1. Introduction
The onset of the SARS-CoV-2 pandemic in 2019 underscores the importance of expeditiously developing reagents to comprehend better antigen properties, viral pathogenesis, and host reactions and to facilitate diagnostic advances. To add more tools targeting SARS-CoV-2, previously, Amarasinghe and coworkers expressed and purified 21 recombinant SARS-CoV-2 proteins, carried out antibody (Ab) selections via phage display, and validated binding of the IgGs in vitro by performing Ab selections using phage display techniques [
1] These mAbs were assessed for activity in Western blot (WB) and immunofluorescence (IF) assays utilizing SARS-CoV-2-infected cells. Although these findings suggest that these synthetic antibodies may be used in the investigation of SARS-CoV-2 viral proteins and for the development of novel diagnostic assays for COVID-19, the findings also invite questions about the location of binding and the effect of the highly flexible nonstructural protein (Nsps) antigens on antibody binding.
The Nsps of SARS-CoV-2 are remarkably flexible. Among these proteins, Nsp1 stands out owing to its ability to satisfy many functions in betacoronaviruses (b-CoVs), including SARS-CoV-2. Nsp1 can inhibit cellular mRNA translation, redirect the translational machinery to viral RNA templates, induce cell cycle arrest in the G0/G1 phase, and degrade cellular messenger RNAs [
2,
3]. Additionally, Nsp1 proteins may play a vital role in the innate immune response, downregulating the expression of virus-specific genes and shutting down host translation. The multifunctional nature of Nsp1 requires it to exhibit structural flexibility to interact effectively with various viral and host factors, enabling it to carry out efficiently its diverse functions. Therefore, understanding the structural flexibility of Nsps is highly important for deciphering their mechanisms of action and devising targeted interventions against SARS-CoV-2.
Typically, the interaction of an antibody and an antigen leads to the stabilization of the antigen. Although “localized destabilization” was reported in some instances [
4], it is uncommon. Thus, antibody binding that reports on the flexibility of Nsp1 would be of high interest. In this study, we characterized the binding of two antibodies (Fabs 15497 and 15498) with Nsp1 by using hydrogen-deuterium exchange mass spectrometry (HDX-MS).
The standard bottom-up HDX-MS process involves these step [
5,
6,
7,
8,
9,
10]: (1) incubation with D
2O (labeling), (2) quenching of the HDX by adding acidified denaturant, (3) digestion of the Nsp1 and/or antibody with acid-stable proteases, (4) chromatographic separation of peptides followed by mass spectrometric measurement of their isotope clusters, and (5) semiautomatic data analysis. Proteins in the solution are mixed into a D
2O-based buffer in the first step, allowing labile hydrogens to exchange with deuterium from the solvent. Both backbone (-CON−H) and some side-chain hydrogens exchange, but HDX of protein side chains is not measured owing to their rapid exchange in and out following the quench. Amide hydrogens in the backbone, however, exchange slowly (minutes to hours), allowing them to be detected with MS. HDX is catalyzed by both acid and base, giving rise to a minimum exchange rate occurring at pH around 2.5, motivating the choice of an acid quench. Using a combination of proteases ensures the generation of small- to medium-sized peptides over most of the protein, offering detailed, good spatial resolution. Semi-automated data analysis software such as HDExaminer 2.5.1 calculates the mass shift due to deuterium incorporation. The software examines the isotopic distribution of a peptide or protein in its undeuterated state and establishes its centroid. Comparing this with the shifted centroid at later time points reveals the most probable extent of HDX. The results of the analysis are pictured as a kinetics graph plotting deuterium uptake (Y-axis) against time (X-axis). In most HDX experiments, researchers conduct differential footprinting, comparing two states (e.g., free versus ligand-bound or wild type versus mutant). Deuterium differences are computed by subtracting HDX values between these states, uncovering both local and allosteric impacts of the binding.
Our previous HDX findings regarding Nsp1 show that approximately one-third of the protein lacks a compact, well-defined structure consistent with its flexibility and posing challenges for crystallizing the full-length Nsp1. This observation aligns with the crystal structure of Nsp1 [
11,
12] , which had to be taken of a truncated version spanning residues from 10 to 127. Notably, we chose HDX for this study because HDX-MS is rapid, solution-based, sensitive to protein dynamics and flexibility and does not depend on protein size or complexity.
The question we ask is whether antibody binding can capture Nsp1 structural dynamics. We report here that Nsp1 binding traps a flexible, high-energy state of Nsp1, a phenomenon that may play a role in the development of therapeutic strategies targeting flexible proteins. Added flexibility increases entropy and would be expected to adversely diminish the ability of a protein to carry out its many functions.
