjian, R.P.; Arber, D.A.; Reichard, K.; Hsi, E.D.; Bagg, A.; Rogers, H.J.; Geyer, J.; Darbaniyan, F.; et al. Myelodysplastic/myeloproliferative neoplasms-unclassifiable with isolated isochromosome 17q represents a distinct clinico-biologic subset: A multi-institutional collaborative study from the Bone Marrow Pathology Group. Mod. Pathol. 2022, 35, 470–479. [Google Scholar] [CrossRef]
- Van Egeren, D.; Escabi, J.; Nguyen, M.; Liu, S.; Reilly, C.R.; Patel, S.; Kamaz, B.; Kalyva, M.; DeAngelo, D.J.; Galinsky, I.; et al. Reconstructing the Lineage Histories and Differentiation Trajectories of Individual Cancer Cells in Myeloproliferative Neoplasms. Cell Stem Cell 2021, 28, 514–523.e9. [Google Scholar] [CrossRef] [PubMed]
- Sousos, N.; Leathlobhair, N.; Karali, C.S.; Louka, E.; Bienz, N.; Royston, D.; Clark, S.-A.; Hamblin, A.; Howard, K.; Mathews, V.; et al. In Utero Origin of Myelofibrosis Presenting in Adult Monozygotic Twins. Nat. Med. 2022, 28, 1207–1211. [Google Scholar] [CrossRef]
- Yarbro, J.W. Mechanism of action of hydroxyurea. Semin. Oncol. 1992, 19 (Suppl. 9), 1–10. [Google Scholar] [PubMed]
- Antonioli, E.; Guglielmelli, P.; Pieri, L.; Finazzi, M.; Rumi, E.; Martinelli, V.; Vianelli, N.; Randi, M.L.; Bertozzi, I.; De Stefano, V.; et al. Hydroxyurea-related toxicity in 3,411 patients with Ph’-negative MPN. Am. J. Hematol. 2012, 87, 552–554. [Google Scholar] [CrossRef]
- Alvarez-Larrán, A.; Kerguelen, A.; Hernández-Boluda, J.C.; Pérez-Encinas, M.; Ferrer-Marín, F.; Bárez, A.; Martínez-López, J.; Cuevas, B.; Mata, M.I.; García-Gutiérrez, V.; et al. Frequency and prognostic value of resistance/intolerance to hydroxycarbamide in 890 patients with polycythaemia vera. Br. J. Haematol. 2016, 172, 786–793. [Google Scholar] [CrossRef]
- Barosi, G.; Birgegard, G.; Finazzi, G.; Griesshammer, M.; Harrison, C.; Hasselbalch, H.; Kiladijan, J.; Lengfelder, E.; Mesa, R.; Mc Mullin, M.F.; et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: Results of a European LeukemiaNet (ELN) consensus process. Br. J. Haematol. 2010, 148, 961–963. [Google Scholar] [CrossRef]
- Alvarez-Larrán, A.; Pérez-Encinas, M.; Ferrer-Marín, F.; Hernández-Boluda, J.C.; Ramírez, M.J.; Martínez-López, J.; Magro, E.; Cruz, Y.; Mata, M.I.; Aragües, P.; et al. Risk of thrombosis according to need of phlebotomies in patients with polycythemia vera treated with hydroxyurea. Haematologica 2017, 102, 103–109. [Google Scholar] [CrossRef]
- Barbui, T.; Tefferi, A.; Vannucchi, A.M.; Passamonti, F.; Silver, R.T.; Hoffman, R.; Verstovsek, S.; Mesa, R.; Kiladjian, J.-J.; Hehlmann, R.; et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: Revised management recommendations from European LeukemiaNet. Leukemia 2018, 32, 1057–1069. [Google Scholar] [CrossRef]
- Andes, W.A.; Noveck, R.J.; Fleming, J.S. Inhibition of Platelet Production Induced by an Antiplatelet Drug, Anagrelide, in Normal Volunteers. Thromb. Haemost. 1984, 52, 325–328. [Google Scholar] [CrossRef]
- Birgegård, G. The Use of Anagrelide in Myeloproliferative Neoplasms, with Focus on Essential Thrombocythemia. Curr. Hematol. Malig. Rep. 2016, 11, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.R.; Martinelli, V.; Rinaldi, P.; Ciancia, R.; del Vecchio, L.; Pane, F.; Nucifora, G.; Rotoli, B. GATA1 is overexpressed in patients with essential thrombocythemia and polycythemia vera but not in patients with primary myelofibrosis or chronic myelogenous leukemia. Leuk. Lymphoma 2008, 49, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Bywater, M.; Lane, S.W. Paving the way to improve therapy for Myeloproliferative Neoplasms. Nat. Commun. 2022, 13, 5025. [Google Scholar] [CrossRef]
- Saleiro, D.; Wen, J.Q.; Kosciuczuk, E.M.; Eckerdt, F.; Beauchamp, E.M.; Oku, C.V.; Blyth, G.T.; Fischietti, M.; Ilut, L.; Colamonici, M.; et al. Discovery of a signaling feedback circuit that defines interferon responses in myeloproliferative neoplasms. Nat. Commun. 2022, 13, 1750. [Google Scholar] [CrossRef] [PubMed]
- Gisslinger, H.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Rossiev, V.; Dulicek, P.; Illes, A.; Pylypenko, H.; et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): A randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020, 7, e196–e208. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.-J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients With Myelofibrosis. JAMA Oncol. 2018, 4, 652. [Google Scholar] [CrossRef]
