1. Introduction
Notoriously, atherosclerotic cardiovascular diseases (ASCVDs), including coronary disease, ischemic heart disease, and hypertension, remain the leading cause of morbidity and mortality worldwide [
1]. Reverse cholesterol transport (RCT) is the process by which cholesterol is transported from peripheral tissues (including arterial wall foam cells in atherosclerotic plaques) to the liver by high-density lipoprotein (HDL) and then to the bile for intestinal excretion [
2]. Previous animal and human studies have confirmed the inverse relationship between RCT and atherosclerosis, with the development of atherosclerosis being delayed when the RCT process is facilitated and, conversely, the impaired RCT process will result in accelerated atherosclerosis [
3]. Therefore, it is reasonable to conclude that targeted improvements in the RCT process will slow the progression of atherosclerosis. To achieve this purpose, three conceptual strategies have been proposed: (1) promote macrophage cholesterol efflux, (2) improve the ability of HDL to accept or transport cholesterol, and (3) improve hepatic cholesterol uptake and biliary/intestinal excretion [
4]. Of these, macrophage cholesterol efflux is considered the first and rate-limiting step of RCT [
5]. Therefore, the most effective strategies for reducing ASCVDs may target macrophage cholesterol efflux in RCT [
2,
6].
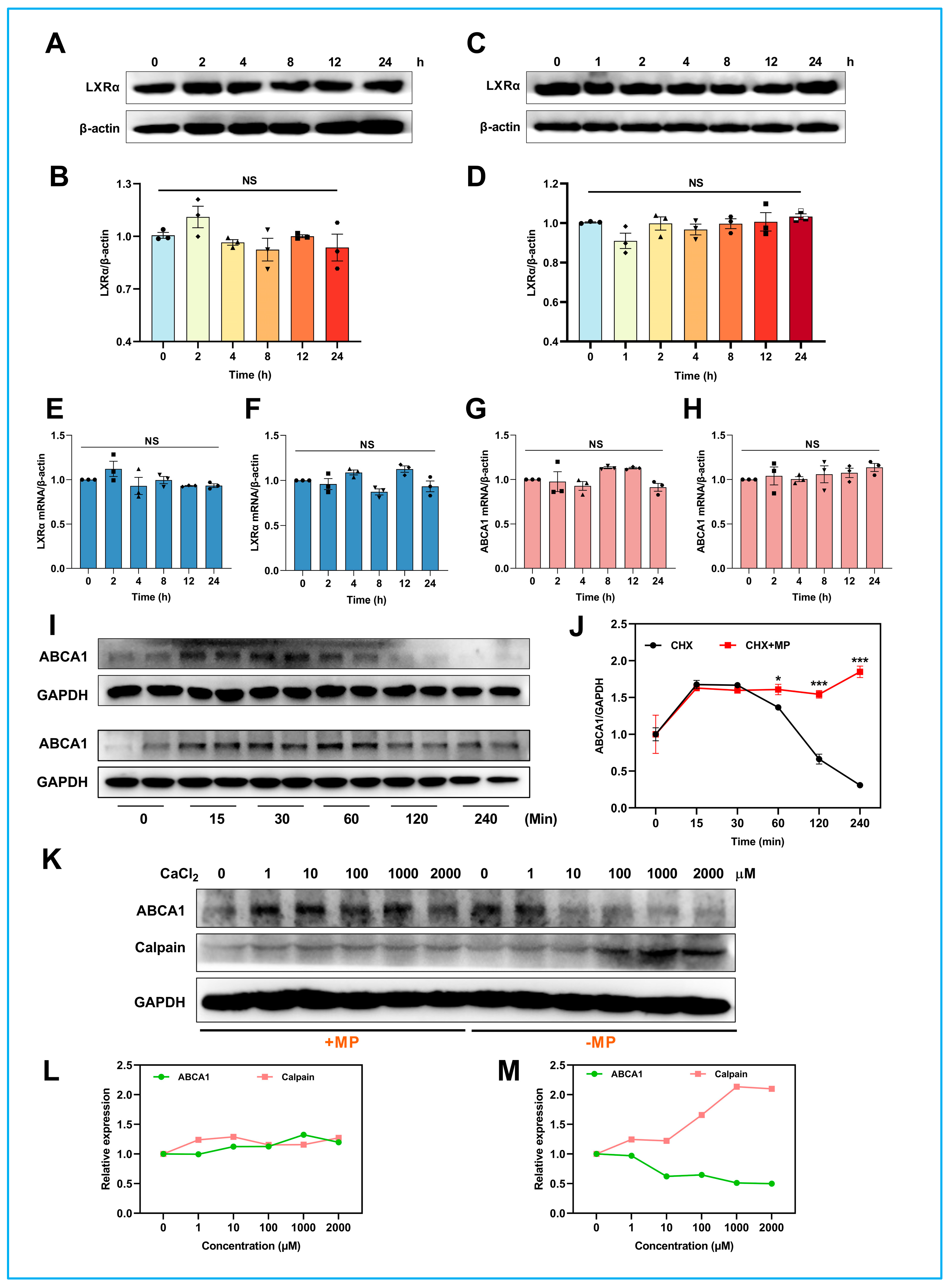
ATP-binding cassette transporter A1 (ABCA1) and ATP-binding cassette transporter G1 (ABCG1) belong to the ATP-binding cassette (ABC) transporter superfamily, were found to be responsible for the mediation of the cholesterol efflux in macrophages [
7]. In particular, ABCA1 plays a crucial role in macrophages and is thought to be a primary regulator of cholesterol efflux [
8,
9]. It promotes the removal of excess cholesterol, thus preventing them from being converted into foam cells and preventing atherosclerosis [
7,
10]. ABCA1 is one of the target genes of the liver X receptor (LXR), a key sterol-sensitive transcription factor in macrophages that regulates intracellular cholesterol [
11]. Surplus intracellular cholesterol induces LXR to bind to the four-nucleotide (DR-4) element of the ABCA1 promoter to drive the ABCA1-mediated efflux pathway, resulting in protection against cholesterol toxicity and maintenance of cholesterol homeostasis [
12,
13]. In addition to LXR-mediated transcriptional regulation, post-translational regulatory mechanisms also play a vital role in regulating ABCA1 expression [
14]. For instance, calpain-mediated proteolytic degradation is considered one of the most important regulatory mechanisms of ABCA1 functions [
15]. Calpains, a family of calcium-activated cysteine proteases, are reportedly present in all human tissues and regulated by intracellular calcium concentration [
16]. Since calcium ions are essential for various biological processes in all living cells, the levels of free calcium ions in the cytoplasm and extracellular fluid of resting cells must be tightly controlled [
17]. There are two major sources of calcium ions in the cytoplasm: the extracellular space and intracellular stores (sarcoplasmic reticulum or endoplasmic reticulum). Voltage-gated calcium channels primarily regulate the former, and the latter is controlled mainly by the ryanodine receptor (RyR) and inositol trisphosphate receptor channels [
18,
19].
On the other hand, a sequence rich in proline, glutamic acid, serine, and threonine (PEST sequence) located in the nucleotide-binding domain 1 (NBD1) of ABCA1 helps to target calpain to ABCA1 on the cell surface, resulting in degradation of ABCA1 by calpain [
8,
20]. Interestingly, the above effects of the PEST sequence require Thr-1286 and Thr-1305 phosphorylation. Molecules that dephosphorylate the PEST motif can consequently stabilize the ABCA1 located on the plasma membrane [
21]. According to the above studies, it is reasonable to conceive that both inhibition of calcium ions in the cytoplasm and dephosphorylation of PEST sequences could retard calpain-mediated proteolytic degradation of ABCA1, increase cellular cholesterol efflux, improve RCT function, and prevent atherosclerosis.