3. Discussion
Identifying the critical regulatory segment and understanding its effects on Nsp1 afford valuable insights into the functional and structural modulation of the protein. Antibody 15948 captures in binding to Nsp1 a higher energy state of the antigen much like a catalytic antibody [
16] selectively stabilizes a high-energy transition state. This intriguing finding prompts the question of whether antibodies can leverage their binding energy, typically responsible for the catalytic ability of enzymes, to catalyze chemical transformations in their targets, going beyond their conventional role of merely serving as labeling molecules.
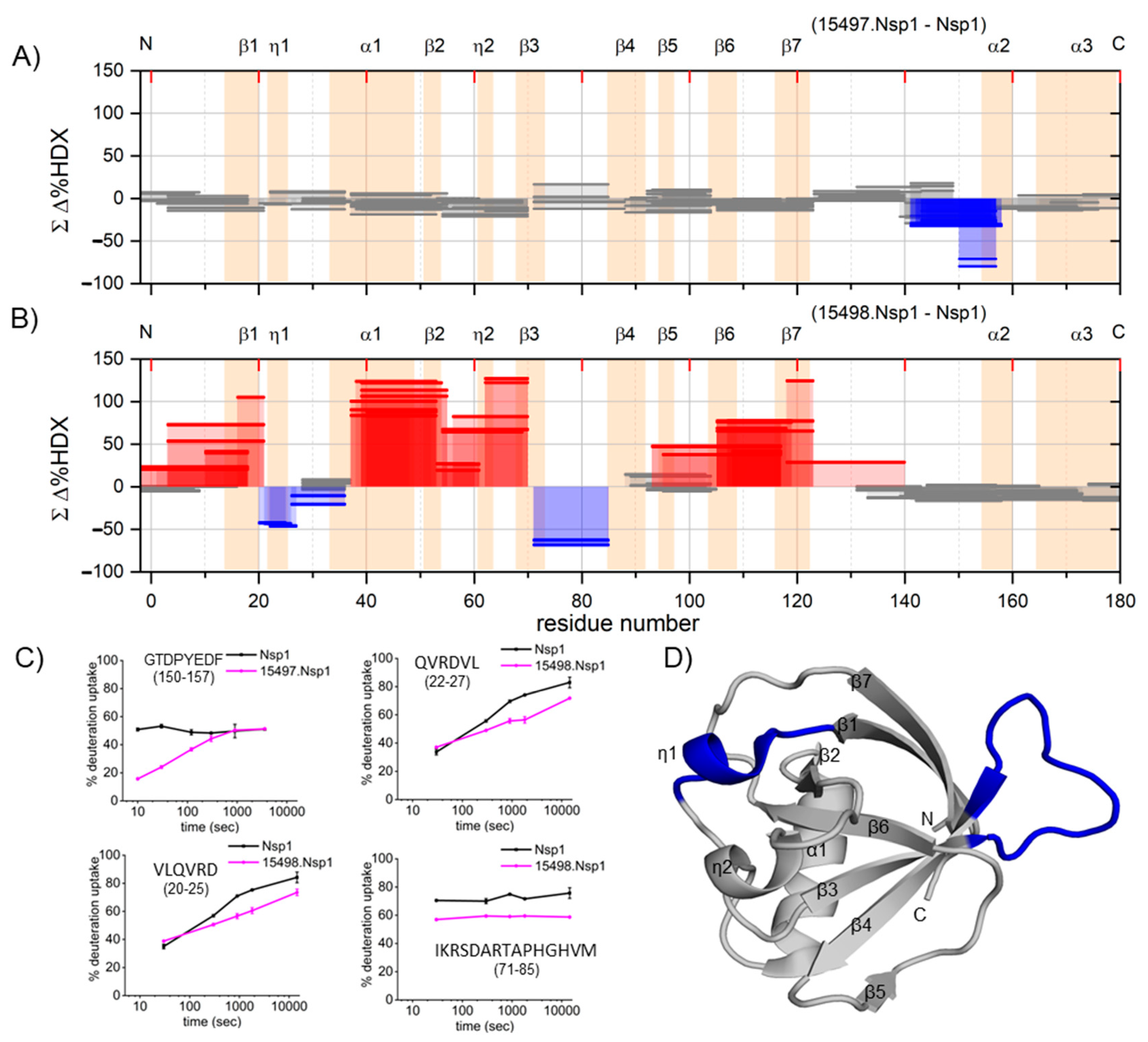
The Woods’ plot and supplementary kinetic plots (
Figure S5) demonstrate increased HDX in most peptides spanning the secondary structure, excluding those at the binding site. In the context of 3D structure, the entire N-terminal domain experiences extensive conformational alterations, spanning regions both proximal and distal to the epitope, resulting in trapping a high-energy state.
