1. Introduction
Critical limb ischemia (CLI), the most severe manifestation of peripheral arterial disease (PAD), is characterized by rest pain, ischemic ulceration, non-healing wounds, and foot gangrene, and is associated with a 6-month mortality rate of 20% and a 5-year mortality rate of 50% [
1,
2]. In addition, CLI is associated with a high risk of lower limb amputation, in approximately 10% to 40% of patients [
3]. The treatment of CLI usually involves a combination of medical management, minimally invasive procedures, and surgical interventions to improve blood flow to the affected limb, relieve pain, promote wound healing, and prevent amputation [
4,
5]. Endothelial cell (EC) dysfunction is one of the key mechanisms contributing to tissue ischemia in CLI [
6]. As a potential treatment for CLI that addresses EC dysfunction, ASCs have shown promise, but further research is required to gain a deeper understanding of the mechanisms involved and to determine the best administration, dosing, and outcomes for CLI [
7].
Both in vivo and in vitro studies have shown that ASCs can improve healing rates and shorten healing times in critical limb ischemia [
8,
9,
10]. ASCs are mesenchymal stromal cells (MSCs) that are obtained from abundant adipose tissue, adherent on plastic culture flasks, can be expanded in vitro, and have the capacity to differentiate into multiple cell types [
11]. Although discovered only 20 years ago, stem cell therapy using ASCs has been extensively used to treat several diseases [
12]. ASCs have recently gained significant attention in the realm of regenerative medicine due to their accessibility, abundance, and multilineage differentiation potential [
13]. Clinically, ASCs have been explored for various applications. One of the hallmark applications of ASCs is in wound healing, where their ability to modulate inflammation and enhance angiogenesis has been harnessed to promote tissue regeneration [
14]. In orthopedics, ASCs have shown promise in the treatment of osteoarthritis, demonstrating potential for cartilage repair and anti-inflammatory effects [
15]. Cardiovascular medicine has also tapped into the potential of ASCs for treating conditions like myocardial infarction, given their capacity to promote neovascularization and inhibit cardiomyocyte apoptosis [
16]. Furthermore, ASCs are being investigated for their immunomodulatory properties in graft-versus-host disease and autoimmune disorders [
17].
In a hypoxic environment, EC dysfunction and endoplasmic reticulum (ER) stress are closely related and can contribute to the progression of the disease [
18]. A better understanding of the link between ER stress and endothelial dysfunction in CLI opens up potential therapeutic avenues, which could help reduce endothelial dysfunction and thus help to restore blood flow [
19]. Hypoxia disrupts calcium homeostasis and induces ER dysfunction and protein folding [
20]. Consequently, the accumulation of numerous misfolded proteins in the ER lumen generates ER stress [
18]. Eukaryotic cells have evolved an unfolded protein response (UPR) to ensure the fidelity of protein folding and prevent the accumulation of unfolded or misfolded proteins [
21]. Mammalian cells recruit three UPR signaling cascades, initiated by three ER-localized protein sensors: inositol-requiring enzyme 1α (IRE1α), double-stranded RNA-dependent protein kinase (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [
19,
22,
23]. Under normal physiological conditions, these sensors are associated with abundant ER chaperone BIP (immunoglobulin-heavy-chain-binding protein, also known as HSPA5 and GRP78) and remain inactive [
19]. Their activation reduces the accumulation of unfolded proteins and accelerates the rate of protein folding in the ER lumen [
22]. The downstream transcriptional programs of the UPR act to restore proteostasis. IRE1α increases the protein-folding capacity of the ER, assists in endoplasmic-reticulum-associated protein degradation (ERAD), and limits the entry of new protein molecules (regulated Ire1-dependent decay [RIDD]); PERK induces the transcription of genes and escapes translation inhibition under ER stress; and ATF6 increases the ER capacity to activate transcriptional programs and direct the misfolded proteins for degradation (ERAD) [
19,
22,
23]. UPR-induced genes include those that increase the protein-folding capacity of the ER and mediate its expansion by increasing the biogenesis of ER and lipid components [
23]. However, if restoration of proteostasis fails and ER stress is unabated, the UPR signaling switches to a pro-apoptotic mode, a process known as the terminal UPR [
22]. In light of all these data, we investigated if and how ASCs modulate hypoxia-induced ER stress by regulating Ca
2+ homeostasis in vitro and in vivo.
In view of this, our current study first uses both an in vitro assay and a murine in vivo model of acute-on-chronic ischemia to investigate the role of ASCs on potential hypoxic injuries to endothelial cells. Moreover, we especially focus on the role of UPR and Ca2+ homeostasis in endothelial cells.