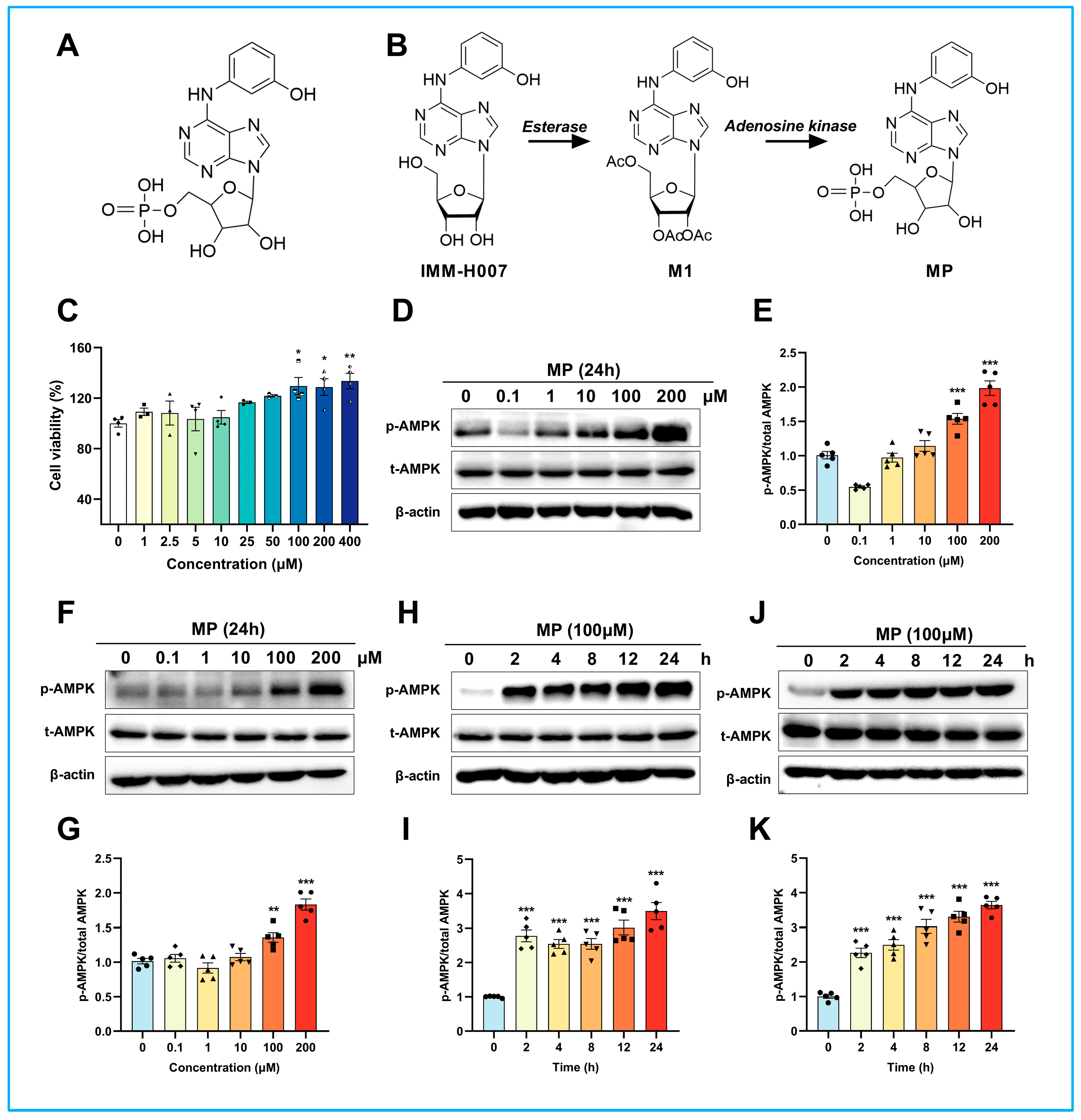
IMM-H007 (2′,3′,5′-tri-O-acetyl-N6-(3-hydroxyphenyl) adenosine), an adenosine derivative derived from cordycepin, has previously been reported to be an agonist of AMP-activated protein kinase (AMPK) that can repress inflammatory atherosclerosis [
22]. AMPK is a highly conserved serine/threonine protein kinase widely expressed in higher eukaryotes. Our previous study found that IMM-H007 promotes ABCA1-mediated RCT by inhibiting the calcium-activated calpain activity rather than altering LXR expression, ultimately reducing atherosclerotic plaque formation in hypercholesterolemic mice [
23]. Noteworthily, our later investigation manifested that the atheroprotective properties of IMM-H007 depend on the pharmacological activation of AMPK [
24]. Moreover, our pharmacokinetic results indicated that IMM-H007 is a prodrug that can be rapidly metabolized to its deacetylated metabolite in the blood by esterase after oral administration, then transported into the cell via equilibrative nucleoside transporter (ENT) and finally phosphorylated to ((2R,3S,4R,5R)-3,4-dihydroxy-5-(6-((3-hydroxyphenyl) amino)-9H-purin-9-yl) tetrahydrofuran-2-yl) methyl dihydrogen phosphate (phosphorylated metabolite, MP) by adenosine kinase [
25]. In this case, whether MP is the actual molecule responsible for the promotion of ABCA1-mediated RCT by IMM-H007 and the accompanying mechanisms needs further study.
This study aims to determine whether MP is the actual molecule responsible for the pharmacological activities exerted by IMM-H007 and to elucidate the underlying mechanisms by which MP facilitates ABCA1-mediated RCT function. By virtue of multiple experiments, we have confirmed that MP is the actual active form of IMM-H007. MP stimulates AMPK Thr172 phosphorylation in a concentration- and time-dependent manner, consistent with previous results with IMM-H007. These activations of AMPK are due to the fact that MP can bind directly to AMPK with a moderate affinity and resultantly alter its spatial conformation. Furthermore, subsequent experiments revealed that MP promotes functional ABCA1 expression by inhibiting calpain-mediated proteolytic degradation rather than transcriptional regulation and that this process is dependent on AMPK activation. In more detail, we found that the mechanism by which MP retards calpain-mediated proteolytic degradation is by suppressing calcium-activated calpain activity but not by dephosphorylating the PEST sequence in ABCA1. Collectively, MP is a direct agonist of AMPK and a promising candidate molecule for strengthening ABCA1 stability, increasing macrophage cholesterol efflux, and preventing atherosclerosis.