This seems to be a novel observation as we were unable to find any precedents for antibody binding destabilizing nearly the entire structure of the antigen. Another possible explanation is the antibody binding destabilizes the native form of the antigen, but this seems less likely because bonding usually brings stability, not instability, to a molecule or to a complex.
Our previous temperature-dependent HDX study addressing the dynamics of full-length Nsp1 revealed that approximately one-third of the protein is unstructured, while the remaining two-thirds exhibits moderate compactness. Additionally, Nsp1 is not highly thermally stable, with a melting temperature of ~44 °C [
15]. This suggests that the native structure of Nsp1 is readily modulated and that the energy barrier between the folded and unfolded states is not substantial, providing a small equilibrium population of the unfolded protein to interact with the antibody. Equilibrium causes more of the population to unfold and bind. The noncontiguous binding site spanning amino acid residues 20–27 and 73–85 can be viewed as a “hotspot” that becomes exposed and flexible in the higher energy state, and that hotspot binds to an antibody to stabilize the open state. Conversely, antibody 15497 exhibits the usual protection accompanying antigen/antibody binding.
These findings underscore the dynamic nature of the Nsp1 protein and its ability to undergo conformational changes. The HDX data show that, in principle, when antibodies interact with an antigen, the outcome can be stabilization or destabilization of the antigen structure [
4]. A previous study demonstrated that dAbs may attach to the IBR of HOIP protein, promoting an alternative conformation that is more open compared to its standard solution-state structure, consequently leading to local destabilization or capturing a high-energy state [
5]. In our study, we observed that the effect of 15498 is particularly pronounced, occurring over most of the structure of Nsp1.
Both HDX-MS and SEC-MALS unveil important characteristics of 15498 and its interactions with Nsp1. Whereas SEC-MALS provides valuable insights, it does not possess the requisite sensitivity to detect subtle conformational changes captured by HDX-MS. To validate the specific residues contributing to this conformational destabilization in Nsp1, we recommend conducting high-resolution studies such as X-ray crystallography and Cryo-EM in future investigations. HDX-based observations, however, stand as compelling and noteworthy in their own right.
The results also provide insight into antibody recognition and its impact on binding the high-order structure of an antigen. Further investigations into the functional implications of this hotspot can contribute to a better understanding of the immune response against Nsp1. Overall, the concept of developing an antibody that targets a transition state or a high-energy structure underscores the potential for conferring a catalytic effect on the corresponding chemical reaction. This exciting prospect paves the way for exploring the use of antibodies not only as catalysts but as probes of high-order structure, particularly for highly flexible proteins that serve many functions. The implications of such a discovery could offer new opportunities in immunology and biocatalysis, with applications in therapeutics and diagnostics. The possible application in therapeutics stems from the increase in entropy that is locked into the antibody/antigen complex and that may mitigate undesirable functions of a protein, especially for proteins that are multifunctional viral proteins.
4. Materials and Methods
4.1. Protein Purification
Nsp1 protein was purified according to recently published methods [
13].
4.2. Fabs Selection and Characterization
The selection of Fab-phage clones, based on their binding to Nsp1, was conducted following previously described methods. Antibody variable domains of the selected clones, which exhibited binding to the Nsp1 antigen in Fab-phage ELISA, were sequenced by PCR amplification and the Sanger method, and unique clones were identified. The estimated affinities of the individual Nsp1 antigen-binding clones were determined as dissociation constants (
Kd) for 15497 and 15498 to be 0.9 nM and 1.4 nM, respectively, as measured by BLI [
16]. The variable domain genes for clones exhibiting higher binding affinity (namely 15497 and 15498) were then cloned into vectors for expression.
4.3. IgG Expression and Purification
The selected variable domains were amplified by PCR and subcloned into pSCSTa-hIg1 and pSCST1-hk vectors. Expression constructs were then transfected into HEK293F cells using Fectopro (101000014, Polyplus, Illkirch-Graffenstaden, France), following the manufacturer’s instructions. The cells were incubated with shaking at 125 rpm at 37 °C for 4–5 days. After incubation, the cells were lysed, and the supernatant was collected after centrifugation. The lysate was applied to a Protein-A affinity column for purification. IgG proteins were eluted by using 100 mM glycine at pH 2.0 and neutralized with 2 M Tris at pH 7.5. The eluted proteins were then buffer exchanged into PBS at pH 7.4, concentrated, and analyzed by Western blotting. The IgGs were further characterized for their binding kinetics and immunofluorescence [
16].
4.4. Hydrogen Deuterium Exchange Mass Spectrometry
HDX-MS was performed to measure the binding site and any structural perturbations of Nsp1 with antibodies 15497 and 15498. Both epitope and paratope mapping experiments were conducted as described elsewhere [
12].