3. Discussion
In this study, we have demonstrated that in the ApoE
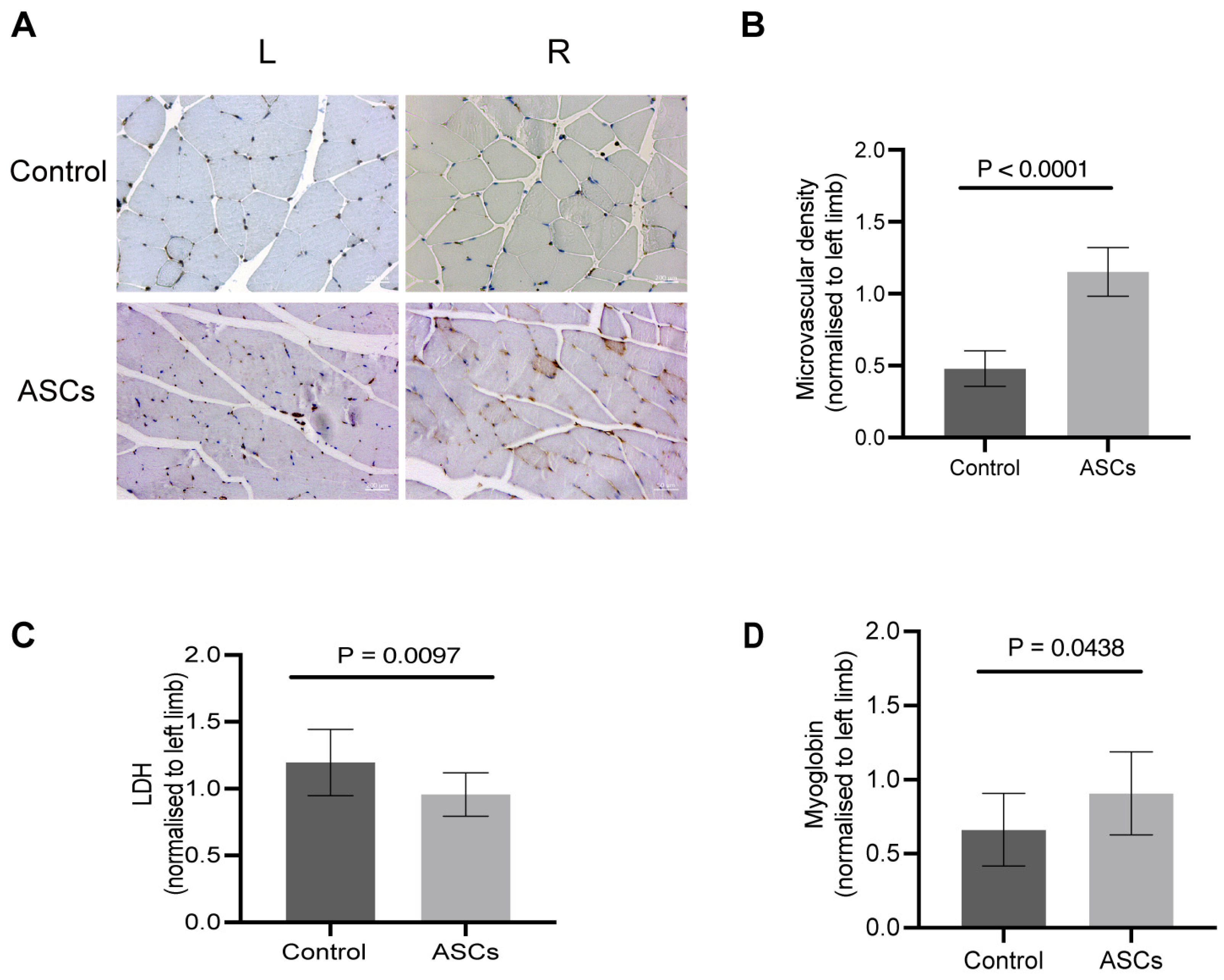
−/− PAD mice model, the xenogeneic ASC treatment improved functional recovery within seven days after surgery. A week after the generation of critical limb ischemia, the ASC-treated mice lost significantly less weight in comparison to the control mice. The histological analysis of the microvascular density in bilateral gastrocnemius muscle of ASC-treated mice showed enhanced angiogenesis. Considering that capillaries supply the nutrients (oxygen, glucose, etc.) and dispose of cellular waste products, microvascular density (MVD) is a crucial prognostic factor for PAD [
24]. Patients with PAD and poor prognosis usually show low MVD. In our ApoE
−/− PAD mice model, the MVD ratio of the ASC treatment group was significantly higher than that of the control group, indicating that ASC treatment enhanced angiogenesis. ASC therapy–induced neovascularization is conducive to restoring metabolic homeostasis, thereby improving functional recovery. Additionally, the average LDH levels of the muscles in the ASC group were lower than those of the muscles in the control group; moreover, these results were statistically significant, indicating that ASCs could improve metabolic homeostasis. In this study, we measured LDH in homogenized muscle tissues rather than the conventional serum-based approach. This method allowed us to directly assess muscle cell damage at the tissue level, capturing immediate LDH releases from injured cells. Moreover, the use of xenogeneic transplantation in our study, injecting human ASCs into mice, bridges the critical gap in interspecies cell therapy dynamics and its therapeutic implications, enabling novel approaches in regenerative medicine.
To study the underlying cell biological mechanisms, we established an in vitro hypoxic environment that reflected the levels of cellular, biochemical, and molecular hypoxia responses. We applied a chemically induced hypoxia model instead of a gas model which requires a hypoxic chamber being aware that this can mimic only a minor part of gas hypoxia [
25]. A commonly used chemical to induce hypoxia is CoCl
2, which strongly stabilizes HIF-1α and HIF-2α under normoxic conditions for several hours in a dose- and time-dependent manner [
26].
In the mimicked hypoxic environment, the MTT analysis revealed that CoCl2 significantly reduced the cell viability of HUVECs, whereas ASC-CM and ASC-CO reversed this effect, indicating that ASCs promoted the metabolic activity of ECs under hypoxia. Migration assays revealed that CoCl2 reduced the migration rate. Again, ASCs indirectly or directly accelerated the migration rate of ECs in the ASC-CO group. The results of the endothelial tube formation assay showed that the number of meshes, and junctions was significantly reduced following CoCl2 treatment. Correspondingly, the effect was significantly reversed by ASC-CM and ASC-CO following CoCl2 treatment. Furthermore, CoCl2 increased the levels of ROS; this effect was reversed by ASC-CM and ASC-CO therapy in 24 h. The therapeutic effect of ASC-CM was exerted earlier than that of ASC-CO. ASC-CO conditioned medium produced delayed effects, which may be attributed to the time taken by the ASCs to exert a paracrine effect on ECs in the co-culture.
Thus, in summary, ASCs accelerated the migration, tube formation ability, and metabolic activity of ECs while they reduced the production of ROS under hypoxia. This indicates that they may promote the angiogenesis ability of ECs. Our results therefore showed that ASCs, directly or indirectly, promoted the survival and the angiogenic functions of ECs under hypoxia.