3. Discussion
To date, atherosclerosis remains a prominent risk factor for various cardiovascular diseases, imposing a substantial burden on human society [
32]. This underscores the significance of identifying a candidate molecule with a potential atheroprotective effect. In this study, we present a promising molecule, MP, identified as a phosphorylated metabolite of IMM-H007. In the first half of this study, we observed that MP exhibits a direct and modest binding affinity to AMPK, subsequently promoting its phosphorylation. This finding, consistent with the effects observed with IMM-H007, supports the hypothesis that MP is the active molecule responsible for IMM-H007’s anti-atherosclerotic effects. Furthermore, the latter part of our investigation focused on elucidating the potential mechanism by which MP enhances the stability of the ABCA1 protein following AMPK activation. Specifically, we observed that activated AMPK not only downregulates the Cav1.2 channel, impeding the influx of extracellular Ca
2+ and thereby reducing intracellular Ca
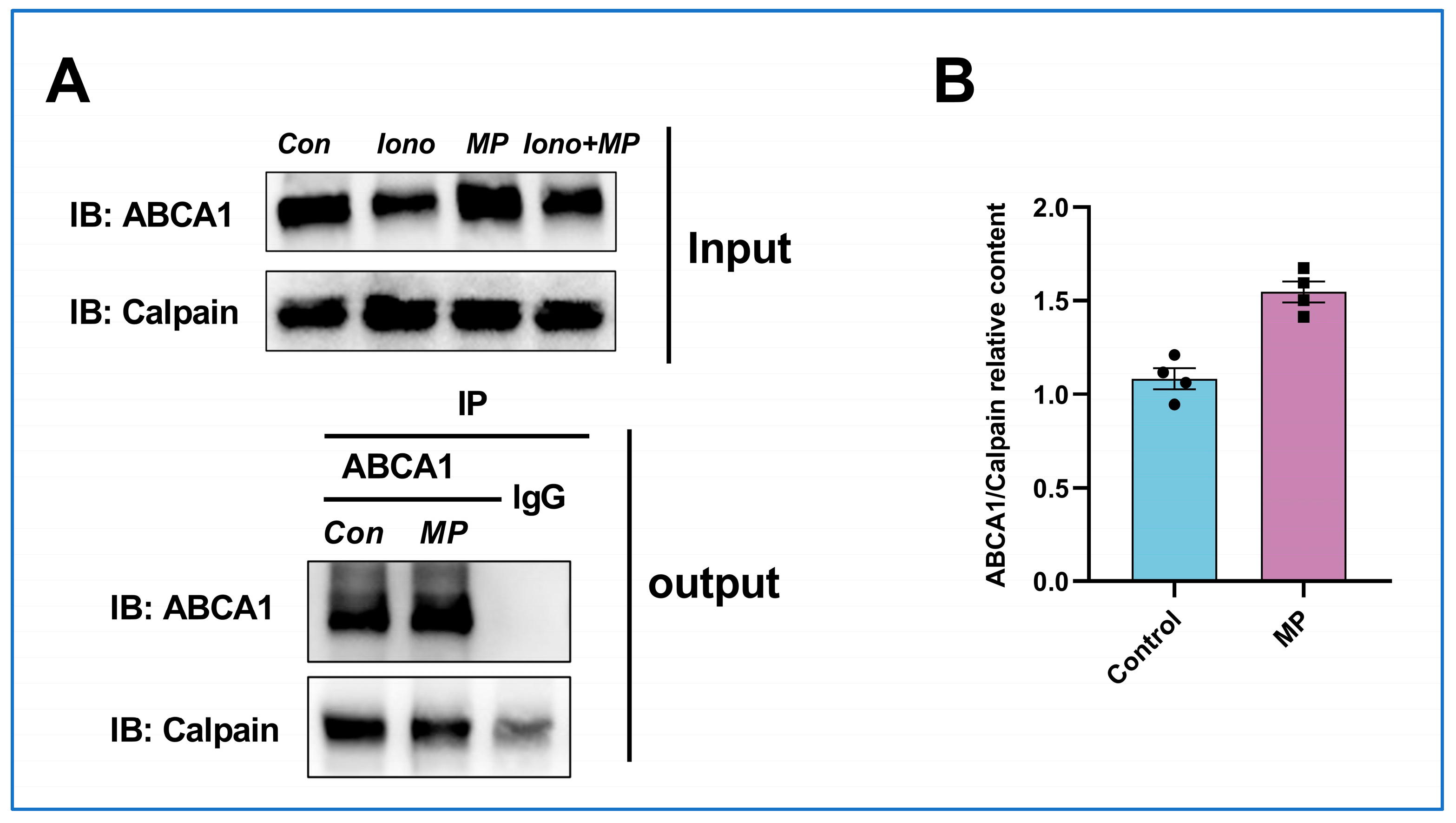
2+ levels and inhibiting calcium-activated calpain activity, but also weakens the interaction between calpain and ABCA1. These events culminate in the deceleration of calpain-mediated degradation of ABCA1. More importantly, gaining a deeper understanding of the mechanism by which MP promotes the functionality of ABCA1 holds the potential to provide insights into strategies for the prevention and amelioration of atherosclerosis.
Our prior investigations have established the rapid metabolism of IMM-H007 in the bloodstream, indicating that the biological effects attributed to IMM-H007 are not a result of its own actions. Furthermore, previous studies have independently shown that IMM-H007 effectively activates AMPK and inhibits ABCA1 degradation. However, the causal relationship between IMM-H007's inhibition of ABCA1 degradation and its activation of AMPK remains unclear [
23,
24]. Building upon these findings, this study was specifically designed to address two fundamental questions: What is the source of the observed effects? And how are these effects generated?
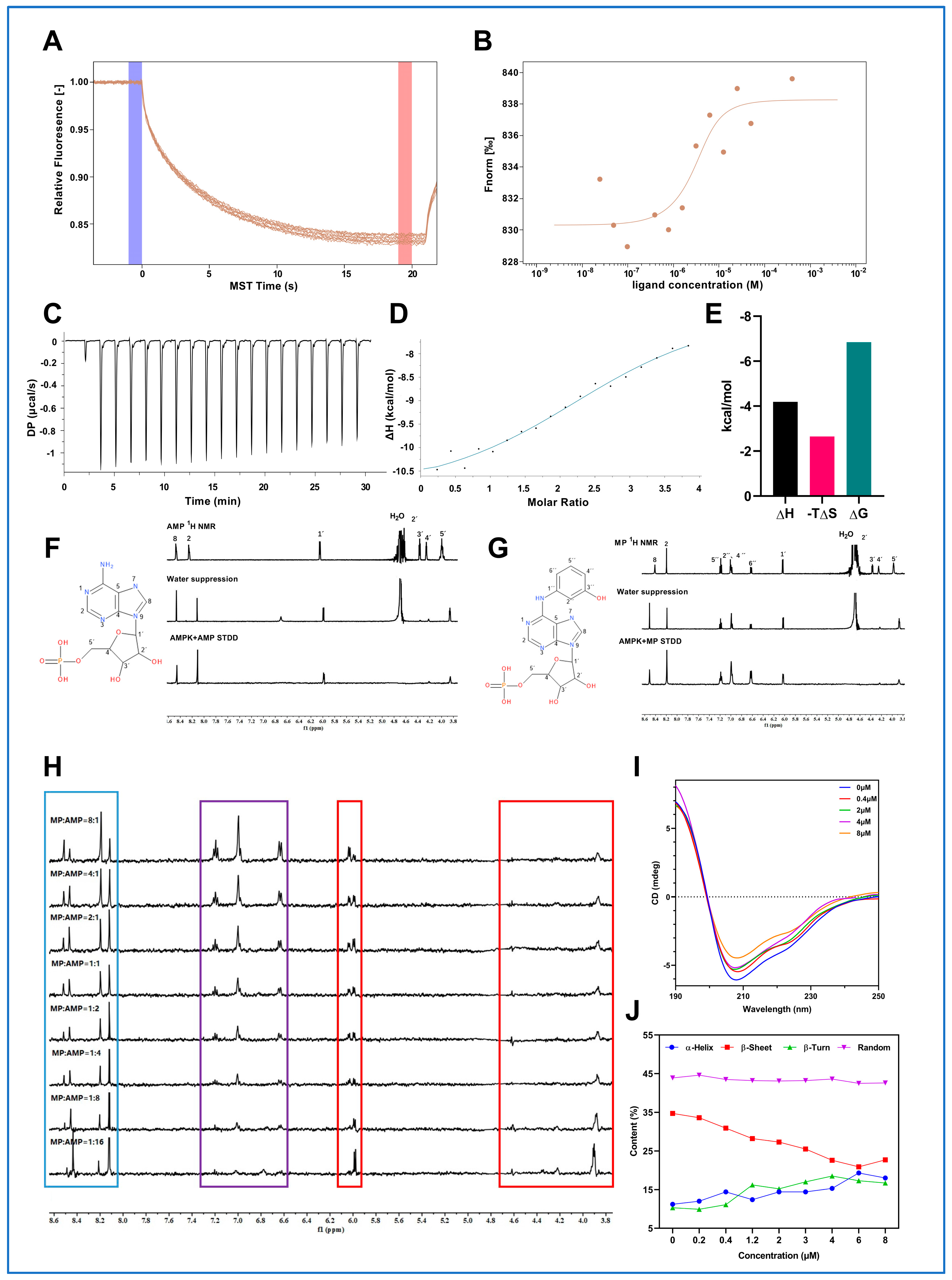
To address the initial question of “What is the source of the observed effects?” we employed pharmacokinetics [
25] and molecular biology. In this paper, we unveil the moderate direct interaction between MP and AMPK and the associated biological effects of MP on AMPK activation at both the molecular and cellular levels. We observed that the affinity of MP for AMPK ranges between 1–10 μM, which is significantly lower compared to other direct agonists of AMPK, such as PF-06409577 (0.005 μM) [
33], 991 (0.06 μM) [
34], A769662 (0.5 μM) [
34], but similar to that of AMP (20 μM) [