- Stivala, S.; Codilupi, T.; Brkic, S.; Baerenwaldt, A.; Ghosh, N.; Hao-Shen, H.; Dirnhofer, S.; Dettmer, M.S.; Simillion, C.; Kaufmann, B.A.; et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J. Clin. Investig. 2019, 129, 1596–1611. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Verstovsek, S.; Cervantes, F.; Barosi, G.; Reilly, J.T.; Dupriez, B.; Levine, R.; Le Bousse-Kerdiles, M.-C.; Wadleigh, M.; Campbell, P.J.; et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk. Res. 2007, 31, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, J.D.; Puda, A.; Malcovati, L.; Berg, T.; Hofbauer, M.; Stukalov, A.; Klampfl, T.; Harutyunyan, A.S.; Gisslinger, H.; Gisslinger, B.; et al. Clinical significance of genetic aberrations in secondary acute myeloid leukemia. Am. J. Hematol. 2012, 87, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Alchalby, H.; Zabelina, T.; Stübig, T.; van Biezen, A.; Bornhäuser, M.; Di Bartolomeo, P.; Beelen, D.; Cahn, J.Y.; Dreger, P.; Schroyens, W.; et al. Allogeneic Stem Cell Transplantation for Myelofibrosis with Leukemic Transformation: A Study from the Myeloproliferative Neoplasm Subcommittee of the CMWP of the European Group for Blood and Marrow Transplantation. Biol. Blood Marrow Transplant. 2014, 20, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Polverelli, N.; Farina, M.; D’Adda, M.; Damiani, E.; Grazioli, L.; Leoni, A.; Malagola, M.; Bernardi, S.; Russo, D. How We Manage Myelofibrosis Candidates for Allogeneic Stem Cell Transplantation. Cells 2022, 11, 553. [Google Scholar] [CrossRef]
- Meng, X.; Gao, J.Z.; Gomendoza, S.M.T.; Li, J.W.; Yang, S. Recent Advances of WEE1 Inhibitors and Statins in Cancers With p53 Mutations. Front. Med. 2021, 8, 737951. [Google Scholar] [CrossRef]
- Ajufo, H.O.; Waksal, J.A.; Mascarenhas, J.O.; Rampal, R.K. Treating accelerated and blast phase myeloproliferative neoplasms: Progress and challenges. Ther. Adv. Hematol. 2023, 14, 20406207231177282. [Google Scholar] [CrossRef]
- Ramanathan, G.; Fleischman, A.G. The Microenvironment in Myeloproliferative Neoplasms. Hematol. Oncol. Clin. 2021, 35, 205–216. [Google Scholar] [CrossRef]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Mutation Category | Relative Frequency (%) |
| ASXL1 | 36 |
| TET2 | 18 |
| SRSF2 | 18 |
| U2AF1 | 16 |
| Class | Frequencies of Common Somatic Mutations | Cytogenetic Abnormalities | Incidence Rate in the US /100,000 |
| Chronic myelomonocytic leukemia | TET2 (60%), SRSF2 (50%), ASXL (40%), NKRAS and JAK2 (40%) | Trisomy 8 (+8), −Y, (monosomy 7 and del7q), trisomy 21 (+21) [76] | 0.6 (0.57–0.63) |
| Atypical chronic myeloid leukemia (aCML) | SETBP 1 (25–40%), ETNK1 (8%), ASXL1, EZH2, TET2, SRSF2, and N/KRAS | Trisomy 8, del(20q), −7/7q-, or isochromosome 17q [i17[q)] [77] | 0.060 (04–0.62) |
| MDS/MPN with SF3B1 mutation and thrombocytosis | SFB3 (89.2%), JAK 2, | Trisomy 8 | Less than 1% of all new MDS/MPN cases |
| MDS/MPN NOS | ASXL1 (29–56%), TP53 (8–9%), SRSF2, SETBP1, JAK2V617F, NRAS, and TET2 | Trisomy 8, monosomy 7, deletion 7q, and deletion 20q. | 0.07 (0.006–0.009) |
| Phenotypic Stage | Blast Percentage | Comment |
| MPN CP | Less than 10% of blasts | May have a static course or progress to advanced disease depending on genetic risk profile |
| MPN AP | 10 to 19% of blasts | |
| MPN BP | Greater than 20% of blasts |
| Class | Examples |
| JAK kinase inhibitors | Ruxolitinib, fedratinib |
| Combined JAK 2 and FLT3 inhibitors | Pacritinib–fedratinib |
| Cytoreductive agents | Hydroxyurea |
| Interferons | Ropeginterferon alfa-2b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmud, M.; Vasireddy, S.; Gowin, K.; Amaraneni, A. Myeloproliferative Neoplasms: Contemporary Review and Molecular Landscape. Int. J. Mol. Sci. 2023, 24, 17383. https://doi.org/10.3390/ijms242417383
Mahmud M, Vasireddy S, Gowin K, Amaraneni A. Myeloproliferative Neoplasms: Contemporary Review and Molecular Landscape. International Journal of Molecular Sciences. 2023; 24(24):17383. https://doi.org/10.3390/ijms242417383
Chicago/Turabian StyleMahmud, Muftah, Swati Vasireddy, Krisstina Gowin, and Akshay Amaraneni. 2023. "Myeloproliferative Neoplasms: Contemporary Review and Molecular Landscape" International Journal of Molecular Sciences 24, no. 24: 17383. https://doi.org/10.3390/ijms242417383
APA StyleMahmud, M., Vasireddy, S., Gowin, K., & Amaraneni, A. (2023). Myeloproliferative Neoplasms: Contemporary Review and Molecular Landscape. International Journal of Molecular Sciences, 24(24), 17383. https://doi.org/10.3390/ijms242417383