ER, a ubiquitous organelle, is responsible for the synthesis, proper folding, maturation, and assembly of proteins before these are further processed by the Golgi apparatus. Stable ER Ca
2+ concentration or homeostasis is crucial to maintaining cellular functions. ER Ca
2+ depletion causes misfolding or unfolding of proteins, resulting in their accumulation within the ER lumen. This in turn causes ER stress and activates the UPR [
27,
28]. UPR is a normal adaptive and protective mechanism to reduce the rate of protein synthesis, increase the folding ability of proteins, and help misfolded or unfolded proteins to enter cellular degradation pathways [
23]. Nevertheless, non-resolved ER stress can cumulatively cause cell death [
19,
22]. Mungai et al. and Gusarova et al. demonstrated the existence of this pathway under hypoxia in osteosarcoma cells and alveolar epithelial cells [
29,
30]. We therefore investigated how CoCl
2-induced hypoxia affects the Ca
2+ homeostasis in HUVECs and how this is thereafter regulated by ASCs. It is well known that hypoxia reduces the ER Ca
2+ restoring ability by increasing the release of ER Ca
2+. In parallel, an influx of extracellular Ca
2+ is induced, consequently increasing the cytoplasmic Ca
2+ concentrations [
31]. The overloading of cytoplasmic Ca
2+ subsequently induces cell dysfunction and apoptosis, indicating prolonged or severe hypoxia [
21]. The imbalance in the interaction between cytoplasmic Ca
2+ and ER Ca
2+ is a sign of ER stress, which may affect the survival of the endothelium.
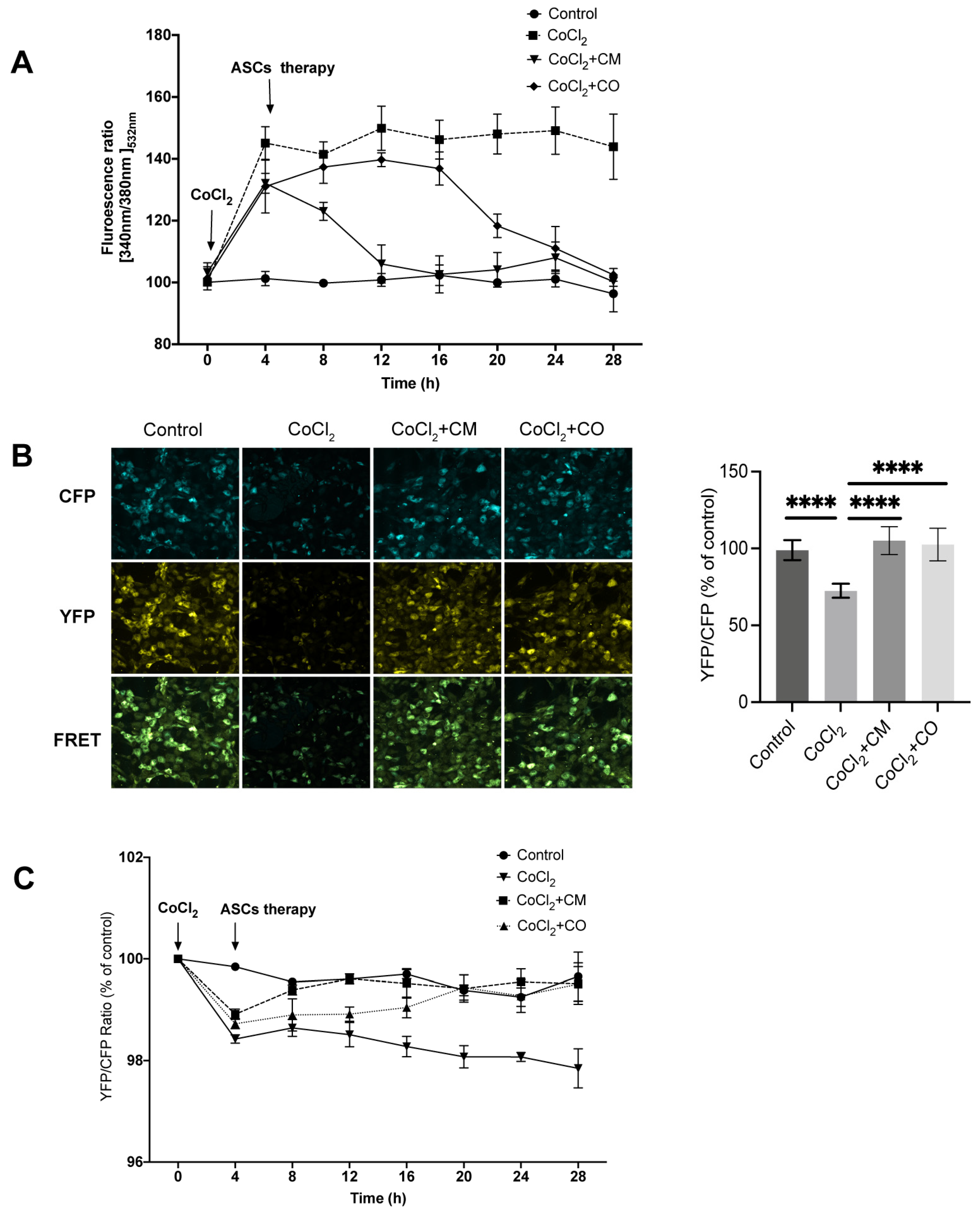
We used fluorescent sensor assays and functional microscopy to analyze the FRET ratio to detect Ca2+ levels. After 4 h of CoCl2 treatment, the HUVECs had a significantly reduced FRET ratio, reflecting a decrease in ER Ca2+ under hypoxia. ASC-CM and ASC-CO treatment for 24 h reversed this effect, with the therapeutic effect of ASC-CM occurring earlier than that of ASC-CO. The results of FRET microscopy showed that the FRET ratio of HUVECs treated with CoCl2 was significantly lower than that of control cells. Dynamic changes in cytoplasmic Ca2+ were detected by fluorescence using Fura-2AM. Compared with the control group, the Fura-2 ratio of the CoCl2 group increased significantly after 4 h of CoCl2 treatment due to CoCl2-induced elevation in cytoplasmic Ca2+. However, the therapeutic effect of ASCs reduced the ratio in the following 24 h, although the impact from ASC-CM was displayed earlier than that of ASC-CO. These results implied that CoCl2 mimicking a hypoxic pathological condition of ECs results in a continuous decrease in ER Ca2+ and an increase in cytoplasmic Ca2+ due to reduced ER Ca2+ restoring ability. The ASC-conditioned medium and adipose co-culture group reversed the above calcium imbalance under the same conditions. These results also indicated that stem cells regulate ER stress.