35]. Surprisingly, these direct AMPK agonists with high affinity remain in the preclinical stage, serving as experimental tools. Therefore, we speculate that stronger in vivo activation of AMPK may not necessarily be advantageous. Due to the fact that AMPK has up to 12 protein isoforms and is widely distributed in the body, stronger affinity may potentially lead to more side effects, thereby restricting their further development. In contrast, MP exhibits a comparable affinity to that of AMP, an endogenous agonist of AMPK. Hence, we believe that MP's more moderate binding mode may facilitate its progression into further clinical studies. Collectively, these findings confirm that MP is the source of the observed effects. However, this section of the study has certain limitations. For instance, the biophysical methods utilized to characterize the interactions between MP and AMPK were rudimentary. The exhaustive conformational alterations accompanying this binding process and the structure-function relationships are areas that require further elucidation through the application of advanced structural biology techniques.
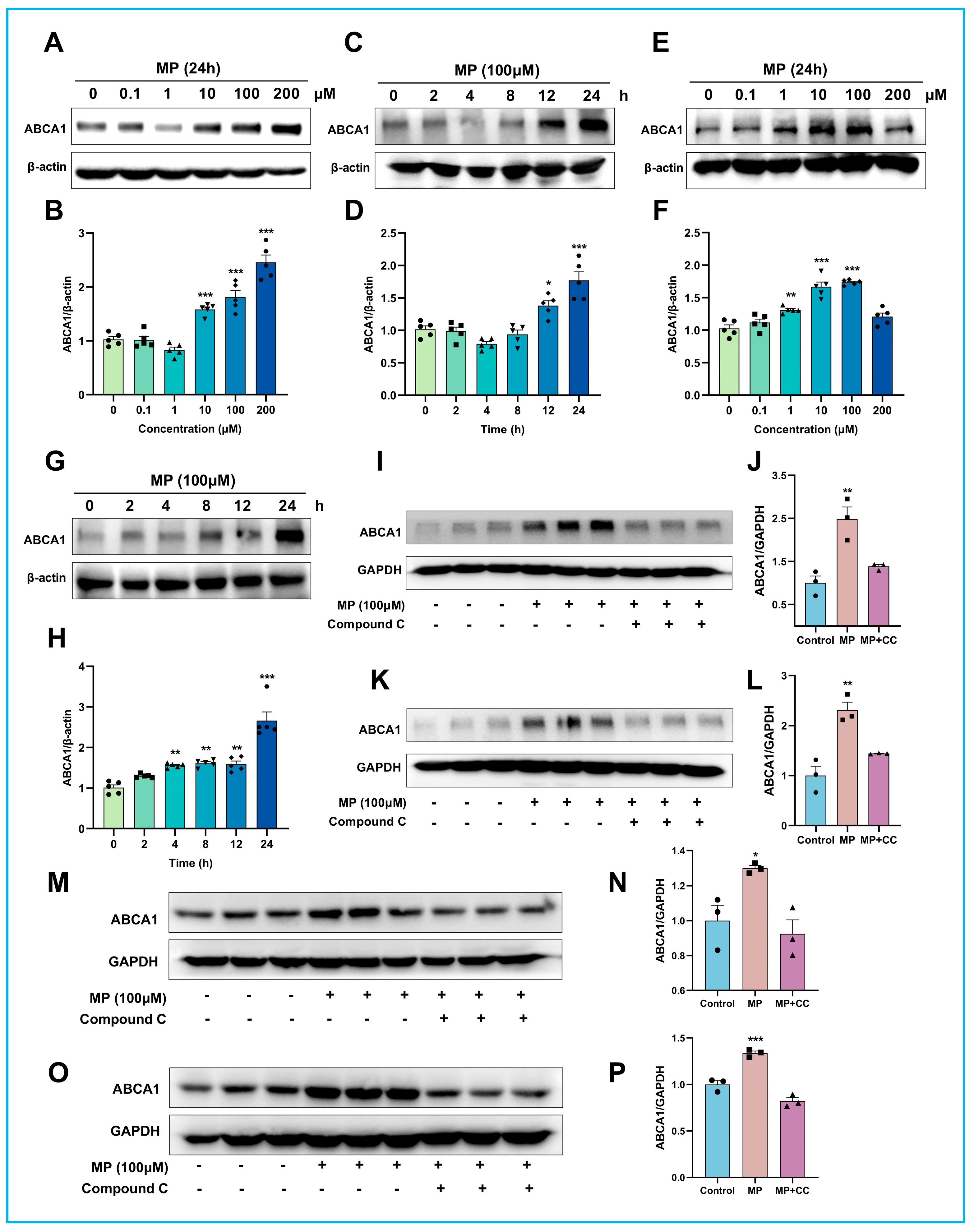
Secondly, we addressed the second question “how are these effects generated?” through cell culture-based studies. AMPK is widely recognized as crucial for the sensitive regulation of cellular energy metabolism, the regulation of ABCA1 expression, and the acceleration of cholesterol efflux from macrophages [
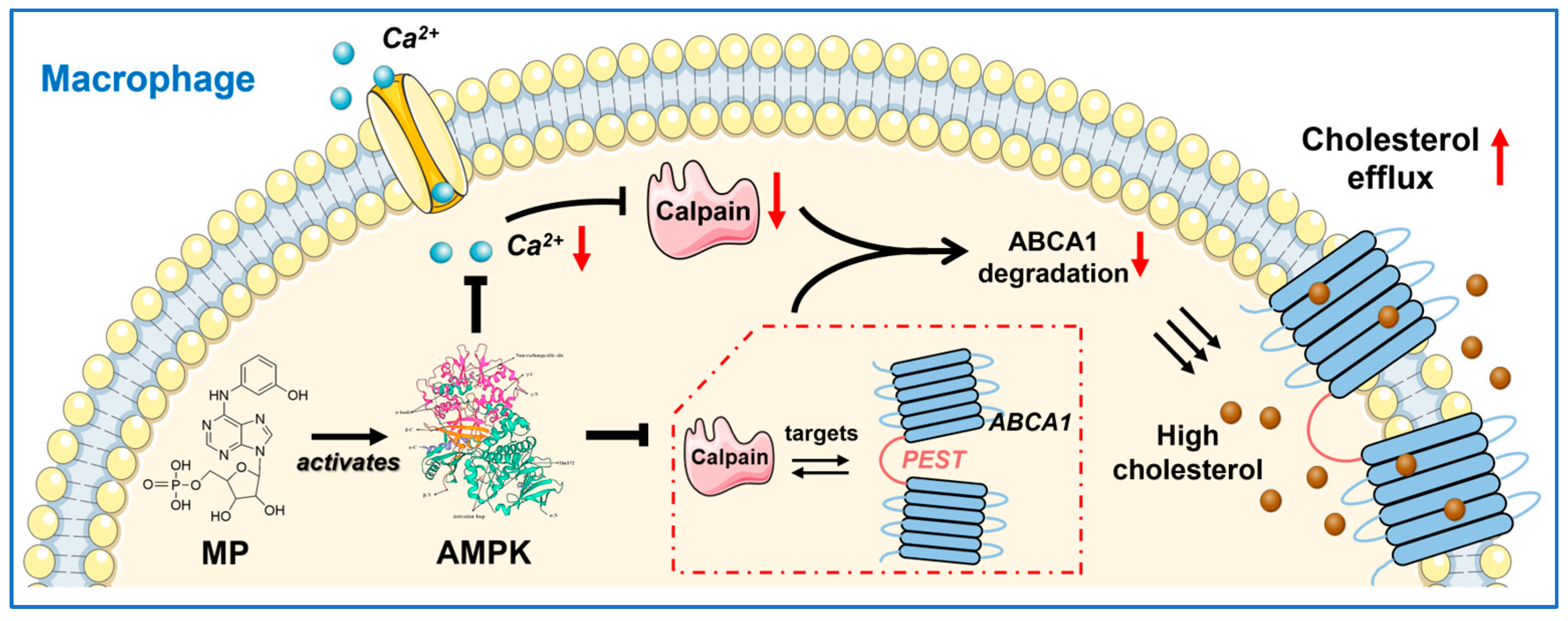
31]. In this study, we not only validated the notion that AMPK activation is fundamental to MP's promotion of ABCA1 expression but also elucidated the cascade between AMPK activation (initial event) and the subsequent reduction in ABCA1 degradation (endpoint event) (
Figure 7). Given that ABCA1 is the key mediator facilitating macrophage cholesterol efflux, constituting the first and rate-limiting step in RCT [
36,
37], the findings presented here may suggest a potential candidate molecule for cholesterol-lowering drugs targeting ABCA1. However, several issues still require attention in future research. For instance, (1) current research on the biological effects of MP is limited to the cellular level, and in vivo studies are necessary to further characterize its performance; (2) the mechanism by which MP inhibited the interaction of calpain with ABCA1 differs from the previous conclusion that promoted the dephosphorylation of PEST sequences [
20], and there may be a new mechanism that remains to be explored.
In summary, we have developed an AMPK agonist, MP, that not only inhibits extracellular Ca
2+ influx by targeting the Cav1.2 channel but also lowers intracellular Ca
2+ concentration. This, in turn, inhibits calpain-mediated ABCA1 degradation. Additionally, it weakens the interactions between calpain and ABCA1, as illustrated in
Figure 7. Collectively, these effects synergistically contribute to the reduction in ABCA1 degradation.
4. Materials and Methods
4.1. Reagents and Antibodies