4.5. Epitope Mapping of Nsp1 with Fabs 15497 and 15498
Epitope mapping experiments used HDX-MS to investigate the differential behavior of Nsp1 in its bound and unbound states. In the unbound state, continuous HDX was conducted by diluting Nsp1 10-fold into a deuterated PBS buffer. The HDX was allowed to proceed for several time points (10, 30, 120, 300, and 3600 s) on ice. To quench the HDX, the deuterated samples were immediately mixed with 4 M guanidine-hydrochloride, 200 mM TCEP, pH 2.5. Fungal XIII solution (10 μL of 10 mg/mL) was pre-mixed with the quench solution, and enzymatic digestion was allowed to occur for 2 min. The samples were then flash-frozen in liquid nitrogen and stored at −80 °C until analysis via MS. Prior to injection into the LC/MS system, the frozen samples were thawed quickly. Online digestion was performed using a custom-packed pepsin column (2 × 20 mm). Simultaneous peptide trapping and desalting were carried out on a ZORBAX Eclipse XDB C8 column (2.1 mm × 15 mm) by using 0.1% formic acid aqueous solvent at a flow rate of 200 μL/min delivered by an HPLC pump. The valves, tubes, and analytical column were kept chilled, while the pepsin column was maintained at room temperature during the HDX measurements. Peptides were separated on a reversed-phase C18 column 2.1 mm × 50 mm, 2.5 µm X select-CSH) with the gradient; the organic solvent B (acetonitrile with 0.1% formic acid) was increased from 5% to 80% over 15 min. Following chromatographic separation, the peptides were analyzed with a Bruker Maxis HM Q-TOF mass spectrometer. The HDX data obtained were processed and analyzed using HDExaminer software (version 2.5.1, Sierra 433 Analytics, Inc., Modesto, CA, USA). The key parameters considered during data analysis by HDExaminer included (1) excluding deuterations in the first two residues, where back exchange is particularly rapid, (2) ensuring the elution time window of the peptides remains within 0.50 min of the non-deuterated window, (3) recognizing that proline does not undergo exchange, and (4) noting the absence of an assessment of back exchange corrections in the analysis (back exchange considerations are less important for differential measurements than for single determinations). HDExaminer computes the centroid of an isotope cluster using experimental isotope distribution (A, A + 1, A + 2, … where A is the monoisotopic mass). When set to “Theoretical isotope clusters”, HDExaminer 2.5.1 determines the centroid of the isotope pattern, including the deuteriums, accurately aligns it with the experimental data, and outputs the extent of HDX.
For the bound state, Nsp1 and antibody 15497 were mixed in a molar ratio of 1:2 on ice and incubated for 30 min before initiating the HDX process. The experimental setup and molar ratios were kept consistent for the second antibody, 15498, for its epitope mapping experiments. All HDX experiments were conducted in duplicate. The antibody stock was initially prepared in PBS buffer. Quench solution and deuterated buffers were then pre-prepared in bulk, frozen in liquid N2, and stored at −80 °C. Upon thawing, these solutions were promptly utilized for experiments. Different time points for both bound and unbound states were collected on the same day, flash-frozen in liquid N2, and stored at −80 °C. Subsequently, samples from both states were processed and analyzed via mass spectrometry on the same day. Prior to initiating the HDX reactions, the pH of the quench solution and deuterated PBS buffers were re-checked for consistency. Nsp1 samples were subjected to one freeze–thaw cycle for bound and unbound state HDX measurements.
4.6. Paratope Mapping of 15497 and 15498 with Nsp1
The HDX experimental setup for paratope mapping was the same, except the concentration of TCEP in the quenching solution was higher (4 M guanidine-hydrochloride, 500 mM TCEP, pH 2.5). The antigen was added in excess to the antibody at a ratio of 1:2.
4.7. Peptide Mapping and HDX Data Analysis
Before initiating epitope mapping, peptide mapping of Nsp1 was performed. A stock solution of 50 μM Nsp1 was prepared in PBS (pH 7.4) buffer and used for peptide mapping in the LC/MS/MS mode. The cycle time was 3 s in the Bruker Maxis HM Q-TOF mass spectrometer. The MS/MS data were analyzed utilizing Byonic and Byologic (Protein Metrics, San Carlos, CA, USA).
To ensure accuracy and minimize false positive results, a reversed protein sequence control was implemented during peptide identification. Statistical analysis was performed to evaluate the differences in deuterium uptake between the bound state (Nsp1 + antibodies) and the unbound state of Nsp1. Cumulative differences in hydrogen–deuterium exchange (HDX) between the two states were calculated, and those exceeding the significance limit were considered statistically significant. The propagated error for each peptide was determined as the square root of the sum of squared standard deviation values across all time points for both states. Differences greater than the propagated error were considered statistically significant [
15].

{kind=link}
{kind=link}
{kind=link}
{kind=link}