Hypoxic stress induces global gene expression changes by altering the cell’s metabolic and angiogenic pathways and restoring oxygen homeostasis, thereby promoting cell survival [
32,
33]. A failure of these repair and adaptive mechanisms causes cells to modify their gene expression profiles and induce programmed cell death [
32,
34,
35]. However, these changes are accompanied by the deregulation of mitochondrial and ER functions, reflected by perturbations in protein folding and trafficking. Erratic protein folding activates another specific stress response pathway; the UPR promotes cellular survival by restoring endoplasmic and mitochondrial homeostasis via distinct signaling networks [
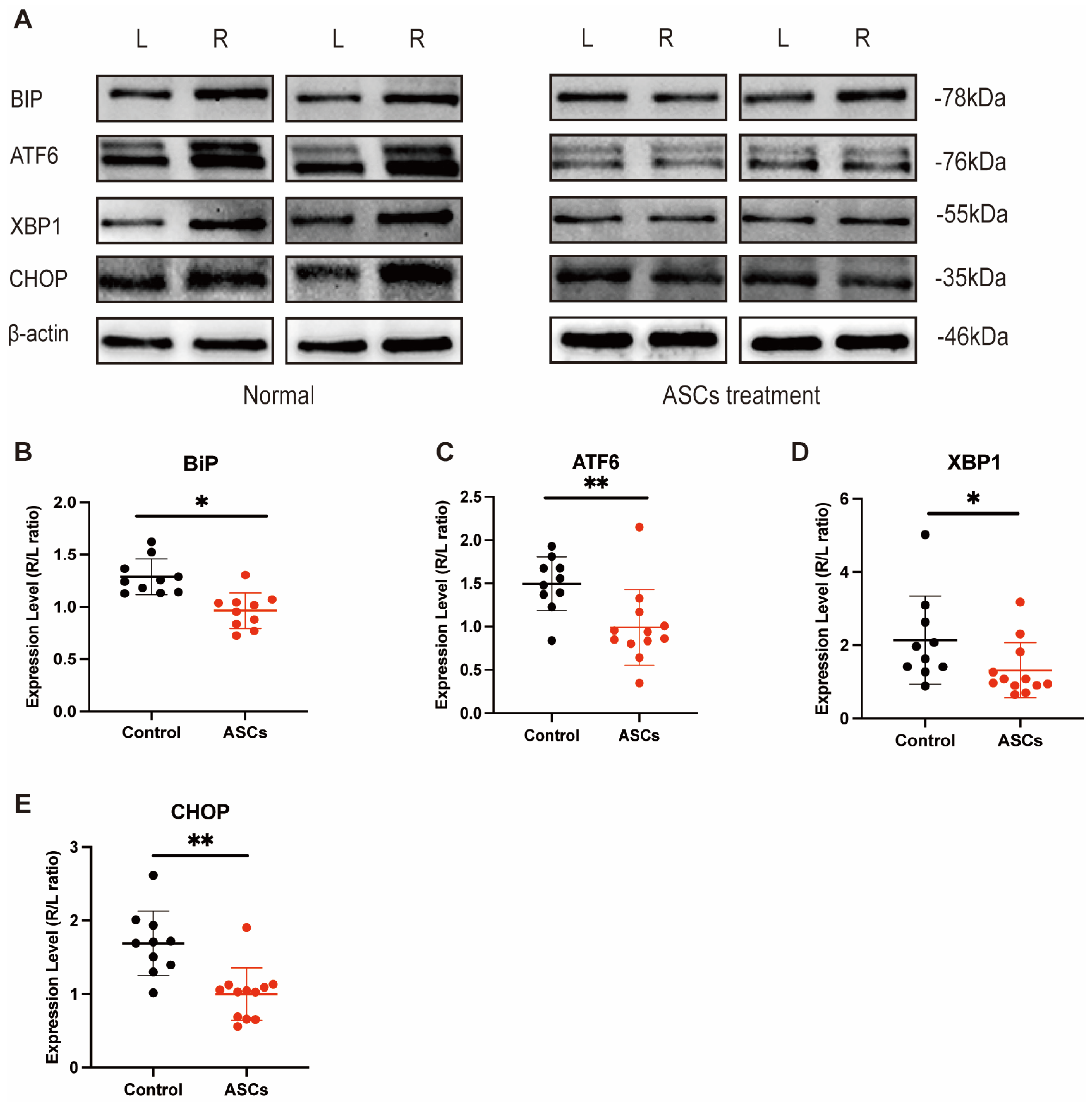
21]. Critical changes in mitochondrial functions occur during hypoxia, causing elevated ROS levels. Furthermore, the proper folding of mitochondria-encoded, as well as the import and corresponding refolding of mitochondrial nucleus-encoded proteins, is crucial for the proper functioning of this organelle. Hence, prolonged hypoxia eventually results in perturbations in mitochondrial protein folding and activation of a related specific stress response mechanism known as the mitochondrial UPR. To better understand the role of UPR in HUVECs under hypoxia we here investigated the expression of BiP, which initiates the UPR by dissociating three proteins, namely PERK, IRE1α, and ATF6, from the ER lumen. Our in vitro and in vivo results revealed that all ER stress-associated proteins (BIP, ATF6, CHOP, and XBP1) are activated once tunicamycin is processed. However, only BIP, ATF6, and XBP1 are activated under hypoxia. We observed that the expression of these proteins was reduced in the stem cell treatment group. Stem cells effectively alleviated endothelial dysfunction under hypoxic conditions by strengthening ATF6 and initiating a transcriptional program to restore ER homeostasis, induce BIP expression, promote protein chaperones and lipid synthesis, stimulate ER degradation, and enhance N-glycosylation of XBP1 by increasing the ER’s folding capacity, as well as increasing the expression of chaperones and proteins involved in ER-associated degradation (by ER degradation-enhancing α-mannosidase-like protein [EDEM]) and vesicular trafficking. The inactivation of CHOP could potentially be attributed to the fact that the CoCl
2 concentration used was insufficient to activate it, leading to cell death.