MP was provided by the Institute of Material Medica, Chinese Academy of Medical Sciences (higher than 99.5% purity by HPLC). Reagents for cell culture, including Dulbecco’s modified Eagle’s medium (DMEM), RPMI 1640 medium, phosphate buffer saline (PBS) buffer (1×), penicillin-streptomycin mix (100×), and phorbol 12-myristate 13-acetate (PMA) were purchased from Beijing Solarbio Science & Technology Co., Ltd.(Beijing, China). Dimethylsulfoxide (DMSO) and Bovine Serum Albumin (BSA) were purchased from Sigma. Fetal bovine serum (FBS) and trypsin were purchased from Corning Incorporated (Shanghai, China). TRIzol reagent was obtained from ThermoFisher Scientific (Waltham, MA, USA). Reagents for preparation of recombinant full-length AMPK heterotrimeric complexes, including agar, isopropyl-beta-D-thiogalactopyranoside (IPTG), ethylene diamine tetraacetic acid (EDTA), protease inhibitor cocktail, and coomassie brilliant blue powder (CBB) were provided from Beyotime Biotechnology (Shanghai, China). Tryptone and yeast extract were purchased from ThermoFisher Scientific (Waltham, MA, USA). Sodium chloride, imidazole, and phenylmethanesulfonyl fluoride (PMSF) were purchased from Shanghai Macklin Biochemical Technology Co., Ltd. (Shanghai, China). Glycerol, chloropropanol, ethanol, and acetic acid were purchased from Beijing InnoChem Science & Technology Co., Ltd. (Beijing, China). Ampicillin and chloramphenicol were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) was obtained from Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China). Deoxyribonuclease I (DNase I) was provided from Gene-Protein Link Biotech (Beijing, China). Tris•Hcl (pH 8.0) was purchased from M&C Gene Technology (Beijing), LTD. (Beijing, China) Reagents for Electrophysiological recordings, including zeocin, G418, and hygromycin B, TEA-Cl, CsCl, EGTA, Na2-GTP, Mg-ATP, CsOH were provided from ICE BioScience Co., Ltd. (Beijing, China). Inhibitors, including Compound C, SKF96365, BAPTA-AM, 2-Aminoethyl Diphenylborinate (2-APB), and MG-132 were purchased from Selleck Chemicals (Shanghai, China). Ionomycin was obtained from Beyotime Biotechnology (Shanghai, China). Reagents for phosphorylation sites analysis, including DL-dithiothreitol (DTT), iodoacetamide (IAM), formic acid (FA), acetonitrile (ACN), methanol, were purchased from Sigma (St. Louis, MO, USA). Protease was purchased from Promega (Madison, WI, USA). Ultrapure water was prepared from a Millipore purification system (Billerica, MA, USA).
The list of antibodies is as follows, anti-ABCA1 (ab18180, Abcam, Cambridge, UK), anti-Calpain (MA3-940, ThermoFisher Scientific, Waltham, MA, USA), anti-AMPK (D63G4, CST, Danvers, MA, USA), anti-p-AMPK (D4D6D, CST), anti-LXRα (BM5134, Boster, CA, USA), anti-β-actin (K200058M, Solarbio, Beijing, China), anti-GAPDH (D16H11, CST), and anti-ABCA1 for Co-IP (ab307534, Abcam). HRP-conjugated secondary antibodies (A0216 and A0208, Beyotime, Shanghai, China).
4.2. Cell Culture
J774A.1 and THP-1 cells were provided from the BeNa Culture Collection. J774A.1 cells were cultured in DMEM plus 10%FBS, penicillin (100 U/mL), and streptomycin (100 U/mL). THP-1 cells were cultured in suspension in RPMI 1640 medium supplemented with 10% FBS. PMA (100 ng/mL) was used to induce THP-1 cells to differentiate into macrophages. J774A.1 and THP-1 cells were maintained in a 37 °C incubator with 5% CO2. Cells used for the following experiments were 5–9 passages, reaching 80–90% confluence. All experiments were repeated three times.
4.3. Cell Viability Assay
The J774A.1 cells were equally seeded in a 96-well plate (1 × 10
5 cell/mL) and cultured for 24 h. After that, cells were treated with different concentrations (1, 2.5, 5, 10, 25, 50, 100, 200, 400 μM) of MP for 24 h to explore the suitable intervention concentration. Then, their viability was evaluated using CCK-8 (Gene-Protein Link Biotech, Beijing, China). Briefly, 100 μl serum-free medium plus 10% CCK-8 solution was added to each well and incubated for 2 h at 37 °C. Finally, the absorbance was detected at 450 nm using a microplate reader, and cell viability was calculated as follows:
4.4. Preparation of Recombinant Full-Length AMPK Heterotrimeric Complexes
4.4.1. Escherichia coli Culture and Protein Expression
The expression plasmid of full-length Rattus norvegicus AMPKα1β1γ1 was constructed in our previous work. A 6×His tag was introduced at the N-terminal end of the α-subunit to facilitate purification. The expression plasmid was transformed into Transetta (DE3) chemically competent cells (TransGen Biotech Co., LTD. Beijing, China), and transformants were selected on LB agar plates containing ampicillin (100 μg/mL) and chloramphenicol (33.3 μg/mL). Single colonies were used to inoculate 5 mL of LB medium containing 100 μg/ml ampicillin and 33.3 μg/mL chloramphenicol. A conical flask containing 1L LB medium supplemented with 100 μg/mL ampicillin and 33.3 μg/mL chloramphenicol was inoculated with 2 mL of the above overnight culture for large-scale expression and purification. Then, the Escherichia coli was grown in a shaker incubator with 220 rpm at 37 °C until the A600 reached 0.6. Protein expression was induced by 500 μM IPTG. At the same time, the incubator temperature was reduced to 16 °C, and the Escherichia coli was grown for an additional 18 h.
4.4.2. Protein Extraction
After expression, Escherichia coli was harvested and resuspended in 10 ml lysis buffer [20 mM Tris-HCl (pH 8.0), 150 mM NaCl, protease inhibitor cocktail, 1 mM PMSF, 5% glycerol]. The Escherichia coli suspension was sonicated on ice for 2 min at 50% power. Then, 100 μL DNase I was put in the suspension and placed on ice for 30 min. Insoluble material was removed by centrifugation at 18,000× g in a Backman centrifuge for 30 min at 4 °C.
4.4.3. Protein Purification
The supernatant was filtered using a 0.45 μm filter and then loaded on 5 mL His-tag protein purification beads (Solarbio, Beijing, China). Then, wash buffer [20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 100 mM imidazole, protease inhibitor cocktail, 1mM PMSF, 5% glycerol] was used to wash five times to remove the non-specifically bound substances. After that, a gradient elution procedure was applied to obtain the expected protein. The detailed imidazole elution concentrations were 150 mM, 200 mM, 300 mM, 400 mM, 500 mM, 500 mM, and 500 mM, elution buffer volume was 10 mL. Subsequently, fractions containing the expected protein were pooled based on 8% SDS-PAGE and CBB analysis and concentrated to 1 ml using an ultrafiltration tube.
Apart from affinity chromatography, we used HiLoad 16/600 Superdex 200 pg (Cytiva life sciences, Logan, UT, USA) to purify the target protein further. Elution buffer [20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, protease inhibitor cocktail, 0.5 mM PMSF, 1.5 mM TECP, 5% glycerol] was used to elute the expected protein, and elution volume was 180 mL. Fractions were collected manually according to the A280 signal and then pooled based on 8% SDS-PAGE and CBB analysis. Lastly, the protein was concentrated using an ultrafiltration tube and then measured the final concentration, stored in a −80 °C refrigerator.
4.5. MST Analysis
We used a MonolithTM RED-NHS second-generation protein-labeling kit (Nano Temper, South San Francisco, CA, USA) to label the protein samples. First, 100 μL purified AMPK protein (10 μM) was buffer exchanged using the labeling buffer NHS and A-column. The protein was then labeled with RED-NHS dye (final concentration 300 μM), incubated at room temperature for 30 min in the dark, and the labeled protein was collected. The tagged protein was then purified using B-column and stored in a −80 °C refrigerator for later use. The specific operation was carried out according to the kit instructions.
For MST detection, MP was diluted into 16 concentration gradients, as follows: 400,000 nM, 200,000 nM, 100,000 nM, 50,000 nM, 25,000 nM, 12,500 nM, 6250 nM, 3125 nM, 1562.5 nM, 781.25 nM, 390.63 nM, 195.31 nM, 97.66 nM, 48.83 nM, 24.41 nM, 12.21 nM. After that, the above MP work solutions were co-incubated with tagged AMPK protein (50 nM) at room temperature for 5 min in the dark, and then samples were loaded in the capillary for subsequent detection. MST data were collected by Mo Pico Microscale thermophoresis (Nano Temper, South San Francisco, CA, USA) and analyzed using MO Affinity Analysis software (3_v01).
4.6. ITC Analysis
Microcal PEAQ-ITC system was employed to reveal whether there was a direct interaction between MP and AMPK and the accompanying thermodynamic parameters. All experiments were conducted at 298.15 K, with 19 injections (the first injection volume was 0.4 μL, and the rest were 2 μL). The concentrations of AMPKα1β1γ1 and MP were 20 μM and 400 μM, respectively. The detailed experimental procedures were performed according to the Instrument program. The K
d, stoichiometry (N), ΔH, and ΔS were calculated from raw data by MicroCal PEAQ-ITC analysis software (v1.41). In addition, the ΔG was calculated using the following equation:
4.7. Nuclear Magnetic Resonance (NMR) Analysis
4.7.1. STD-NMR Experiments
MP and adenosine monophosphate (AMP, TopScience, Shanghai, China) were dissolved in DMSO to obtain a working solution at a concentration of 250 μM. AMPK was prepared in 0.01 M PBS buffer (pH = 7.0) with 10% D2O to give a working solution at a concentration of 7.5 μM. The complex ligand-protein molar ratio was 33.3:1. Samples were subjected to 1H NMR, water suppression 1H NMR, and STD NMR experiments at 25 °C.
4.7.2. Titration Experiments
Titration experiments were applied to explore whether there was a competitive relationship between AMP and MP binding to AMPK. The concentration of MP was fixed at 250 μM, and the concentration of AMP increased sequentially (31.25, 62.5, 125, 250, 500, 1000, 2000, 4000 μM).
4.7.3. Data Collection and Processing
STD-NMR experiments were performed on Bruker Avance 500 MHz NMR spectrometer (Swiss) using the Topspin 3.6.2 suite. The experimental parameters were as follows: pulse sequence was SCREEN_STD, spectral width was 16 ppm, central frequency (o1p, ppm) was 4.7 ppm, 0.73 ppm (off resonance), −40 ppm (on resonance), both relaxation delay and saturation time were set to 2 s, spinlock time (D29, msec) was set to 25 ms. All spectrograms were processed in Topspin 3.6.2 suite, and the STD NMR of protein-ligand was subtracted from the STD NMR of ligand and protein to obtain saturation transfer double difference (STDD NMR).