In summary, we first explored how ASC stems can mediate protection against hypoxic injury, specifically by investigating their role in suppressing terminal UPR. Our findings with human umbilical-vein endothelial cells (HUVECs) are in line with previous studies that demonstrate the therapeutic potential of stem cells in ameliorating hypoxic conditions [
36]. The utilization of 80 μM Cobalt (II) chloride (CoCl
2) in our experiments to induce a hypoxic environment, and the subsequent protective effects observed in stem cells, are consistent with the findings of Ejtehadifar et al. (2015) [
37]. Their study explored the influence of hypoxic conditions on mesenchymal stem cell biology, highlighting the adaptive and protective responses of these cells in low-oxygen environments. This similarity in findings underscores the robustness of mesenchymal stem cells’ protective mechanisms under hypoxia-induced stress conditions [
37]. Furthermore, the interplay between ER Ca
2+ dynamics and angiogenesis has previously been hinted at, but our findings offer a more direct link [
38]. In future works, it will be also intriguing to apply an ASC-conditioned medium into ischemic tissues using a similar in vivo setup.
Overall, our study does offer a fresh potential perspective on the adaptability and reliability of adipose-derived mesenchymal stem cells in mitigating cardiovascular maladies.
Limitations
First, we did not use hypoxic chambers to simulate true hypoxia; instead, we used CoCl2 to mimic hypoxic conditions. Therefore, our results are potentially incomparable with those observed in a truly hypoxic environment. An in vitro hypoxic model could be established using a hypoxic chamber in the future to overcome this shortcoming. Second, we have so far not correlated our results with potential paracrine effects of stem cells, such as growth factors and VEGF tracking, on ECs (which will be the topic of ongoing studies). Third, to investigate the relationship between ER Ca2+ recovery ability and EC angiogenesis function using D1ER and Fura-2 AM, we detected the dynamic changes in ER Ca2+ and cytoplasmic Ca2+ in only in vitro and due to the nature of these sensors—not in vivo. Thus, more studies are warranted in the future to further explore and explain the therapeutic mechanism of stem cells in more depth. Finally, it will be also intriguing to investigate the influence of an ASC-conditioned medium on the fate of ischemic muscular tissue.
4. Materials and Methods
4.1. Chemicals
All chemical reagents were purchased from Sigma Aldrich (Sigma-Aldrich Chemie GmbH, Munich, Germany) unless otherwise indicated. Treatment with 80 µM Cobalt (II) chloride (CoCl2) for a period of 4 h was used to mimic hypoxia in human umbilical-vein endothelial cells (HUVECs). Tunicamycin (TUN) 2.0 μg/mL was used to induce UPR in HUVECs.
4.2. Cell Culture
Human umbilical-vein endothelial cells (HUVECs) were isolated from freshly available umbilical cords provided by the Obstetrics Department of the University Hospital Mannheim. HUVECs isolation was approved by the local ethics committee (Medizinische Ethik-Kommission II, Medizinische Fakultät Mannheim, 2015-581N-MA, Mannheim, Germany) and was performed as previously described [
39]. The cells were grown in basal endothelial cell growth medium (Provitro GmbH, Berlin, Germany) supplemented with 2% fetal bovine serum (Gibco™ Thermofisher Scientific, Sao Paulo, Brazil) without antibiotics. HUVECs from three different donors were pooled, and the experiments were conducted till passage 7 at approximately 80−90% visual confluence. We have mixed cells from every three donors into one group.
ASCs were isolated from the visceral fat as described by S. Kern et al. [
40]. The adipose stem cells were extracted from human adipose tissue, and for each biological replicate samples from at least three independent donors were propagated for experimental use. The study was approved by the Mannheim Ethics Commission II (vote numbers 2010-262 N-MA, 2009-210 N-MA, 49/05 and 48/05). The cells were cultured in Dulbecco’s Modified Eagle’s Medium normal glucose (NG; 1 g/L, PAN Biotech; Aidenbach, Germany) supplemented with 10% FCS (Gibco™ Thermofisher Scientific, Brazil), 1% penicillin/streptomycin (Gibco™ Thermofisher Scientific, Frankfurt, Germany), and 2% L-glutamine (Gibco™ Thermofisher Scientific, Germany). Prior to use, all ASCs were assured to express the typical MSC immunophenotype and to differentiate into adipogenic and osteogenic lineages. The cells were then incubated with fluorescent-conjugated antibodies against CD73, CD90, CD105, and negative markers such as CD34 and CD45 for 30 min in the dark at 4 °C. Following the incubation, cells were washed and resuspended in PBS, and then subjected to flow cytometry analysis. Data acquisition was performed on flow cytometry (BD FACSCalibur) and data analysis was carried out using (FlowJo). ASCs were characterized by the positive expression of CD73, CD90, and CD105, and the negative expression of CD34 and CD45. Both HUVECs and ASCs were maintained at 37 °C in a 5% CO
2-humidified atmosphere. We have mixed cells from every three donors into one group.
4.3. Co-Culture and Conditioned Medium Preparation
Co-culture experiments were performed using ThinCert™ cell culture inserts suitable for 6- and 24-well plates (Greiner Bio-One, Frankfurt, Germany). 2 × 104 HUVECs per cm
2 were seeded in 6- and 24-well plates coated with 1% gelatin, and 0.6 × 104 ASC per cm
2 were seeded into ThinCert™ cell culture inserts (4.5 cm
2/insert, Greiner Bio-One, Germany, 657640) and 24-well plates (0.3 cm
2/insert, Greiner Bio-One, Germany, 657640). Co-cultures with ASCs (ASC-CO) were performed with a medium consisting of an ASC culture medium and an HUVEC culture medium mixed at a 1:1 ratio. The co-cultures were maintained at 37 °C in a 5% CO
2-humidified atmosphere [
41,
42,
43,
44]. For the ASC-conditioned medium (ASC-CM), ASCs were cultured until 70% confluency, the medium was discarded, and the cells were washed twice with PBS, followed by the addition of endothelial cell growth medium (2011101, Provitro, Germany) containing 1% penicillin and streptomycin. After 24 h, culture media were collected and centrifuged at 2500 rpm for 10 min at 4 °C and frozen at −80 °C for later use [
45]. The experimental outline is described in
Figure S3.