4.8. CD Spectroscopy Study
CD spectra were obtained from a JASCO-715 spectropolarimeter (Jasco International Co., Ltd., Tokiwa-machi, Chuo-ku, Japan) using a 1 mm path-length quartz cuvette. Experimental settings were set as follows: Measure range: 190–300 nm; Data pitch: 0.5 nm; Sensitivity: Standard; D.I.T.: 1 sec; Bandwidth: 1.00 nm; Start Mode: Immediately; Scanning Speed: 50 nm/min; CD Detector: PMT; PMT Voltage: Auto. The baseline was obtained using 0.4 μM AMPK solution in 40 mM phosphate buffer (containing 10 mM NaF, pH 7.4). CD titration experiments were performed at a fixed concentration of AMPK (0.4 μM) incubated with MP at increasing concentrations in phosphate buffer (containing 10 mM NaF, pH 7.4). AMPK/MP molar ratios were 1:1, 1:5, 1:10, and 1:20. Each spectrum was obtained by averaging three scans and subtracting the contribution from corresponding blanks. The results were given in mdeg. The percentages of secondary structures were performed utilizing the CD analysis software (v2.12.00 [Build 1]), and secondary structure changes of AMPK at different concentrations of MP were distinguished.
4.9. Total and Membrane Protein Extraction
Total proteins from J774A.1 and THP-1 cells were extracted using RIPA lysis buffer (Gene-Protein Link Biotech, Beijing, China) supplemented with protease and phosphatase inhibitors cocktail (Roche ACCU-CHEK Active, China), with detailed procedures as described in previous work [
38]. Cell membrane proteins were extracted using a membrane and cytosol protein extraction kit (Beyotime, Shanghai, China). Briefly, after washing with PBS, the cells were harvested by centrifugation at 600×
g for 5 min at 4 °C. Then, 500 μL of reagent A (with 1 mM PMSF) was added, and the cells were placed on ice for 15 min. Cells were sonicated for 20 s on ice, and the supernatant was collected by centrifugation at 700×
g for 10 min at 4 °C. Subsequently, the above supernatant was centrifuged at 14,000×
g for 30 min at 4 °C to pellet cell membrane debris. Add 120 μL of reagent B to the tube to resuspend the pellet, and then place it on ice for 10 min. Finally, the cell membrane proteins were collected by centrifugation at 14,000×
g for 5 minutes at 4 °C. All extracted proteins were assayed for concentration using the BCA protein assay kit (ThermoFisher Scientific, Waltham, MA, USA) and then stored at −80 °C for later analysis.
4.10. Quantitative RT-PCR Analysis
Total RNA in J774A.1 and THP-1 cells were extracted using TRIzol reagent. The detailed experimental procedures according to our previous work [
23]. Primers used in current experiments were designed by PrimerBank and checked by the oligonucleotide property calculator, as follows: mouse ABCA1: GCCTGGATCTACTCTGTCGC (Forward) and GCCATTGTCCAGACCCATGA (reverse), human ABCA1: CTCGGTGCAGCCGAATCTAT (Forward) and CACTCACTCTCGCTCGCAAT (reverse), LXRα: AGAGTCTTGGGTCGCCAGTA (forward) and CTGGAGCCCTGGACATTACC (Reverse), β-actin: GAGCACAGAGCCTCGCCTTT (forward) and TCATCATCCATGGTGAGCTGG (reverse). The transcript abundance was normalized to the β-actin mRNA level.
4.11. ABCA1 Degradation Analysis
To investigate whether MP can enhance the stability of ABCA1, the degradation rate of ABCA1 was analyzed in the current experiment. Briefly, THP-1 cells were equably seeded in a 3.5 cm dish (total 5 × 105 cells) and pretreated with either vehicle (RPMI medium with 1% BSA) or 100 μM MP for 24 h. After that, cells were washed with 1 × PBS and then maintained in 1% BSA medium with 20 μg/mL CHX (purchased from CST) for the indicated periods (15, 30, 60, 120, 240 min). The degradation of ABCA1 at various time points was examined by immunoblotting.
4.12. Calpain-Mediated Degradation Analysis
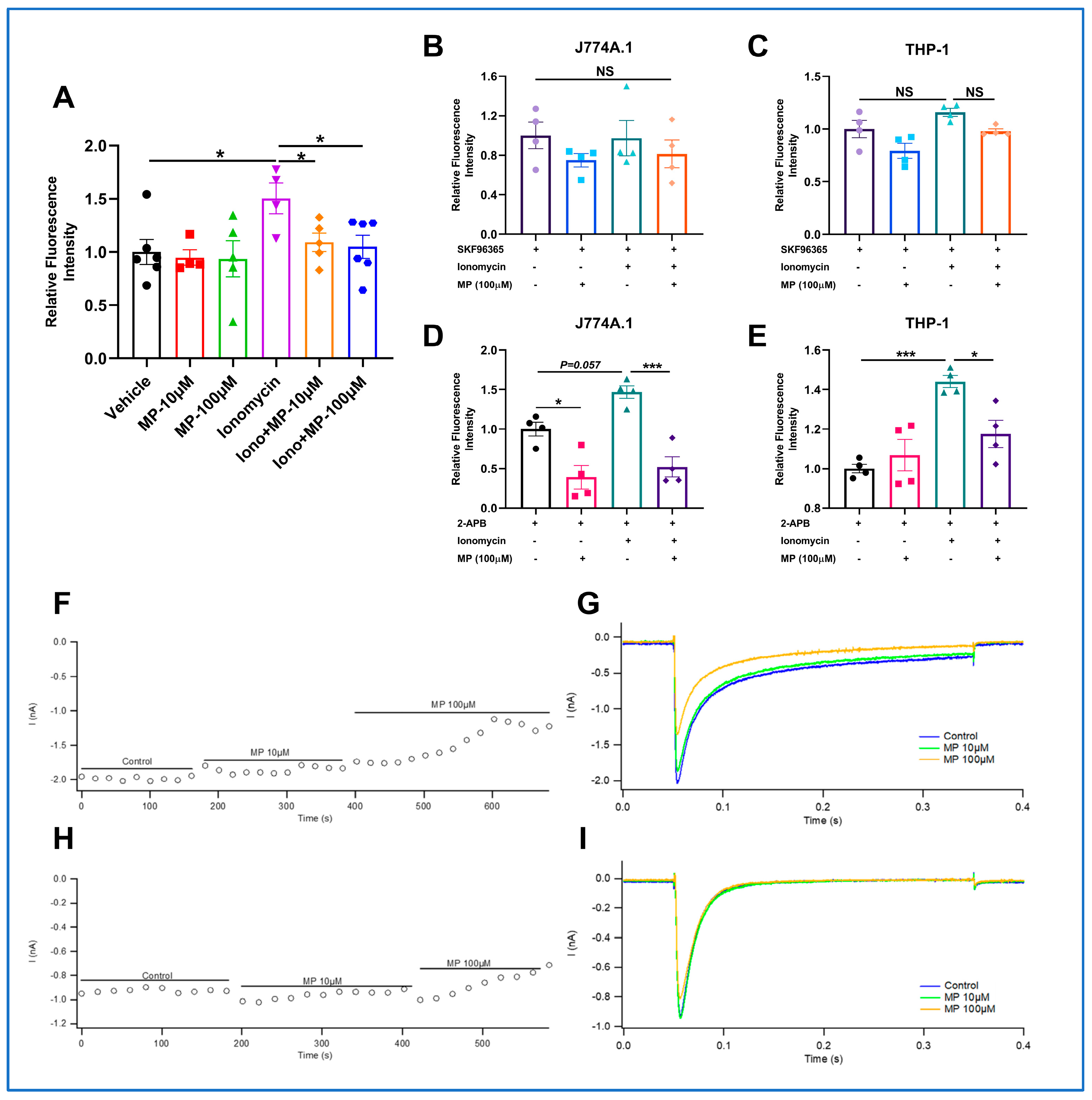
To confirm that ABCA1 can be degraded by calpain-mediated proteolytic pathway, THP-1 macrophages were treated with CHX plus increasing Ca2+ (0, 1, 10, 100, 1000, 2000 μM) with or without MP for 30 min after incubated with 10 μM BAPTA-AM for 3 h. The degradation of ABCA1 and the expression level of calpain were detected by immunoblotting.
4.13. Measuring Intracellular Ca2+ Level
Intracellular Ca2+ level was detected using the Fluo-4 AM kit (Solarbio, Beijing, China). THP-1 macrophages were first loaded with 5 μM of Fluo-4 AM working solution and incubated at 37 °C in the dark for 20 min. Later, quintuple Hank’s Balanced Salt Solution (HBSS, Solarbio, Beijing, China) containing 1% FBS was added and incubated for another 40 min to ensure intracellular transformation of Fluo-4 AM into Fluo-4. After gently washing thrice using HEPES buffer saline (Solarbio, Beijing, China), the fluorescence intensity was quantified using an Enspire® multimode reader (PerkinElmer, Singapore).
4.14. Electrophysiological Recordings
Cells used for electrophysiological recordings were provided by ICE BioSci. CHO cells and HEK cells were stably expressing the Cav1.2 channel and Cav3.2 channel, respectively. CHO cells were maintained in HAM’S/F-12 medium containing 10% FBS, 100 µg/mL Zeocin, 800 µg/mL G418, and 200 µg/mL Hygromycin B. Before patch clamp experiments, cells were separated using 0.25% trypsin-EDTA and treated by tetracycline for 24–72 h. HEK cells were maintained in DMEM containing 10% FBS and 200 µg/mL Hygromycin B. Before patch clamp experiments, cells were separated using 0.25% trypsin-EDTA and cultured in a 24-well plate for 18 h. The extracellular solution for whole-cell current measurement contained 140 mM TEA-Cl, 2 mM MgCl2•6H2O, 10 mM CaCl2•2H2O, 10 mM HEPES, and 5 mM D-Glucose. The pH was adjusted to 7.4 using TEA-OH. The internal pipette solution contained 120 mM CsCl, 1 mM MgCl2•6H2O, 10 mM HEPES, 10 mM EGTA, 0.3 mM Na2-GTP, 4 mM Mg-ATP, the pH was adjusted to 7.2 using CsOH.
The whole-cell patch clamp recordings were conducted using an EPC-10 patch-clamp amplifier (HEKA, Stuttgart, Germany), MP-285 Micromanipulator (Sutter Instruments, Novato, CA, USA), and PatchMaster software (HEKA, Stuttgart, Germany, v2×92). The cell membrane voltage was clamped at −80 mV after forming a whole-cell seal. The clamp voltage was maintained from −80 mV depolarized to +10 mV for 0.3 s. The data acquisition was repeated at 20 s intervals to observe the effect of the MP on the peak Cav1.2 current. Experimental data were collected by the amplifier and stored in the PatchMaster software (HEKA, Stuttgart, Germany, v2×92). The inhibition ratio of MP on the Cav1.2 channel was calculated as follows:
4.15. Co-Immunoprecipitation (Co-IP)
A commercial kit (absin) was applied for Co-IP experiments, and all procedures were performed according to the manufacturer’s instructions. In brief, protein lysates were collected from THP-1 macrophages and washed with 500 μL lysis buffer (containing 5 μL protein A/G agarose beads). The lysates were incubated with anti-ABCA1 antibody or control lgG overnight at 4 °C, followed by incubation with the protein A/G agarose beads for 7 h at 4 °C. Later, the pellets were collected, rinsed thrice with 1 × wash buffer, and resuspended in 1 × SDS loading buffer. The immunoprecipitated proteins were then eluted from the beads by incubation at 95 °C for 5 min. After centrifuging at 14,000× g for 1 min at 4 °C, the supernatant was obtained and stored at −80 °C for further detection.
4.16. Identification of Phosphorylation Modification Sites
4.16.1. Sample Preparation and Analysis
In brief, protein samples of ABCA1 were first extracted through Co-immunoprecipitation and then separated in 8% SDS-PAGE. The target gel containing ABCA1 was cut for subsequent experiments, including decolorization, dehydration, alkylation, digestion (trypsin and chymotrypsin enzymolysis), peptide fragment extraction, and Nano LC-MS/MS Analysis. The detailed procedures and experimental parameters were described in
Supplementary Materials.
4.16.2. Data Analysis
The raw MS files were analyzed and searched against the target protein database based on the species of the samples using Byonic. The parameters were set as follows: the protein modifications were carbamidomethylation (C) (fixed), oxidation (M) (variable), Phospho (S, T, Y) (variable), the enzyme specificity was set to trypsin or chymotrypsin; the maximum missed cleavages were set to 3; the precursor ion mass tolerance was set to 20 ppm, and MS/MS tolerance was 0.02 Da. Only highly confident identified peptides were chosen for downstream protein identification analysis.
4.17. Immunoblotting
The extracted proteins were separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to analyze the protein expression level. Proteins were then transferred to polyvinylidene difluoride (PVDF) membranes and blocked by 5% non-fat milk for 1 h at room temperature. After that, the whole membrane was rinsed with TBST to remove residual milk. Subsequently, bands of target proteins with large differences in molecular weight were cut and incubated with the corresponding primary antibodies, e.g., ABCA1 (254 kDa, 1:250 dilution), Calpain (80 kDa, 1:500 dilution), LXRα (51 kDa, 1:1000 dilution), β-actin (42 kDa, 1:3000 dilution), or GAPDH (37 kDa, 1:2000 dilution). After that, all bands were incubated with the appropriate HRP-conjugated secondary antibody and visualized using ECL reagents (Solarbio).
In addition, for target proteins with very small differences in molecular weight, e.g., AMPK (62 kDa, 1:1000 dilution), p-AMPK (62 kDa, 1:1000 dilution). After visualizing one of the target proteins, we incubated the bands for 30 min at room temperature on a shaker using a stripping solution to remove the bound antibody and re-performed the incubation with the blocking as well as the other antibody.
Finally, we visualized several small membranes cut from the same membrane separately and quantified the gray values of the protein bands to statistically analyze the relative protein expression levels (target protein/β-actin or GAPDH).
4.18. Statistical Analysis
All the abovementioned experiments were repeated three times for reproducibility. Data were processed using GraphPad Prism 8.3.0, and results were displayed as mean ± SD or mean ± error. Student’s t-test evaluated differences between two groups, and differences between three or more groups were evaluated by one-way analysis of variance (ANOVA). Dunnett’s multiple comparisons test and non-parametric tests were utilized in the present study when data were normally distributed or not normally distributed, respectively. Statistical significance was considered when p-value < 0.05. (* p < 0.05, ** p < 0.01, *** p < 0.001).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}