4.4. MTT Assay
2 × 104 HUVECs per cm2 in the log phase were collected and seeded on gelatin-coated 24-well plates. One-day post-seeding cells were first treated with CoCl2 for 4 h and afterward, either cultured in ASC-CM or co-cultured in ASC-CO for another 24 h. MTT dye solution (1 mg/mL in HUVECs medium) was added to the cells for 4 h, and formazan salt crystals were dissolved in 100 μL of MTT solvent (40% of Dimethyl sulfoxide (DMSO, HN47.1, Roth, Germany), 40% of 10%-sodium dodecyl sulfate (SDS, 0183.3, Roth, Germany), 20% of DPBS, and 1.2% of Acetic acid (6755.1, Roth, Germany)). The absorbance was read the next day at 540 nm (reference length = 630 nm) using a multi-mode microplate reader (Tecan Spark, Zurich, Switzerland).
4.5. Scratch Assay
2.6 × 104 HUVECs per cm
2 were seeded into 12-well plates. After overnight incubation, the cells were pre-treated with 80 µM CoCl
2 for 4 h. Using a 200 μL pipette tip, a single straight line was scraped. The wells were washed to remove lose and floating cells, and afterward, ASC-CM and ASC-CO were introduced with the continued presence of 80 µM CoCl
2. Images were acquired using a camera attached to an inverted microscope (DM IRB; Leica, Berlin, Germany) at 0 h, 3 h, 6 h, 12 h, and 24 h. The gap was quantitatively evaluated using the AutoCellSeg as described previously [
46].
4.6. Tube Formation
After 4 h of treatment with 80 µM CoCl2, the HUVECs were diluted with 1% FBS medium to 2.5 × 104 cells/mL and seeded into pre-coated 24-well plates (10,000 cells/well) in a Matrigel basement membrane matrix without phenol red (356237, Corning, NY, USA). Thereafter, the HUVECs were then, respectively, treated with ASC-CM or ASC-CO. After 6 h of incubation, images were acquired using an inverted microscope and analyzed using the Angiogenesis Analyzer software plugin (ImageJ version 1.5).
4.7. Virus Production and Transduction
4.7.1. roGFP3
Lentivirus particles were produced as described previously [
47]. HUVECs were then ftransduced with different concentrations of lentiviral particles for 48 h; the transduction efficiency was assessed by visualization of GFP. In transduced HUVECs, an optimal expression of GFP was observed at 1:100 dilution and thus this lentiviral concentration was used in further experiments. The stability of the expression over passages was also verified by qualitative assessment of GFP under the fluorescence microscope. In up to three passages after transduction, no evident GFP expression changes were observed.
4.7.2. D1ER
D1ER, a genetically encoded FRET-based ER Ca2+ biosensor that contains enhanced yellow fluorescent protein (YFP: Ex 514 nm/Em 527 nm) and enhanced cyan fluorescent protein (CFP: Ex 430 nm/Em 474 nm), was used to study changes in ER Ca2+. It was synthesized by Genewiz from Merck Sigma Aldrich in pUC57 with BamHI and XbaI restriction sites. The pUC57-D1ER was further cloned into pHR’SIN-cPPT-SEW 15 via BamHI and XbaI restriction sites. Lentivirus particles were produced as previously described, and HUVECs were transduced. The ratio of fluorescence resonance energy transfer was calculated by dividing CFP intensity by YFP intensity both detected from D1ER (FRET ratio = YFP/CFP). This ratio represented the dynamic changes in ER Ca2+ concentration.
4.8. Protein Isolation and Western Blotting
Western blotting (WB) was performed to detect the protein expression. The total protein lysate from HUVECs and mice hindlimb muscle tissue was mixed with loading buffer (161-0767, Bio-Rad, Frankfurt, Germany), denatured by incubation at 100 °C for 10 min, loaded onto 10% SDS-PAGE gels, and electrophoresed at 200 V for about 35 min. Afterward, proteins were transferred onto polyvinylidene fluoride (PVDF, 1620177, Bio-Rad, Germany) membranes. Hereafter, the PVDF membranes were blocked for 1 h in Tris-buffered saline with 0.1% Tween 20 (TBST) containing 5% non-fat milk. Membranes were next incubated overnight at 4 °C with primary antibodies against GRP78 (BIP) (1:5000, PA5-34941, Invitrogen, Bartlett, IL, USA), XBP1 (1:1000, PA5-27650, Invitrogen, USA), ATF6 (1:1000, PA5-20215, Invitrogen, Bartlett, USA), GADD153 (CHOP) (1:1000, NB600-1335, Novus Biologicals), phospho-IRE1 alpha (1:1000, PA1-16927, Invitrogen, Bartlett, USA), IRE1 alpha (1:1000, PA1-16928, Invitrogen, Bartlett, USA), and β-actin (1:1000, ab8227, Abcam, Frankfurt, Germany). Following the incubation with primary antibodies, PVDF was washed thrice with TBST (10 min each time) and incubated for 1 h at room temperature with a corresponding horseradish peroxidase (HRP)-conjugated anti-rabbit (Sc-2357, 1:1000, Santa Cruz, München, Germany) or anti-mouse (Sc-51610, 1:1000, Santa Cruz) secondary antibody. The membrane was washed again thrice with TBST (5 min each time) following the detection of immune reactive protein bands using an enhanced luminol reagent (1 min–5 min exposure, Western Lightning Plus-ECL, PerkinElmer, Waltham, MA, USA, 203-17431) and visualized by chemiluminescence (1 min to 5-min exposure). To detect the phosphorylation of IRE1 alpha, the PVDF membrane was stripped using the following protocol: the stripping buffer with 2-mercaptoethanol (M6250, Sigma-Aldrich, Burlington, VT, USA) was heated to 65 °C, and the PVDF membrane was immersed in the stripping buffer for 35 min with agitation. Following this, the PVDF membrane was washed extensively for 10 min in TBST twice. Afterward, it was blocked for 1 h in TBST containing 10% non-fat milk. After stripping, the PVDF membrane was incubated with primary and secondary antibodies and subsequently exposed as described earlier. Densitometric analysis was performed using the NIH ImageJ software (v1.52, Bethesda, MD, USA). All the lanes were normalized to β-Actin; in addition to normalized to β-Actin, the right leg/left leg ratio was used for the WB of the in vivo part.
4.9. Measurement of ER Ca2+
HUVECs transduced with the D1ER sensor were seeded into 1% gelatin-coated 24-well-plates (3.8 × 104 cells per well). 24 h post-seeding cells were treated with 80 μM CoCl2 for 4 h. Thereafter, the cells were either co-cultured with ASCs or cultured in ASC-CM for 24 h. The fluorescence intensity of CFP and YFP was detected by a Tecan multimode microplate reader every 2 h. In addition to live cell measurements, ER Ca2+ was also measured by confocal microscopy. To this end, HUVECs transduced with a D1ER sensor were seeded into 1% gelatin-coated 15 mm coverslips in 24-well-plates (3.8 × 104 cells per well), and the experiment was set up as described above. After 24 h treatment, the cells were fixed with 4% paraformaldehyde (PFA) and imaged by an SP5 microscope system (Leica, Germany) to detect the fluorescence emitted by YFP and CFP. To compare the FRET ratio among the different groups, the images were analyzed by NIH ImageJ version 1.52.
4.10. Measurement of Cyto Ca2+
Fura-2-acetoxymethyl ester (Fura 2-AM, Biotium, Fremont, CA, USA, 50033) was used to detect changes in the Cyto Ca
2+ as previously described [
48]. These experiments were performed in two formats: the cells were treated with 80 μM CoCl
2 for 4 h; after that, they were either co-cultured with ASCs or cultured in ASC-CM under treatment with 80 μM CoCl
2 for 24 h. The dynamic changes of Cyto Ca
2+ were detected by monitoring the fluorescence (Em 535 nm) from Fura 2-AM excited by wavelengths of 340 nm and 380 nm on the multimode microplate reader. The fluorescence intensity of Fura 2-AM was monitored by a Tecan multimode microplate reader every 2 h.
4.11. Measurement of ROS Signal
HUVECs expressing roGFP3 were seeded into 24-well-plates (1 × 106 cells/well). One-day post-seeding cells were first treated with either 80 μM CoCl
2 for 4 h. Afterward, the HUVECs were either co-cultured with ASCs or cultured in ASC-CM for 24 h, and treatment with 80 μM CoCl
2 was continued for 24 h. The dynamic changes to the ratio of fluorescence intensity emitted by roGFP3 were monitored on the Tecan multimode microplate reader as previously described [
47].
4.12. Hind Limb Ischemia
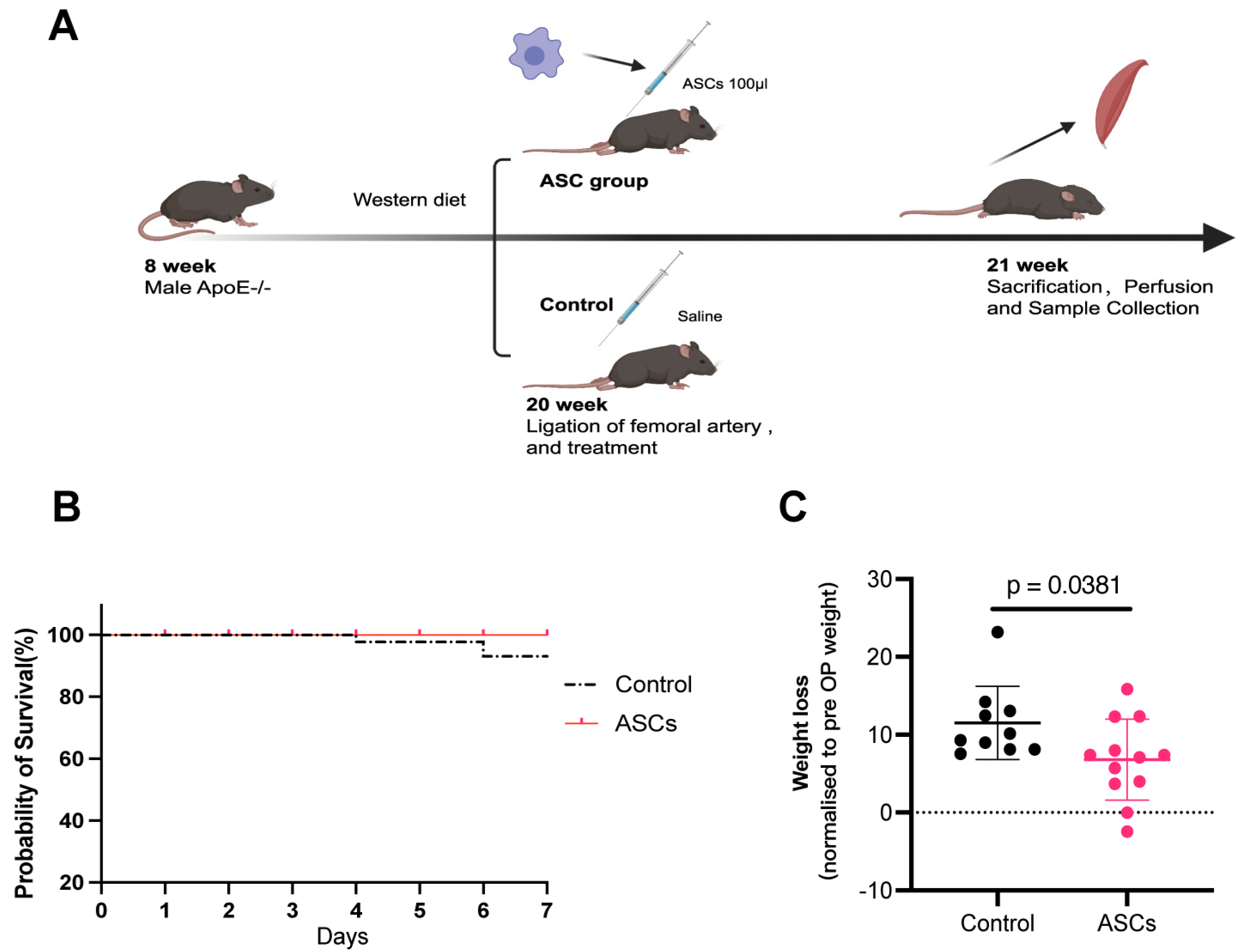
In this study, we employed the atherosclerosis-prone apolipoprotein E-deficient (ApoE
−/−) mice as our animal model. ApoE
−/− mice are a well-established model for atherosclerosis. Especially when fed on a Western diet, the mice spontaneously develop atherosclerotic lesions even when on a standard diet, and these lesions progressively exhibit features closely resembling those of human atherosclerosis. Twenty-four 8-week-old male ApoE
−/− mice were procured from the Charles River and fed a Western diet (5% cholesterol and 21% fat) for 12 weeks. All experimental procedures on animals were performed according to the EC guideline EC 2010/63/EU and have been approved by the local German government authority (35-9185.81/G[1]239/18). The mice had free access to food and water. Their cages, bedding, water bottles, and aspen shelters were changed weekly. Ambient temperature and relative humidity were kept at 22–23 °C and 50–70%, respectively. Room illumination was automatically set on a 12 h day/night lighting cycle, with lights on at 7:00 a.m. At 20 weeks of age, Hind limb ischemia (HLI) was induced as described in our recent report [
49]. Briefly, the mice were anesthetized with a subcutaneous injection (S.C.) of a mixture of midazolam (5 mg/kg, 44856.01.00, Ratiopharm, Ulm, Germany), medetomidine (0.05 mg/mL/kg, 128366-50-7, Cayman Chemical, Ann Arbor, MI, USA), and fentanyl (0.5 mg/kg, 437-38-7, Cayman Chemical, Hamburg, Germany) before all surgical procedures. After 6 min of anesthesia, when mice breathed slowly and stably, hairs on their lower limbs were removed using a depilatory cream (Veet, Heidelberg, Germany). A saline solution was injected subcutaneously to maintain sufficient fluid in the body. Next, the mouse was placed on a 37 °C heating pad and the skin was rubbed with an alcohol scrub. An incision of about 3 to 4 mm was made in the middle of the groin area using pointed-tip forceps and surgical scissors. To increase the surgical field of view, all surgical procedures were performed under a dissecting microscope (Axiovert 100, ZEISS, München, Germany). The superficial femoral artery (FA) was dissected from the femoral vein (FV) and femoral nerve (FN). The FA was ligated proximally and distally and excised. Afterward, the mice were subjected to a double ligation of the femoral artery (DLFA) using Ethicon 7-0 sutures (8776H, PROLENE, Lidingö, Sweden)to simulate an acute-on-chronic model of PAD, as described previously [
49]. After the DLFA, the mice were divided into two groups, which included the control and MSCs treatment groups (
n = 12, for each group) (
Figure 2). The animals in the control group received intramuscular 100 μL normal saline injected into 5 different muscle localizations, while the animals in the treatment group received 100 μL fresh from culture ASCs (1 × 10
8 cells/mL) suspension in the right hind limb (20 μL for each localization) immediately after the DLFA. With regards to postoperative analgesia, Butorphanol (1 mg/kg, S.C., Q8h, Intervet, Unterschleißheim, Germany, 42408-82-2) was given to the mice 24 h after the DLFA. Drinking water was supplemented with metamizole (24 mg/5 mL of water, corresponding to a dose of 200 mg/kg 4 times daily) to maintain the analgesic effect for 2 days following the DLFA procedure. One-day post-operation, magnetic resonance imaging (MRI) scans of the bilateral hind limbs were taken to document the perfusion situation of the proximal and distal femoral artery (FA) (
Figure S2).
4.13. Plasma and Tissue Collection
The animals were sacrificed 7 days after the DLFA. Mice were euthanized by injection using a combined anesthetic (MMF) half an hour before harvesting. Blood was collected from the inferior vena cava. Plasma was prepared by centrifuging the blood at 1000 rpm for 10 min at 4 °C and stored at −80 °C for biochemical analysis. Both sides of the fresh vastus lateralis (VL) and Gastrocnemius (GM) were rapidly removed after perfusion with 4% PFA and fresh samples were snap-frozen in liquid nitrogen and kept at −80 °C for further analysis.
4.14. Histology and IHC Staining
The histological analysis was carried out on the bilateral gastrocnemius muscle (GM) and the aorta with Hematoxylin & Eosin (HE) and CD31 staining via Immunohistochemistry (IHC) as described previously [
49]. ImageJ was used to estimate the percentage of microvascular area (CD31 positive area) in five randomly selected fields of view (×40).
4.15. Lactate Dehydrogenase (LDH) Assay
The relative levels of LDH in the vastus lateralis muscle (VL) tissue were detected using an LDH kit (ab197000, Abcam, Germany) according to the manufacturer’s guidelines. The 40 mg tissue was homogenized using a T18 digital homogenizer (IKA, Wilmington, NC, USA) at the highest speed of 25,000 rpm for 30 s after which it was kept on ice for 10 min. Subsequently, tissue lysate was centrifuged for 5 min at 4 °C at 10,000× g; the supernatant was then collected and added into 96-well-plates (each measuring 50 μL). Hereafter, 50 μL reaction mix supplied by the kit was added to each sample. Then, the fluorescence intensity at 535 nm/587 nm (Ex/Em) was immediately measured by a multimode microplate reader.
4.16. Myoglobin Assay
The relative level of Myoglobin (Mb) in the VL tissue was detected using the Mb kit (ab210965, Abcam, Germany) according to the manufacturer’s instructions. Briefly, 50 μL of tissue lysate supernatant obtained from homogenization of VL was added to each well of a 96-well plate and incubated at room temperature for 1 h. After washing, 100 μL of 3,3′,5,5′-tetramethylbenzidine developing solution was added to each well. The plate was incubated for 10 min and the stop solution (50 μL per well) was added. Absorbance was measured at 450 nm on a multi-mode microplate reader.
4.17. Statistical Analysis
Statistical analyses were performed using GraphPad Prism 8 (Graphpad Software, San Diego, CA, USA). The results for the different experimental groups were expressed as ± SD. The differences between groups were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc correction analysis. Throughout the analysis, a p-value < 0.05 was considered statistically significant.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}